- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Electron Microscopy Sample Preparation Protocol Enabling Nano-to-mesoscopic Mapping of Cellular Connectomes and Their Habitats in Human Tissues and Organs

(*contributed equally to this work) Published: Vol 9, Iss 14, Jul 20, 2019 DOI: 10.21769/BioProtoc.3298 Views: 8456
Reviewed by: Khyati Hitesh ShahJose Martinez HernandezRajesh D Gunage
Original research article
The authors used this protocol in:

Dec 2016
Advertisement





Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Multibeam scanning electron microscopy (multiSEM) provides a technical platform for seamless nano-to-mesoscale mapping of cells in human tissues and organs, which is a major new initiative of the U.S. National Institutes of Health. Such cross-length-scale imaging is expected to provide unprecedented understanding of relationships between cellular health and tissue-organ as well as organismal-scale health outcomes. For example, understanding relationships between loss in cell viability and cell network connectivity enables identification of emergent behaviors and prediction of degenerative disease onset, in organs as diverse as bone and brain, at early timepoints, providing a basis for future treatments and prevention. Developed for rapid throughput imaging of minute defects on semiconductor wafers, multiSEM has recently been adapted for imaging of human organs, their constituent tissues, and their respective cellular inhabitants. Through integration of geospatial approaches, statistical and network modelling, advances in computing and the management of immense datasets, as well as recent developments in machine learning that enable the automation of big data analyses, multiSEM and other cross- cutting imaging technologies have the potential to exert a profound impact on elucidation of disease mechanisms, translating to improvements in human health. Here we provide a protocol for acquisition and preparation of sample specimen sizes of diagnostic relevance for human anatomy and physiology. We discuss challenges and opportunities to integrate this approach with multibeam scanning electron microscopy workflows as well as multiple imaging modalities for mapping of organ and tissue structure and function.
Keywords: Scanning electron microscopyBackground
Connectomics refers to the mapping of cellular connectivity in a physiological context, e.g., in organs and organ systems (Sporns et al., 2005; Lichtman et al., 2008; Knothe Tate, 2017). Seamless, multiscale and multimodal imaging provides a platform for interdisciplinary research and the trans-dimensional linking of biological structure and function (Sporns et al., 2005; Lichtman et al., 2008; Eberle et al., 2015; Hageman et al., 2016; Knothe Tate et al., 2016a, 2016b and 2016c; Pereira et al., 2016; Knothe Tate, 2017; National Institutes of Health, 2018; https://www.humanconnectome.org/). The U.S. National Institutes of Health's (NIH) Connectome Project, launched in 2009, aimed to map "the human brain...to connect its structure to function and behavior" (https://www.humanconnectome.org/). In 2018, NIH debuted a program to promote cross-cutting research mapping the cells within the human body (National Institutes of Health, 2018). Connectome maps of brain and other tissues of the body have typically been restricted to small organisms due to challenges associated with cross-scale imaging from nano- to mesoscopic length scales. New imaging technology platforms enable such seamless scale-crossing image acquisition, and current barriers include biophysical hurdles intrinsic to specimen preparation as well as digital hurdles inherent to massive data management, analysis, and curation to maximize sharing across platforms and among geographically far flung users.
Currently, there is a great imperative not only to study the cells themselves but also to refocus at the intersection of and across multiple length scales encompassing the cellular networks that inhabit complex biosystems comprising tissues, organs, and organ systems. The status of these organ connectomes provides a powerful early indicator of degenerative changes and disease states over the lifespan of the organism. Three-dimensional multiscale imaging methods allow unprecedented study of structure and function across length scales; from organ to tissue, cells to molecules (Anderson et al., 2008; Hageman et al., 2016; Knothe Tate et al., 2016; Pereira et al., 2016).
Traditional transmission electron microscopy (TEM), commonly used to visualize cellular ultrastructure, provides superior resolution and contrast. However, this is contingent on the often-laborious preparation of specimens into ultrathin sections (no more than 100 nm in thickness) for electron transparency.
