- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

In vitro RNA Cleavage Assays to Characterize IRE1-dependent RNA Decay
(*contributed equally to this work) Published: Vol 9, Iss 14, Jul 20, 2019 DOI: 10.21769/BioProtoc.3307 Views: 8267
Reviewed by: Alexandros AlexandratosIndranil MalikJoe Zhang
Original research article
The authors used this protocol in:

Dec 2018
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
The kinase/RNase IRE1 is a key effector of the cellular response to endoplasmic reticulum stress. The RNase activity of IRE1 can be measured in cells or in the test tube. Here we describe a protocol for the in vitro cleavage and analysis of RNA substrates of IRE1. The method consists of the in vitro transcription, purification and re-folding of IRE1 substrate RNAs followed by their cleavage using recombinant cytosolic kinase/RNase domains of IRE1 and the separation of the resulting fragments by denaturing polyacrylamide gel electrophoresis. This protocol allows the study of the cleavage kinetics of IRE1’s RNA substrates in vitro.
Keywords: IRE1Background
Accumulation of un(mis)folded proteins in the endoplasmic reticulum (ER) causes ER stress and activates the unfolded protein response (UPR), an adaptive mechanism that maintains ER homeostasis (Reviewed in Karagöz et al., 2019). IRE1, a transmembrane ER stress sensor/transducer with cytosolic kinase and RNase activities, governs the most conserved UPR signaling arm, and it is found from yeasts to metazoans (Mori, 2009). ER stress leads to IRE1 oligomerization and activation of its C-terminal cytosolic RNase domain. IRE1 preserves ER homeostasis in two ways. First, by its most studied mechanism, IRE1’s RNase domain cleaves an intron found in the XBP1 mRNA to initiate an unconventional splicing event that activates the transcription factor XBP1 (Yoshida et al., 2001; Calfon et al., 2002; Peschek et al., 2015). Active XBP1S (“S” for spliced) controls the upregulation of genes that improve protein processing capacity of the ER (Lee et al., 2003; Acosta-Alvear et al., 2007). Second, IRE1 degrades ER-bound mRNAs in a process known as regulated IRE1-dependent decay (RIDD), to reduce the ER protein folding load or to initiate specific cytoprotective responses (Hollien and Weissman, 2006; Hollien et al., 2009; Bae et al., 2019). IRE1’s RNase output has been measured on a global scale through microarray-based gene expression analysis in wild-type and in IRE1-deficient cells (Hollien and Weissman, 2006; Han et al., 2009; Hollien et al., 2009; So et al., 2012). While this type of approach lends itself to the discovery of potential IRE1 substrates, it does not allow evaluating the direct engagement of IRE1 with potential substrates. Moreover, such global approaches can be costly and technically demanding. We and others have measured IRE1’s RNase output in mammalian cells using XBP1 mRNA splicing and RIDD reporter constructs (Iwawaki et al., 2004; Iwawaki and Akai, 2006; Sidrauski et al., 2013; Mendez et al., 2015; Moore and Hollien, 2015; Peschek et al., 2015), or by directly assessing the mRNA levels of endogenous spliced XBP1 mRNA or of validated RIDD targets by TaqMan and/or RT-PCR assays (Acosta-Alvear et al., 2007; Hollien et al., 2009; Sidrauski et al., 2013; Mendez et al., 2015; Peschek et al., 2015). While useful, these techniques do not allow precise time-resolved measurements of RNA processing by IRE1. Precise kinetic measurements of IRE1’s RNase activity require examining RNA cleavage in the test tube. We and others have measured the kinetics of XBP1 mRNA cleavage by IRE1 using short RNA substrates containing XBP1 mRNA stem-loops recognized by IRE1 (Korennykh et al., 2009; Lu et al., 2014; Mendez et al., 2015; Peschek et al., 2015; Acosta-Alvear et al., 2018). In