The emergence of multibeam scanning electron microscopy (multiSEM), with 61 or 91 parallel beams, is heralded by a new age of organ connectomics and presents a suitable platform for imaging large areas at nanometer resolution (Eberle et al., 2015; Knothe Tate et al., 2016a and 2016c; Hageman et al., 2016; Pereira et al., 2016; Knothe Tate, 2017). The imaging rates and resolutions of multiSEM far supersede single-beam SEM, imaging an area of 1 cm2 at 4 nm pixel size in less than 3 h. It is able to image specimens reaching 10 cm in diameter at up to 4 nm resolution. Despite advances in the imaging technologies, biological tissue specimen preparation has remained unchanged for decades, and hinders translation to cutting-edge imaging modalities (multiSEM).
This protocol addresses the difficulties associated with specimen preparation workflows enabling multiSEM, as well as multimodal and correlative imaging methods. While having been implemented successfully in peer-reviewed publications (Figure 1), details of the protocol have been requested by the medical and scientific community alike, providing impetus for its publication (Knothe Tate et al., 2004; Eberle et al., 2015; Knothe Tate et al., 2016b; Hageman et al., 2016; Pereira et al., 2016).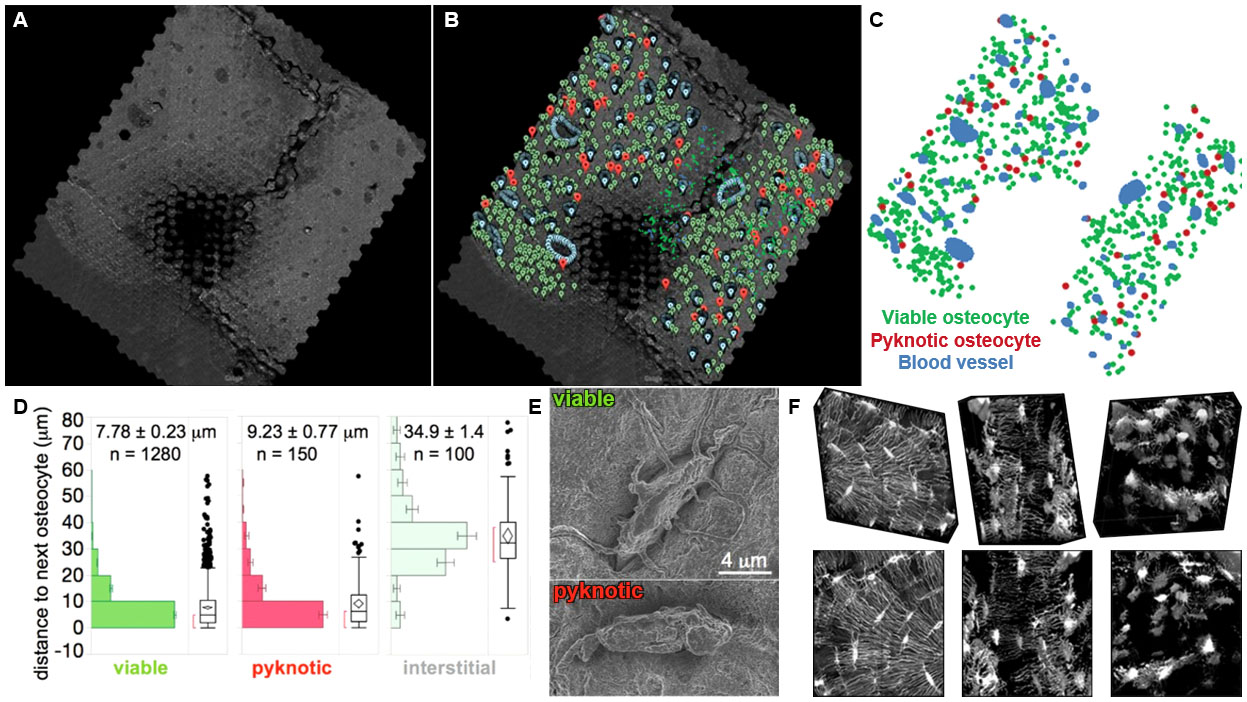
Figure 1. Cellular epidemiology studies in large scale maps comprising stitched, scanning electron microscopy images. These high-resolution images are obtained using multiSEM and demonstrate the feasibility of the approach that greatly expands beyond fields of view previously acquired using confocal scanning microscopy. A. Geonavigational methods and the Google Maps Javascript API were utilized to create navigable maps of osteocytes within the human hip imaged using multiSEM and spanning the nano- to mesoscale. The world's first map of the human hip was created by imaging a sample approximately 2.4 x 2.4 mm. Imaging using a 61-parallel-beam Zeiss MultiSEM 505 prototype yielded over 55,000 SEM images, which were stitched into a single image, and rendered in a pyramidal structure for seamless zooming into and out of the map using Google Maps API. The dark area in the middle of the sample is an artifact of automated polishing, a sample preparation step that was later changed to precision CNC milling and hand polishing (see Procedure section). B-C. Google Maps Javascript API enabled the geographic coordinates of physiologically relevant landmarks to be recorded. Blood vessel edges, viable osteocytes and pyknotic (sick) osteocytes were marked with blue, green and red pins respectively. D. The position of each pin could be exported for post hoc analyses, e.g., to test the hypothesis that cell viability correlates to blood vessel proximity, path lengths of viable, pyknotic (sick) and interstitial cells to blood vessels were calculated and compared. E. MultiSEM images of a viable cell, with projections emanating outwards into the matrix and