cells, however, IRE1 is presented with cognate RNA structures embedded in full-length mRNAs. Since RNA folding is influenced by the size of the molecule and local sequence composition (Cheng et al., 2015), non-natural short RNA substrates containing putative IRE1 cleavage sites may contribute to false positive or false negative results in in vitro RNA cleavage assays. Radioactive in vitro cleavage assays for IRE1 RNA substrates of ~400-500 nucleotides in length have been performed (Lu et al., 2014; Tam et al., 2014), and while undoubtedly useful, the use of radioactivity could be limiting for many laboratories. With these considerations in mind, we have developed a non-radioactive protocol for analyzing the cleavage of long RNA substrates of IRE1 in vitro (Figure 1). Our method can be performed in the presence and absence of purified ribosomes to account for IRE1 activity in IRE1-ribosome complexes (Acosta-Alvear et al., 2018), it can be deployed to study the RNase activity of IRE1 and its consequences in the context of maintaining cell viability (Lu et al., 2014), and it can be useful for assessing the specificity of recently developed IRE1 inhibitors (Cross et al., 2012; Harnoss et al., 2018).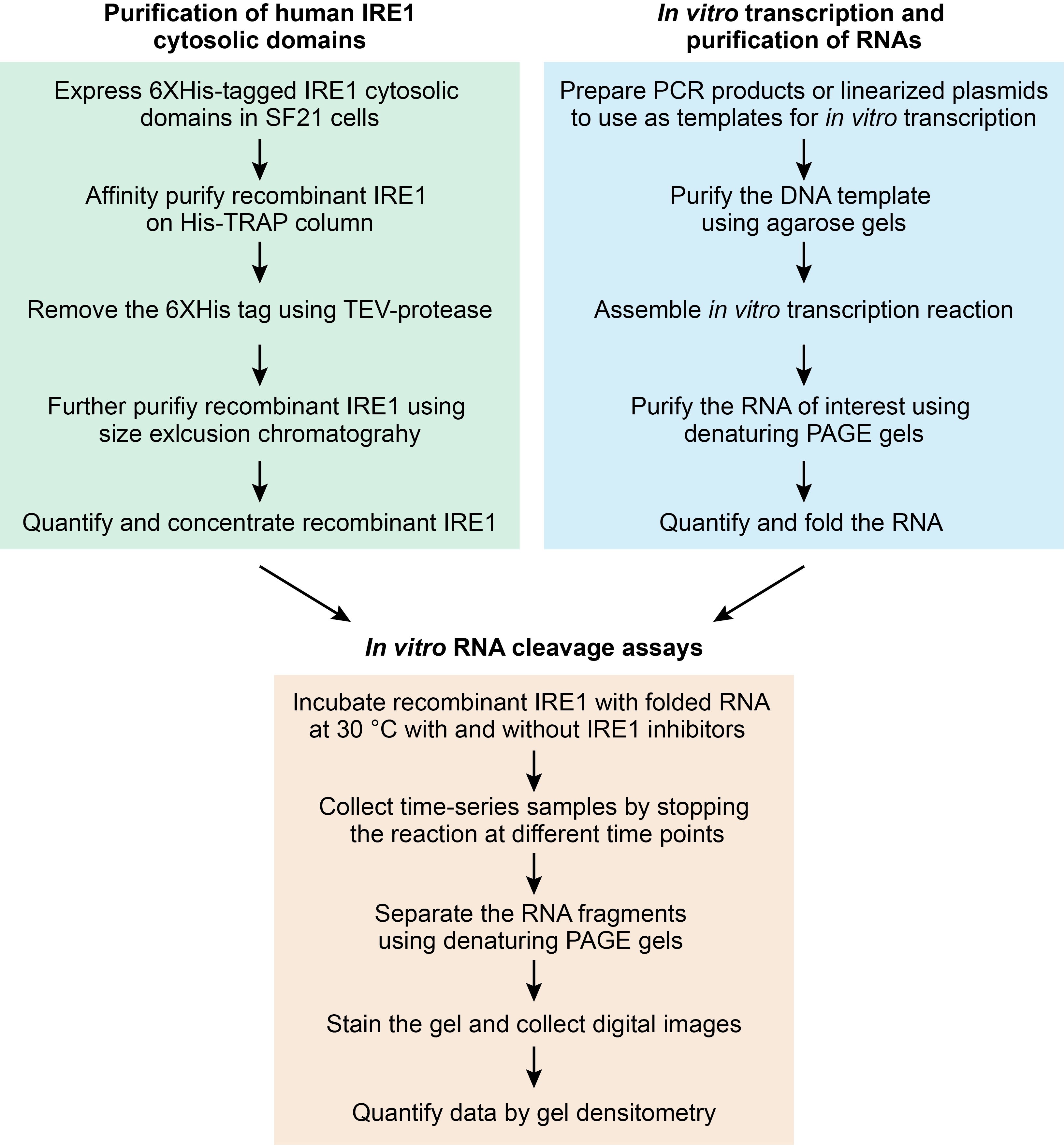
Figure 1. Protocol workflow
Materials and Reagents
- Purification of recombinant cytosolic domains of human IRE1
- Superdex 200 10/300 GL size exclusion chromatography column (GE Healthcare, catalog number: 17517501)
- 50 ml screw-cap conical tubes (various suppliers)
- 5 ml HisTrap HP histidine-tagged protein purification column (GE Healthcare, catalog number: 17524801)
- Size exclusion chromatography columns HiLoad® 16/600 Superdex® 200 (GE Healthcare, catalog number: 28989335), or similar
- Amicon Ultra-15 centrifugal filters (30 kDa MW cut-off; Mfr. Millipore, catalog number: 903008)
- SF21 cells (SFM adapted, Invitrogen, catalog number: 11497-013)
- Sf-900TM II SFM (Gibco, Thermo Fisher Scientific, catalog number: 10902096)
- Fetal bovine serum (FBS), heat inactivated (Gibco, Thermo Fisher Scientific, catalog number: 10082147)
- Antibiotic-antimycotic solution (100x), stabilized (Sigma-Aldrich, catalog number: A5955)
- Baculovirus expression construct encoding N-terminal hexahistidine tagged IRE1 of human origin bearing a 43 amino acid long portion of a linker domain located between the epitope tag and the kinase domain (IRE1-KR43, amino acids 521-977) as described in Li et al., 2010
- cOmplete EDTA-free protease inhibitor cocktail tablets (Mfr. Roche. Sigma-Aldrich, catalog number: 11873580001)
- TCEP-HCl (Sigma-Aldrich, catalog number: 75259)
- Tobacco-etch virus (TEV) protease (recombinantly expressed and purified from E. coli)
- HEPES (Sigma-Aldrich, catalog number: H3375)
- KCl, 1 M solution (Sigma-Aldrich, catalog number: 60142)
- MgCl2, 2 M solution (Sigma-Aldrich, catalog number: 68475)
- Imidazole (Sigma-Aldrich, catalog number: I5513)
- SF21 cell lysis buffer (see Recipes)
- IRE1 dialysis buffer (see Recipes)
- In vitro transcription, purification and refolding of IRE1 substrate RNAs
- Plastic cling-wrap
- Spin-X centrifuge tube filters, pore size 0.45 μm (Mfr. Corning Costar. Sigma-Aldrich, catalog number: CLS8162)
- Non-stick 1.5 ml RNase-free tubes (Mfr. Applied Biosystems., catalog number: AM12450)
- Non-stick 0.5 ml RNase-free tubes (Mfr. Invitrogen., catalog number: AM12350)
- Sterile No. 20 G syringe needles (Mfr. Beckton-Dickinson, catalog number: 305176)
- pUC19 (Addgene plasmid #50005 or Thermo Scientific, catalog number: SD0061)
- T7 oligonucleotide 5′-TAATACGACTCACTATAG(N14-21)-3′; where N corresponds to bases on the target gene of interest to amplify by PCR (Integrated DNA Technology)
- Phusion high-fidelity DNA polymerase (New England Biolabs, catalog number: M0530S)
- dNTPs solution (New England Biolabs, catalog number: N0447S)
- RNaseZAP RNase decontamination solution (Invitrogen, catalog number: AM9780)
- QIAquick PCR & gel cleanup kit (QIAGEN, catalog number: 28506)
- DNA clean and concentrator-5 kit (Zymo Research, catalog number: D4013)
- HiScribe T7 high yield RNA synthesis kit (New England Biolabs, catalog number: E2040S)
- RNase-free DNase I (New England Biolabs, catalog number: M0303S)
- Novex 6% TBE-Urea PAGE gels (Invitrogen, catalog number: EC6865BOX)
- Novex TBE-Urea 2x sample buffer (Invitrogen, catalog number: LC6876)
- Novex TBE-Urea 5x running buffer (Invitrogen, catalog number: LC6675)
- 3 M NaOAc pH 5.5, RNase-free (Mfr. Invitrogen, catalog number: AM9740)
- RiboRuler low range RNA ladder (Thermo Scientific, catalog number: SM1833)
- 80% ethanol in RNase-free water
- 70% ethanol
- Pure isopropanol
- RNase-free water (Ambion, catalog number: AM9930)
- Recombinant RNasin RNase inhibitor (Promega, catalog number: N251A)
- EDTA (Sigma-Aldrich, catalog number: EDS)
- HEPES (Sigma-Aldrich, catalog number: H3375)
- NaCl (Sigma-Aldrich, catalog number: S3014)
- MgCl2, 2 M solution (Sigma-Aldrich, catalog number: 68475)
- RNA gel extraction buffer (see Recipes)
- RNA re-suspension buffer (see Recipes)
- In vitro cleavage assay of in vitro transcribed RNAs with purified recombinant IRE1 cytosolic domains and denaturing urea-PAGE of IRE1-generated RNA fragments
- Non-stick 1.5 ml RNase-free tubes (Mfr. Applied Biosystems, catalog number: AM12450)
- Non-stick 0.5 ml RNase-free tubes (Mfr. Invitrogen, catalog number: AM12350)
- Purified recombinant IRE1 cytosolic kinase/RNase domains (IRE1-KR43, 50 nM to 5 µM per reaction)
- Purified, refolded substrate RNA (25-100 ng per reaction)
- TCEP-HCl (Sigma-Aldrich, catalog number: 75259)
- Proteinase K solution (Mfr. Invitrogen., catalog number: AM2546)
- IRE1 inhibitor III, 4µ8C (CAS 14003-96-4; Calbiochem. Mfr. EMD Millipore. Sigma-Aldrich, catalog number: 212512)
- Novex 6% TBE-Urea PAGE gels (Invitrogen, catalog number: EC6865BOX)
- Novex TBE-Urea 2x sample buffer (Invitrogen, catalog number: LC6876)
- Novex TBE-Urea 5x running buffer (Invitrogen, catalog number: LC6675)
- SYBR Gold nucleic acid gel stain (Invitrogen, catalog number: S11494)
- RiboRuler low range RNA ladder (Thermo Scientific, catalog number: SM1833)
- HEPES (Sigma-Aldrich, catalog number: H3375)
- NaCl (Sigma-Aldrich, catalog number: S3014)
- MgCl2, 2 M solution (Sigma-Aldrich, catalog number: 68475)
- Glycerol (Sigma-Aldrich, catalog number: G5516)
- Urea (Sigma-Aldrich, catalog number: 51456)
- SDS (Sigma-Aldrich, catalog number: L3771)
- EDTA (Sigma-Aldrich, catalog number: EDS)
- Bromophenol blue (Sigma-Aldrich, catalog number: B8026)
- Xylene cyanol FF (Sigma-Aldrich, catalog number: X4126)
- 2x RNA cleavage buffer (see Recipes)
- Stop solution (see Recipes)
Equipment
- Pipettes
- -80 °C freezer
- Sterile No. 11 scalpels (Mfr. Graham-Field, Fisher Scientific, catalog number: 08-927-5B)
- Cell homogenizer (Avestin Emulsiflex-C3, or similar)
- Superspeed fixed angle rotor (SS-34, Mfr. Sorvall., Thermo Scientific, catalog number: 28020TS, or similar)
- Refrigerated benchtop centrifuge (Beckman Coulter Allegra 6R, or similar) equipped with a horizontal swinging bucket rotor (Beckman GH-3.8 or similar), and rotor adaptors for 15 ml and 50 ml conical tubes
- Benchtop microcentrifuge (Eppendorf 5424, or similar)
- ÄKTA pure protein purification system (GE Healthcare, catalog number: 29018224 ), or similar fast-performance liquid chromatography [FPLC] system)
- Thermomixer C, equipped with a 1.5 ml tube block (Eppendorf, catalog number: 2231000574, or similar)
- Thermocycler (BioRad C1000, S1000 or similar)
- Microvolume spectrophotometer (Thermo Scientific, NanoDrop ND-2000; or similar)