pyknotic cell, no projections observed. F. The role of cell health and connectivity on tissue health was first observed in 3D images of osteocyte networks from (left) healthy, (middle) osteoporotic and (right) severely osteoporotic cortical bone from the femoral head of human patients, using laser scanning confocal microscopy. A shortcoming of this approach is that volumes of interest are limited to e.g., 274 x 274 x 50 microns. Figures adapted and used with permission. (Knothe Tate et al., 2004; Eberle et al., 2015; Knothe Tate et al., 2016c; Hageman et al., 2016; Pereira et al., 2016).
Presented in this protocol is a detailed workflow for sample preparation of sizeable tissue specimens (up to 10 cm in diameter, Figure 2) of human and large mammalian origin. The method has been tested in human samples from the femoral neck and head of subjects undergoing hip replacement (all human samples collected and used in development of this protocol complied with all relevant ethical regulations of the Institutional Review Board of the Cleveland Clinic, IRB #12-335) as well from sheep (Ovis aries) femur and guinea pig (Cavia porcellus) knee to verify multimodal imaging (all animal tissue samples used in development of this protocol derived from studies that adhered to protocols approved through the respective Institutional Animal Care and Use Committees [IACUC] of the Canton of Grisons, Switzerland and Case Western Reserve University, in adherence with the Animal Research: Reporting of In Vivo Experiments [ARRIVE] guidelines). Here we adapted methods typically used for TEM and SEM tissue sample preparation to enable across length scales, from nano- to mesoscopic. The methods integrate techniques including precision CNC-milling and chemical etching methods developed for atomic force microscopy to enable three dimensional and multimodal imaging. This method can be applied to macroscopic human tissues and tissues of large mammals and can be integrated into a multimodal imaging workflow, in particular correlative light electron microscopy (CLEM).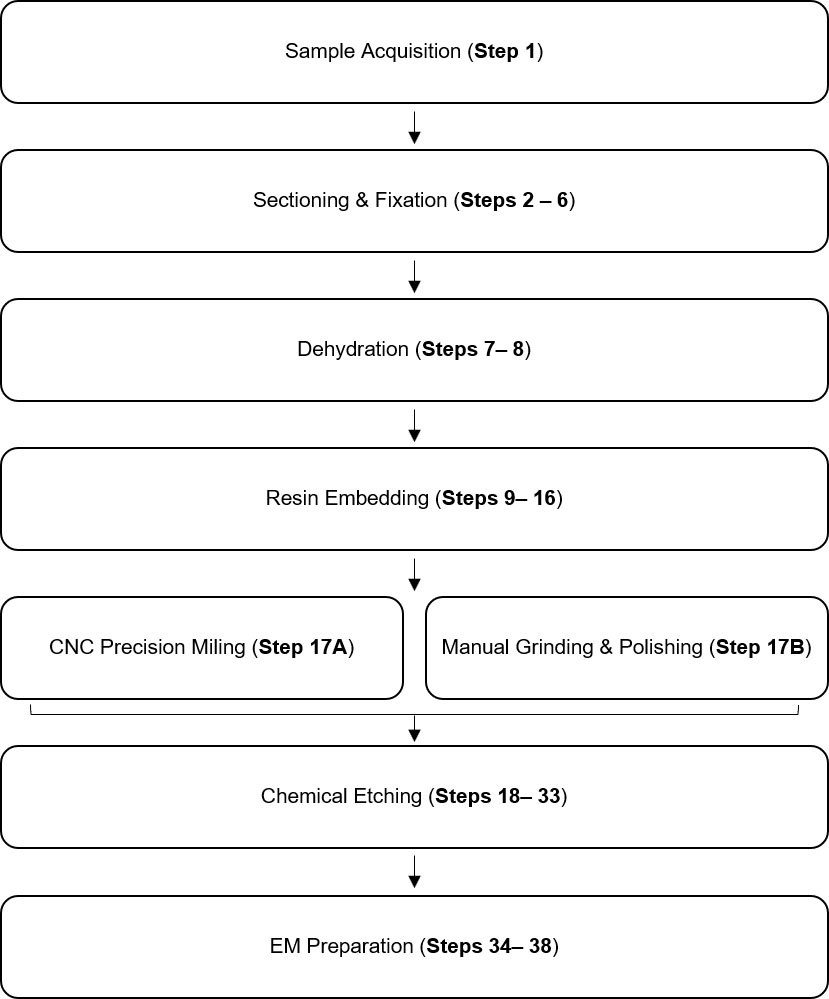
Figure 2. Workflow for acquisition of multiSEM data using mesoscopic samples
Comparison with other methods
The key advantages of this method for biological specimen preparation are that it is streamlined yet maintains the flexibility to be applied to any tissue specimen using readily available materials (typically used in atomic force microscopy, EM and laser confocal microscopy specimen preparation). However, this flexibility and ease of implementation has its drawbacks, as the method is not entirely self-contained and the sample must be moved from platform to platform, if light microscopy is used in addition to EM. Advantageously, the use of heavy metal stains (uranyl acetate and lead citrate) are eliminated, as carbon sputtering provides sufficient contrast enhancement in the workflow. This further simplifies the process, avoiding the use of hazardous chemicals and their associated risks. Similarly, osmium tetroxide (OsO4) is not used in the fixation contrast as carbon sputtering alone provides sufficient contrast. This provides further advantages in context of multimodal, correlative microscopy, as OsO4 imparts a yellow hue throughout the specimen, impairing any form of light microscopy (Figures 2-4).