- XCell SureLock mini-cell electrophoresis system (Invitrogen, catalog number: EI0001)
- Electrophoresis power supply (BioRad PowerPac basic power supply, catalog number: 1645050; or similar)
- Gel imaging system (BioRad ChemiDoc XRS+, catalog number: 1708265; or similar)
- UV transilluminator (UVP, catalog number: 95042001; or similar)
Software
- ImageJ image processing and analysis software (National Institutes of Health and Laboratory of Optical and Computational Instrumentation, University of Wisconsin at Madison, Madison, WI, USA; https://imagej.nih.gov/ij/)
Procedure
- Purification of recombinant cytosolic domains of human IRE1
- Grow SF21 cells in SF-900 II SFM media supplemented with 10% FBS and 1x antibiotic-antimycotic solution (100 U/ml penicillin, 100 µg/ml streptomycin, 250 ng/ml amphotericin B) at 28 °C in Erlenmeyer flasks, shaking at 150 rpm.
- Infect SF21 cells at mid logarithmic growth phase at a density of 1 x 106-2 x 106 cells per ml with high titer baculoviral stock.
- Collect the SF21 cells 72-96 h after infection in 50 ml conical tubes and pellet them by centrifugation at 4 °C, at 160 x g for 5 min, using a refrigerated benchtop centrifuge (Beckman Coulter Allegra 6R or similar). At this point you can either flash freeze the cell pellet and store it at -80 °C, or continue with the purification of the protein.
- Suspend the pellet from 250 ml of SF21 cells (around 5 ml dry volume) in 20 ml ice-cold SF21 cell lysis buffer. Keep the tube on ice.
- Lyse the cells by passing them once through the homogenizer. Set the Emulsiflex homogenizer at 16,000 psi. Collect the homogenized cells in ice cooled tubes; keep the homogenates on ice.
- Centrifuge the lysate at 30,500 x g for 40 min at 4 °C in SS-34 rotor to remove cell debris.
- Equilibrate the 5 ml HisTrap HP column with SF21 cell lysis buffer using the FPLC system, which is kept in a cooling cabinet or cold room.
- Load the clarified cell lysate onto the 5 ml HisTrap HP column at a flow rate of 2 ml/min to ensure efficient binding of the protein.
- Wash the bound protein with 20 column volumes of lysis buffer at a flow rate of 5 ml/min.
- Elute the protein with an imidazole gradient (0 to 500 mM) in 15 column volumes in the SF21 cell lysis buffer. Analyze a small sample of each collected fraction by SDS-PAGE to define the fractions in which the protein eluted. Pool the fractions containing the purified protein.
- Incubate the purified IRE1-KR43 with TEV protease (250 μl of 0.5 mg/ml) to remove the hexahistidine tag overnight (12-16 h) at 4 °C during dialysis against the IRE1 dialysis buffer.
- Load the protein onto a HisTrap HP column equilibrated with the IRE1 dialysis buffer after TEV cleavage to remove impurities as well as the fraction of IRE1-KR43 still containing the hexahistidine tag.
- Further purify the protein by running the HisTrap HP column eluate on a HiLoad 16/600 Superdex 200 column (GE Healthcare) which has been equilibrated with the dialysis buffer with 2 column volumes according to the manufacturer’s instructions.
- Concentrate the purified IRE-KR43 protein using Amicon Ultra centrifugal filters to 50 μM and flash freeze in 25 μl aliquots. Store at -80 °C.
- In vitro transcription, purification and refolding of IRE1 substrate RNAs
- Prepare templates for in vitro transcription. Assemble multiple parallel reactions to increase yield. The DNA templates can be either PCR products prepared with Phusion polymerase (a high-fidelity DNA polymerase that generates blunt ends), or linearized pUC19 plasmids cut at the 3′ end of the sequence encoding the RNA of interest. We routinely use 1 μg of gel purified DNA templates for in vitro transcription. To account for loses during purification, start with at least 2 μg of PCR products or 2 μg of plasmid for digestion.
Note: Design oligonucleotides to use as primers to amplify the sequence encoding your RNA of interest from high-quality cDNA obtained from the cell of your choice or from available cDNA clones. Use the T7 oligonucleotide 5′-TAATACGACTCACTATAG(N14-21)-3′ as your forward primer and a gene-specific reverse primer. If you want to clone the templates, engineer restriction enzyme recognition sites compatible with the multiple cloning site of pUC19 into your oligonucleotides. Use recognition sites for enzymes that generate blunt-ends or 5′-overhangs on the 3′-end of your cloned DNA of interest. If you are planning on using linearized plasmids as templates for the in vitro transcription reaction, digest the plasmids to completion. - Purify the templates from Step B1 above from 1% agarose gels. Use the QIAquick PCR & gel cleanup kit following the manufacturer’s recommendations. Elute DNA from each column with 30 μl of elution buffer.
Note: Do not skip the gel purification step. Unincorporated oligonucleotides, off-target amplicons and circular plasmids all contribute to the synthesis of heterogenous RNAs. - Concentrate and further clean-up the gel-purified DNA templates using the Zymo DNA clean and concentrator-5 kit following the manufacturer’s recommendations. Elute DNA from each column with 6 μl of elution buffer. Quantify your template concentration and purity using a microvolume spectrophotometer and adjust the DNA concentration to 500 ng/μl.
Note: Highest transcription rates are obtained with highest purity templates. Expect A260/A280 ratios of ~1.8 and A260/A230 ratios > 2.0. - Transcribe RNA using 1 μg of DNA template as input with the HiScribe T7 high yield RNA synthesis kit in 20 μl reactions, for 2 h at 37 °C, following manufacturer’s recommendations.
Notes:- While the reactions are running, pre-run a Novex 6% TBE-Urea PAGE gel for 20 min in 1x Novex TBE-Urea running buffer.
- Other pre-cast TBE-urea PAGE gels can be used as an alternative to Novex gels with compatible electrophoresis systems. While we routinely do not cast our own TBE-Urea gels, denaturing TBE-urea PAGE gels prepared in the laboratory could also be used.
- Stop the reactions by adding 2 μl DNase to the reactions, pipette gently to mix and incubate for an additional 15 min at 37 °C.
- Add 1 volume of Novex TBE-Urea 2x sample buffer to the reactions, mix well by pipetting and heat at 80 °C for 3 min, then cool the samples immediately on ice. Load your samples on the Novex 6% TBE-Urea PAGE gel from Step B4a “NOTE”. Load 2 μl of diluted RiboRuler low range RNA ladder on the first well. Run the gel using the XCell SureLock mini-cell electrophoresis system at 100 V (constant voltage) for 50-60 min. For longer RNAs run the gel for a longer time (around 90 min) to achieve better separation. The dark blue bromophenol blue dye front corresponds to a fragment length of 25 nt according to the manufacturer’s instructions.
Notes:- Before loading your samples, pipette buffer vigorously into each well of the gel to remove urea buildup. Load your samples on the rinsed, clean wells. Use a single gel per transcription reaction; if using a 10-well gel, load ~5 μl of sample per well. Do not overload the gel.
- Dilute the RiboRuler low range RNA ladder in RNase free water to load 10-20 ng RNA per band. The undiluted RNA ladder contains 140 ng of each RNA transcript.
- Disassemble the XCell SureLock mini-cell, remove the gel and dismantle the gel cassette carefully, leaving the gel mounted on one of the plastic plates. Stain the gel with SYBR Gold (diluted as per manufacturer’s recommendations in 1x TBE) by submerging the gel in the buffer for 20 min at room temperature.
Note: Avoid touching the gel with gloved fingers as it will leave marks. We have found that staining the gel without agitation yields better results. Cover the gels to prevent exposure to light while staining. - Carefully remove the gel from the backing plate and place it on plastic cling-wrap. Wipe the stage of the UV transilluminator with RNaseZAP RNase decontamination solution and follow by wiping the surface with 70% ethanol. Place the gel on the plastic cling wrap on the transilluminator and visualize the RNA. Use a sterile No. 11 scalpel to cut around the band of interest. Collect the band of interest in a non-stick 0.5 ml RNase-free tube. Use one tube for every 2-3 lanes of gel (max. gel slice per tube should not exceed 1 mm x 15 mm x 5 mm).
Notes:- It is possible to have too much RNA loaded on the gel, in which case expect to see “ghosts” around the band of interest (regions where the dye accumulates around the band). Such event does not compromise the RNA extraction downstream.
- Do not drag the tip of the scalpel over the transilluminator stage. Instead place the blade edge parallel to the surface and press down around the band. No. 11 scalpels have straight blade edges.
- Nest each 0.5 ml tube containing the gel fragments inside a non-stick 1.5 ml RNase-free tube. Carefully pierce the bottom of the 0.5 ml tube with a sterile 20 G syringe needle. Do not pierce the larger 1.5 ml tube. Spin in a benchtop microcentrifuge at maximum speed for 3 min to force the gel through the hole. Transfer any residual gel from the 0.5 ml tube to the holder tube. Discard the 0.5 ml tubes.