This protocol is ideal for studies involving tissue samples from human or large mammals (up to 10 cm in diameter) where the organization of cells and tissues are of primary interest. While the resultant data encodes great structural detail, it is also extremely large (> 10 TB) and requires an appropriate and robust data management system or pipeline for efficient analytics and interpretation, which is outside the scope of traditional laboratory infrastructure. Considering the magnitude of data, it is improbable that analyses can be conducted using conventional means. This presents a disadvantage as well as an opportunity, i.e., through incorporation of cutting edge approaches including deep learning and machine learning, the economic and time costs of manual methods can be eliminated, providing a path to full automation in the future (Nathanson et al., 2019).
Figure 3. Seamless multi-scale imaging of hip joint tissue, prior to etching step. A. Transmitted light image of OsO4 stained femoral head section, with greatest penetration of contrast agent (darkest) in cartilage (white dotted square) and marrow spaces of the subchondral bone (black dotted square). B. Subchondral bone exhibits less staining than cartilage and is highly cellular. Area corresponds approximately to black dotted square (in A). C. The tidemark, delineating the interface of unmineralized (rust-brown) and mineralized (orange) cartilage is visible in the upper right corner. Defects in the uncalcified cartilage are visible as light "cracks" running orthogonal to the tidemark line. Area corresponds approximately to the white dotted square (in A). Regarding suboptimal quality of images: these images are taken of a 300 μm thick, EPON® embedded, undecalcified section without prior mounting and cover slipping, using an inverted epifluorescent microscope with 1.6x (A) and 10x (B, C) dry objectives. Specimens were not mounted, or cover slipped due to plans to image subsequently using multiSEM. After completion of multiSEM studies, microradiography and renewed optical imaging can be carried out. D. Laser scanning confocal image of specimen shows autofluorescence (light areas) of subchondral bone and marrow spaces (ellipsoids) as well as cartilage (dark), with two visible cartilage defects (bright cracks, arrows). E. Large scale, tiled multiSEM image of same area with single field of view showing (sub-) cellular resolution of bone (F) and blood (G) cells without additional etching. H. Traditional SEM studies on specimens containing soft tissue reveal intertwining muscle fiber architecture.