- Add ~3 gel volumes of RNA gel extraction buffer to the crushed gel in the holder tubes. Incubate for one hour at room temperature with constant agitation at 1,000 rpm in a thermomixer.
- Remove tubes from thermomixer. Vortex the gel slurry and recover it completely using a cut P1000 tip. Transfer the slurry to a Spin-X centrifuge tube filter. Be careful when transferring as gel pieces can clog the pipette tip. Spin the Spin-X centrifuge tube filters at max speed in a benchtop microcentrifuge for 3 min. The filtrate containing your RNA will be at the bottom of the filter tube and the gel pieces will stay on the filter.
- Transfer the filtrate to a fresh non-stick nuclease free 1.5 ml microcentrifuge tube. Pool all filtrates for the same RNA (up to ~0.5 ml aliquots). Add 1/9 of the volume of 3 M NaOAc pH 5.5 and 1 volume of isopropanol and incubate for at least 30 min at -80 °C to precipitate the RNA.
- Spin at maximum speed in a benchtop microcentrifuge at 4 °C for 30 min top pellet the RNA. Remove the supernatant being careful not to disturb the pellet. Wash the RNA pellet twice with 1 ml of 80% ice-cold ethanol prepared in RNase-free water.
- Remove ethanol from the pellet. Spin the tubes to collect trace ethanol from the walls of the tube and remove as much as possible of the remaining ethanol with a pipette. Air-dry the RNA pellets for at least 10 min at room-temperature. Make sure all ethanol has evaporated before proceeding. Resuspend the pellets in 20 μl RNase-free water or RNA resuspension buffer by gentle pipetting.
- Fold the RNAs from Step B14 by incubating them for 5 min at 95 °C in a thermocycler and then slowly cool the samples (1 °C/min) until they reach 25 °C. Keep the samples on ice and use immediately for in vitro cleavage assays or store them at -80 °C for future use.
Note: Before setting up the in vitro cleavage assays, measure the RNA concentration and purity using a microvolume spectrophotometer. Expect to obtain A260/A280 ratios ~2.0 and A260/A230 ratios > 2.0.
- Prepare templates for in vitro transcription. Assemble multiple parallel reactions to increase yield. The DNA templates can be either PCR products prepared with Phusion polymerase (a high-fidelity DNA polymerase that generates blunt ends), or linearized pUC19 plasmids cut at the 3′ end of the sequence encoding the RNA of interest. We routinely use 1 μg of gel purified DNA templates for in vitro transcription. To account for loses during purification, start with at least 2 μg of PCR products or 2 μg of plasmid for digestion.
- In vitro cleavage assay of in vitro transcribed RNAs with purified recombinant IRE1 cytosolic domains and denaturing urea-PAGE of IRE1-generated RNA fragments
- Assemble the in vitro cleavage reactions in non-stick 1.5 ml RNase-free tubes on ice.
- Assemble a master mix for your entire reaction time-series. Each time point has a final volume of 10 μl. Add the following components (in order; per reaction):
- 2x RNA cleavage buffer
- 15-100 ng of PAGE-purified in vitro transcribed RNA
- 50 nM to 5 μM recombinant IRE1-KR43
- Place the tube containing the master mix in a thermomixer set at 30 °C* and incubate for predetermined amounts of time, ranging from 30 s to 90 min depending on the RNA you are studying. Do not agitate.
Notes:- Plan your time series. *You may use a dry bath or water bath set at 30 °C.
- Include a control for cleavage specificity by supplementing one duplicate reaction (the one corresponding to the longest time point) with 4μ8C to a final concentration of 10 μM. To inhibit IRE1, pre-incubate 10 μM IRE1-KR43 with 20 μM 4μ8C for 30 min on ice.
- For each time point take out 7 μl from the master mix and stop the reactions by adding 1 μl of proteinase K solution (20 μg) and incubating for 10 min at 37 °C. Add 1 reaction volume of stop solution, mix gently and heat the samples to 80 °C; incubate for an additional 5 min, then cool the samples immediately on ice to prevent re-folding or base-pairing of the denatured RNA.
- Load 5 μl of reaction for each data point (~1/3 of the total volume per time point) on a Novex 6% TBE-Urea PAGE gels. Load diluted RNA ladder on the first well of the gel as indicated in step B6 “NOTE b”). Run the gel at 100 V (constant voltage) for 50-60 min (or longer if you are analyzing longer RNAs) using the XCell SureLock mini-cell electrophoresis system.
Note: Before loading your samples, thoroughly rinse each well of the gel to remove urea buildup. Load your samples on the rinsed, clean wells. - Disassemble the XCell SureLock mini-cell, remove the gel and dismantle the cassette gel carefully, leaving the gel mounted on one of the plastic plates. Stain the gel with SYBR Gold (diluted as per manufacturer’s recommendations in 1x TBE) by submerging the gel in the buffer for 20 min at room temperature.
Note: Avoid touching the gel with gloved fingers, as it will leave marks. Cover the gels to prevent exposure to light while staining. - Place the stained gel on the stage of a gel imaging system and capture digital images using various exposure times. Record the exposure times for each photograph. Use the longer exposure time that gives the best contrast without saturated pixels. Representative examples of typical results are shown in Figure 2.

Figure 2. TBE-urea PAGE gels showing the IRE1-dependent cleavage of select IRE1 targets, BLOC1S1 (left) and SEC61A1 (right). 5 μM IRE1-KR43 was used to cleave 25 nM of RNA substrates for the indicated times. The IRE1 inhibitor 4µ8C was used as a control for specificity.
Data analysis
The digital images of the gels can be densitometrically quantified and analyzed using ImageJ Software.
- Adjust the image brightness and contrast and remove background prior to the analysis.
- Draw boxes of equal size around the bands corresponding to the uncleaved RNA and calculate the area under the curve (pixel density).
- Normalize your numbers to the uncleaved control and calculate the fraction of remaining uncleaved RNA. To determine observed rate constants, plot these values as a function of time and calculate the best curve fit using your choice of scientific graphing and analysis software.
Recipes
Notes:
- For all recipes, use molecular biology grade reagents or pre-made solutions (available from multiple suppliers). Stock solutions should be prepared in RNase-free water.
- To pH a 0.5 M or 1 M HEPES stock solution, use KOH pellets, add slowly while stirring the solution and monitor the pH continuously.
- *We have not observed significant differences when using Mg(OAc)2 as opposed to MgCl2.
- SF21 cell lysis buffer
20 mM HEPES pH 7.4
600 mM KCl
2 mM MgCl2
10% glycerol
10 mM imidazole
Supplement with freshly added cOmplete EDTA-free protease inhibitor cocktail - IRE1 dialysis buffer
20 mM HEPES pH 7.4
300 mM KCl
2 mM MgCl2
5% glycerol
1 mM TCEP-HCl - RNA gel extraction buffer
300 mM NaOAc
1 mM EDTA
Supplemented with freshly added recombinant RNasin RNase inhibitor - RNA re-suspension buffer
20 mM HEPES pH 7.4
100 mM NaCl
1 mM MgCl2*
Note: We have not observed significant differences when re-folding RNA in this buffer versus refolding in RNase-free water. - 2x RNA cleavage buffer
40 mM HEPES pH 7.4
200 mM NaCl
2 mM MgCl2*
10% glycerol
2 mM TCEP-HCl - Stop solution
10 M urea
0.1% SDS
1 mM EDTA
0.01% xylene cyanol
0.01% bromophenol blue
Note: Bromophenol blue can generate background noise/signal artifacts during imaging when the RNA concentration is low.
Acknowledgments
DAA was supported by an Irvington Postdoctoral Fellowship of the Cancer Research Institute. JP was supported by a Human Frontier Science Program Postdoctoral Fellowship. PW is an Investigator of the Howard Hughes Medical Institute. We thank Aaron Mendez for his help with setting up the purification of human IRE1 Kinase/RNase domains.
Competing interests
The authors declare no competing interests.
References
- Acosta-Alvear, D., Karagoz, G. E., Frohlich, F., Li, H., Walther, T. C. and Walter, P. (2018). The unfolded protein response and endoplasmic reticulum protein targeting machineries converge on the stress sensor IRE1. Elife 7: e43036.
- Acosta-Alvear, D., Zhou, Y., Blais, A., Tsikitis, M., Lents, N. H., Arias, C., Lennon, C. J., Kluger, Y. and Dynlacht, B. D. (2007). XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol Cell 27(1): 53-66.
- Bae, D., Moore, K. A., Mella, J. M., Hayashi, S. Y. and Hollien, J. (2019). Degradation of Blos1 mRNA by IRE1 repositions lysosomes and protects cells from stress. J Cell Biol 218(4): 1118-1127.
- Calfon, M., Zeng, H., Urano, F., Till, J. H., Hubbard, S. R., Harding, H. P., Clark, S. G. and Ron, D. (2002). IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415(6867): 92-96.
- Cheng, C. Y., Chou, F. C., Kladwang, W., Tian, S., Cordero, P. and Das, R. (2015). Consistent global structures of complex RNA states through multidimensional chemical mapping. Elife 4: e07600.
- Cross, B. C., Bond, P. J., Sadowski, P. G., Jha, B. K., Zak, J., Goodman, J. M., Silverman, R. H., Neubert, T. A., Baxendale, I. R., Ron, D. and Harding, H. P. (2012). The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc Natl Acad Sci U S A 109(15): E869-878.
- Han, D., Lerner, A. G., Vande Walle, L., Upton, J. P., Xu, W., Hagen, A., Backes, B. J., Oakes, S. A. and Papa, F. R. (2009). IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 138(3): 562-575.
- Harnoss, J. M., Le Thomas, A., Marsters, S. A., Lawrence, D. A., Lu, M., Chen, Y. A., Qing, J., Totpal, K., Kan, D., Segal, E., Wallweber, H. A., Wang, W., Clark, K., Kaufman, S., Beresini, M., Sandoval, W., Lorenzo, M., Wu, J., Ly, J., De Bruyn, T., Heidersbach, A., Haley, B., Gogineni, A., Weimer, R., Lee, D., Braun, M., Rudolph, J., VanWyngarden, M. J., Sherbenou, D. W., Gomez-Bougie, P., Amiot, M., Acosta-Alvear, D., Walter, P., Ashkenazi, A. (2018). Disruption of IRE1α through its Kinase Domain Attenuates Multiple Myeloma. BioRxiv doi: https://doi.org/10.1101/495242.