Figure 4. Seamless nano- to mesoscale imaging of the human hip, visualized using the Google Maps navigational platform. A. The Google Maps JavaScript API was used to visualize and create navigable maps of an imaged region. The arrowheads point to the eroded surface of the hip joint, toward areas internal to the femoral head. B. A single osteocyte on the sample surface with C. projections (osteocyte processes) into the underlying matrix. This sample was imaged using a 61- parallel-beam Zeiss MultiSEM.
The choice of poly (methyl-methacrylate) (PMMA) over epoxy, results in more resilient samples, with little damage observed after electron beam exposure. MultiSEM requires sample flatness and parallelism to the mount surface to prevent charging artifacts, which ideally requires precision CNC-milling, an unconventional technique for EM sample preparation. Similar results have been achieved with conventional polishing methods.
The protocol does not involve ultra-thin sectioning characteristic of TEM, which is often difficult and time consuming. As a result, use of the method may not perfectly preserve or enable visualization of internal cellular ultrastructure. For ultrastructure studies, TEM resolutions remain superior to SEM. Neither this protocol method nor any electron microscopy method enables live-cell imaging and images represent single points in time. Hence, the method does not capture the nuance and temporal dynamic of complex biological processes.
Novel techniques such as field emission scanning electron microscopy (FESEM) (Tapia et al., 2012) and serial block-face scanning electron microscopy (SBF-SEM) (Mikula and Denk, 2015) allow the three-dimensional visualization of and reconstruction of cellular ultrastructure, however samples are limited to a few millimeters on edge in size. Although the images acquired from this protocol are two-dimensional, they provide a birds-eye-view, at nanometer resolution, of cellular networks and populations over large areas. The method thus enables visualization of tissue architecture not readily achievable using two-dimensional EM.
Experimental Design
For each experiment, we defined the relevant research question and a testable hypothesis, relating independent variables (also referred to as outcome measures) as a function of dependent variables. Exclusion criteria were defined to reduce effects of unrelated disease states that could lead to misinterpretation of results, e.g., metabolic bone disease is excluded in a study of loss of bone density due to disuse.
Power calculations were made to determine sample size for each experimental group. Some novel studies do not have a representative dataset upon which, variance measures can be made and initial sample sizes have to be based on previous experience. Power analyses conducted for previous studies determined the need for a sample size of 5 patients per cohort. Post hoc analyses on these data indicated the robustness of study outcomes; from a translational perspective, even if more than 700 patients were tested to obtain statistical significance, differences (in periosteum derived stem cell regenerative capacity attributable to age and/or disease state) would remain small, due to the small to medium effect size (of age and disease), even if significantly different (Chang et al., 2014).
Specimen Acquisition
The critical phase research requiring human tissue samples is the acquisition of specimens (Figure 5). It is crucial to find a surgeon willing and able to collaborate in the study, acting as not only clinical investigator on the ethics protocol but ensuring the identification and screening of possible participants, alongside ensuring informed consent is obtained for each individual involved. It is crucial all specimens are de-identified, by removing all identifying information such as name, medical patient numbers, etc. by the tissue procurement and/or pathology department of the clinic prior to release of the tissues for scientific research. Age and sex of the patient are retained in sample documentation. Together with date of acquisition, specimens are separately identifiable without access to medical documentation.
In some experimental study designs, samples are given non-obvious identifiers and the key to decoding the identifiers is retained in a secure position by the clinical partner to enable later information from the patient's medical record, e.g., specific health history information, if needed. In obtaining patient consent while also striving to include diverse populations in cohorts, consideration should be made to patient groups whose beliefs may preclude sharing of tissues. It may be possible to encourage their inclusion through awareness of cultural sensitivities and inclusion of language that addresses culture-specific considerations in outreach brochures as well as informed consent documentation.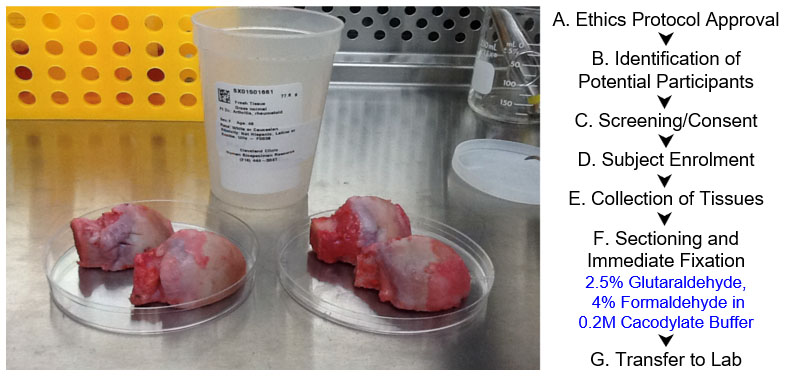
Figure 5. Procedures for acquiring and handling human tissues. Two samples from different patients, acquired on the same afternoon. Femoral heads (see photo, rounded structures divided into two halves) were sectioned along the plane of interest, e.g., coronal or sagittal plane, to allow for infiltration of fixation and embedding chemicals. A-G. Work flow chart for sample acquisition, with chemicals indicated in blue.
Sectioning and Fixation
With samples of size larger than a millimeter on edge, it is important to take extra measures to ensure samples undergo adequate fixation before proceeding. In this protocol, we are limited to diffusion fixation; perfusion of fixatives is not an option in human tissues. The femoral head and neck are sectioned, with the precision cutter, from the proximal and distal end before further sectioning of the medial and lateral portions, transversely across the bone. Following this, 1 mm slices in the plane of interest are taken with the saw microtome, slices at this thickness allow the complete infiltration of glutaraldehyde and paraformaldehyde fixatives in cacodylate buffer in a fume hood. The specimens are then washed in cacodylate buffer before sequential ethanol dehydration (30%, 50%, 75%, 95%).
Contrast Enhancement
Biological specimens for EM typically require post-fixation contrast enhancement to resolve subcellular detail. Contrast enhancing agents such as osmium tetroxide are commonly used but impart exogenous color throughout the entire sample, making it unsuitable for multimodal imaging (Figure 2). Heavy metal staining is another mode of contrast enhancement, whereby uranyl acetate and lead citrate are routinely employed. The use of heavy metals is a more labor intensive and hazardous process, which was not desirable. Additionally, uranyl acetate introduces instability into the sample, making it light and UV sensitive and prone to forming precipitates over time. Instead, we opted to highlight subcellular detail post hoc, with the use of both chemical etching (detailed below and adapted from protocols used for AFM) and carbon sputtering.
Resin Embedding
Conventionally, biological specimens are embedded in epoxy resins for EM. Initial attempts at using epoxy, EPON®, proved unsuccessful as it is typically used for samples that are 1-2 mm on edge. Under inspection with a light microscope, the epoxy embedded specimens exhibited variations in surface topography of up to 100 μm making it unsuitable for multiSEM. The specimens exhibited incomplete and non-uniform resin polymerization, most likely caused by insufficient resin penetration in such large specimens. Additional contributing factors to poor polymerization, include insufficient time for immersion and perfusion, inadequate dilution steps and excessive resin viscosity, all resulting in poor resin infiltration into tissue pores, leaving residual ethanol and propylene oxide within the sample.
Due to the difficulties encountered with EPON® and prior experience with PMMA, we used lower viscosity PMMA resin as the embedding medium. To ensure complete infiltration of the resin into tissue pores and uniformity of the sample surface, dehydration time was increased, and the tissues were bulk embedded under vacuum to enable gradual polymerization.
Planarity and Sample Size
The development of multiSEM enables rapid throughput of specimens that far exceed traditional dimensions, up to 100 x 100 x 30 mm. However, it requires specimens to be flat and parallel to the stage, with a surface flatness less than or equal to 500 nm per 100 μm (peak-to-peak). This minimizes edge effects and specimen damage resultant of increased electron emissions at high points in the surface topography.
Initially, specimens were manually and semi-automatically mechanically ground and polished, with both methods exhibiting varying degrees of success. Automated polishing proved too aggressive, rapidly removing the sample surface. Conversely, manual polishing was not time effective and resulted in the emergence of microscopic cracks in the sample surface, most likely due to the release of residual stresses. These cracks could later cause edge effects along the edges of the crack, interfering with SEM imaging quality. We progressed to micro- CNC milling and manual buffing with acrylic polish, the sample surface was milled with a great level of precision without the introduction of surface cracks. Specimens were precision milled in a CNC-lathe using a carbide insert with the unhoned edge against the sample surface. Following this, the surface was lightly buffed with a soft microfiber cloth, acrylic polish and ultra-pure water.