- Hollien, J. and Weissman, J. S. (2006). Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 313(5783): 104-107.
- Hollien, J., Lin, J. H., Li, H., Stevens, N., Walter, P. and Weissman, J. S. (2009). Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol 186(3): 323-331.
- Iwawaki, T., Akai, R., Kohno, K. and Miura, M. (2004). A transgenic mouse model for monitoring endoplasmic reticulum stress. Nat Med 10(1): 98-102.
- Iwawaki, T., and Akai, R. (2006). Analysis of the XBP1 splicing mechanism using endoplasmic reticulum stress-indicators. Biochem. Biochem Biophys Res Commun 350(3): 709-15.
- Karagöz, G. E., Acosta-Alvear, D. and Walter, P. (2019). The unfolded protein response: detecting and responding to fluctuations in the protein-folding capacity of the endoplasmic reticulum. Cold Spring Harb Perspect Biol. pii: a033886.
- Korennykh, A.V., Egea, P.F., Korostelev, A.A., Finer-Moore, J., Zhang, C., Shokat, K.M., Stroud, R.M., and Walter, P. (2009). The unfolded protein response signals through high-order assembly of Ire1. Nature 457(7230): 687-93.
- Lee, A. H., Iwakoshi, N. N. and Glimcher, L. H. (2003). XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol 23(21): 7448-7459.
- Li, H., Korennykh, A. V., Behrman, S. L. and Walter, P. (2010). Mammalian endoplasmic reticulum stress sensor IRE1 signals by dynamic clustering. Proc Natl Acad Sci U S A 107(37): 16113-16118.
- Lu, M., Lawrence, D. A., Marsters, S., Acosta-Alvear, D., Kimmig, P., Mendez, A. S., Paton, A. W., Paton, J. C., Walter, P. and Ashkenazi, A. (2014). Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 345(6192): 98-101.
- Mendez, A. S., Alfaro, J., Morales-Soto, M. A., Dar, A. C., McCullagh, E., Gotthardt, K., Li, H., Acosta-Alvear, D., Sidrauski, C., Korennykh, A. V., Bernales, S., Shokat, K. M. and Walter, P. (2015). Endoplasmic reticulum stress-independent activation of unfolded protein response kinases by a small molecule ATP-mimic. Elife 4: e05434.
- Moore, K. and Hollien, J. (2015). Ire1-mediated decay in mammalian cells relies on mRNA sequence, structure, and translational status. Mol Biol Cell 26(16): 2873-2884.
- Mori, K. (2009). Signalling pathways in the unfolded protein response: development from yeast to mammals. J Biochem 146(6): 743-750.
- Peschek, J., Acosta-Alvear, D., Mendez, A. S. and Walter, P. (2015). A conformational RNA zipper promotes intron ejection during non-conventional XBP1 mRNA splicing. EMBO Rep 16(12): 1688-1698.
- Sidrauski, C., Acosta-Alvear, D., Khoutorsky, A., Vedantham, P., Hearn, B. R., Li, H., Gamache, K., Gallagher, C. M., Ang, K. K., Wilson, C., Okreglak, V., Ashkenazi, A., Hann, B., Nader, K., Arkin, M. R., Renslo, A. R., Sonenberg, N. and Walter, P. (2013). Pharmacological brake-release of mRNA translation enhances cognitive memory. Elife 2: e00498.
- So, J. S., Hur, K.Y., Tarrio, M., Ruda, V., Frank-Kamenetsky, M., Fitzgerald, K., Koteliansky, V., Lichtman, A.H., Iwawaki, T., Glimcher, L.H., et al. (2012). Silencing of lipid metabolism genes through IRE1α-mediated mRNA decay lowers plasma lipids in mice. Cell Metab 16(4):487-99.
- Tam, A. B., Koong, A. C., and Niwa, M. (2014). Ire1 has distinct catalytic mechanisms for XBP1/HAC1 splicing and RIDD. Cell Rep 9(3): 850-8.
- Yoshida, H., Matsui, T., Yamamoto, A., Okada, T., and Mori, K. (2001). XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107(7): 881-91.
Article Information
Copyright
![]() Karagöz et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Karagöz et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Karagöz, G. E., Peschek, J., Walter, P. and Acosta-Alvear, D. (2019). In vitro RNA Cleavage Assays to Characterize IRE1-dependent RNA Decay. Bio-protocol 9(14): e3307. DOI: 10.21769/BioProtoc.3307.
- Acosta-Alvear, D., Karagoz, G. E., Frohlich, F., Li, H., Walther, T. C. and Walter, P. (2018). The unfolded protein response and endoplasmic reticulum protein targeting machineries converge on the stress sensor IRE1. Elife 7: e43036.
Category
Biochemistry > RNA > RNA-protein interaction
Molecular Biology > RNA > RNA detection
Molecular Biology > Protein > Activity
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.