Chemical Etching and SEM Preparation
As noted above, this protocol does not use heavy metals based post-fixation contrast enhancement. Instead, chemical etching methods adapted from techniques developed for AFM were used to uncover underlying molecular detail and cellular networks (Reilly et al., 2001; Knapp et al., 2002). Dilute hydrochloric acid (HCl) and sodium hypochlorite (NaClO) enable controlled etching of the organic, collagen, and inorganic, apatite composite matrices, revealing cells and projections embedded in the extracellular matrix. This method also allows for selective etching of regions of interest by masking around areas to be etched with electrical tape (geometric shapes) or nail polish (free form regions). Following this step, the sample can be imaged with light microscopy. To increase sample conductance and contrast, the specimen requires a thin carbon coat (20 nm) before it can be imaged using SEM.
Materials and Reagents
- Immersion fixation
- Nitrile gloves (Livingston, catalog number: GLVNTRLC100S)
- Dust-free wipes (Professional Kimtech Science Kimwipes Delicate Task Wipers, Kimberly-Clark Professional, catalog number: 34155)
- Plastic Petri Dishes (Fisher Scientific, catalog number: R80116)
- Parafilm M (Fisher Scientific, catalog number: 13-374-10)
- Aluminium foil (Sigma-Aldrich, catalog number: Z691569-1EA)
- Tissue Samples for fixation
- Glutaraldehyde, 10% aqueous solution (Electron Microscopy Sciences, catalog number: 16100)
- Paraformaldehyde Powder (Sigma-Aldrich, CAS number: 30525-89-4)
- Sodium cacodylate buffer (0.2 M, pH 7.4; Electron Microscopy Sciences, catalog number: 11650)
- Milli-Q ultra-purified water (18.2 MΩcm, Millipore Milli-Q Reference Water Purification System, catalog number: Z00QSV001)
- 16% (w/v) paraformaldehyde solution (see Recipes)
- 2.5% (v/v) glutaraldehyde and 4% (w/v) paraformaldehyde in 0.1 M cacodylate buffer (see Recipes)
- Dehydration and polymer infiltration
- Ethyl alcohol series (ethanol, Rossville Gold Shield, catalog number: 412804, series 50, 70, 95, 100% (v/v))
- Sodium Hydroxide (NaOH) (Sigma-Aldrich, CAS number: 1310-73-2)
- Methyl methacrylate monomer, stabilised (CAS number: 55935-46-1)
- PMMA pellets (Sigma-Aldrich, CAS number: 55935-46-1)
- Calcium chloride anhydrous (pellets, 4-20 mesh) (Fisher Scientific, CAS number: 10043-52-4)
- Benzoyl peroxide powder (Sigma-Aldrich, CAS number: 94-36-0)
- Cleaned Monomer (see Recipes)
- Polymer solution (see Recipes)
- Chemical etching
- Hydrochloric Acid 5 M (HCl, Lowy Solutions, catalog number: UCS-BIO-0035)
- Sodium Hypochlorite 12.5% (NaClO, Chem-Supply Pty Ltd Australia, CAS number: 7681-52-9)
- Methanol (Electron Microscopy Sciences, catalog number: 18510)
- Ethanol, Absolute (Sigma-Aldrich, CAS number: 64-17-5)
- 0.02 M Hydrochloric Acid Solution (see Recipes)
- Sodium Hypochlorite Solution (see Recipes)
- Chemical etching and SEM preparation
- Transfer Pipettes (Thermo Fisher Scientific, catalog number 202-1S)
- Glass Petri Dishes (80 mm diameter)
- Glass Crystallizing dishes (125 mm diameter)
- Lint free cotton swab with anti-static handle (Puritan Medical Products, catalog number: 876-PPC)
Equipment
- Sample sectioning
- Bandsaw (Craftsman)
- Precision Cutter (Isomet 4000 Linear Precision Saw, Buehler)
- Saw Microtome (Leica SP1600 Saw Microtome, Leica Biosystems)
- Electron microscopy system
- Immersion fixation
- Fume hood
- Forceps
- Embedding and Polymerization
- Separatory Funnel (Fisher Scientific, catalog number: 10-437-2B)
- Ring stand (Fisher Scientific, catalog number: S24240)
- Buchner Funnel (lined with filter paper) (Fisher Scientific, catalog number: FB966H)
- Glass jars with lids (Fisher Scientific, catalog number: 02-911-761)
- Mallet
- Surface preparation
- CNC Lathe with Carbide Insert without a honed edge
- Acrylic Polish (Novus Fine Scratch Remover 2)
- Soft Microfiber Cloth
- Semiautomatic Grinder and Polisher (Struers LaboPol-5)
- 1,200-grit Grinding Disk (Struers MD-Piano 1200)
- 500-grit Grinding Disk (Struers MD-Piano 500)
- Polishing Disk for 3 μm suspension (Struers MD-Dac)
- Polishing Disk for 1 μm suspension (Struers MD-Nap)
- 3 µm Diamond Suspension Polishing Compound (Struers DiaPro Dac 3 μm)
- 1 µm Diamond Suspension Polishing Compound (Struers DiaPro Nap 1 μm)
- Chemical etching and SEM preparation
- Evaporative Carbon Coater (JEE-420 Evaporative Coater, JEOL)
- Silver Paste (Electron Microscopy Sciences, catalog number: 12640)
- Ultrasonic Cleaner (Digitech)
Procedure
- Sample sectioning and fixation
- Femoral neck and head samples are sourced from patients who have undergone hip replacement surgery.
- Cut the femoral head into three sections from proximal end to distal end with the precision cutter.
- Cut the medial and lateral portions into three pieces transversely across the bone with the precision cutter.
- Section tissue, with the saw microtome, into 1 mm thick section along the plane of interest, e.g., coronal or sagittal plane, to allow for infiltration of fixation and embedding chemicals.
- Fix the sectioned tissue immediately after with 2.5% glutaraldehyde, 4% paraformaldehyde in 0.2 M cacodylate buffer at room temperature for 5 days.
Note: Tissue must be fully submerged in solution. - Wash sample with cacodylate buffer (0.2 M, pH 7.3) daily for 5 days.
Note: Samples can be stored in cacodylate buffer for up to 2 weeks.
- Dehydration
- Dehydrate the sample in an ethanol series (30%, 50%, 75%, 95%) for 3 days each, changing daily.
- Place the sample in absolute ethanol for 3 days, changing daily.
- Embedding and polymerization
- Fill glass container with polymer.
- Insert specimen with section side of interest facing down.
- Cover with foil and place in explosion-proof refrigerator for 48 h.
- Periodically remove from refrigerator, checking orientation.
- Allow container to remain at room temperature (25 °C) until polymerization occurs (3-7 days). Once polymerization extends slightly above the specimen, check hardness with a teasing needle then place into a 60 °C incubator overnight to complete polymerization.
- Place specimen in plastic bag and strike the glass with a mallet to shatter it.
- Carefully remove block and rinse under running H2O.
- Trim excess plastic from block with bandsaw.
- Surface preparation
Note: Choose option Step D1 if precision-CNC milling is available. Choose option Step D2 if precision CNC milling is not possible or semi-automatic and hand polishing is preferred.- Precision CNC-milling of bulk embedded samples
- Fix the sample onto a CNC-lathe.
- Mill to require depth using a carbide insert without a honed edged facing the sample surface.
- Gently polish the sample surface with a soft microfiber cloth, acrylic polish and ultra-pure H2O.
- Flow ultra-pure H2O over the sample to clear of all polish and residue.
- Gently clean the sample with absolute ethanol and a lint free wipe.
- Ensure the sample is thoroughly dry and free of dust by placing it under a steady stream of inert gas (nitrogen).
- Manual grinding and polishing
- Mount the 500-grit polishing disc on the polishing wheel.
- Start the polishing wheel at 200 rpm and place the surface face down with even pressure.
- Gradually resurface the sample using the 500-grit then lift from the wheel.
- Place the sample in a beaker of H2O and into an ultrasonic cleaner.
- Stop the wheel and then mount the 1,200-grit polishing disc.
- Start the polishing wheel at 200 rpm and place the surface face down with even pressure.
- Gradually resurface the sample using the 1,200-grit then lift from the wheel.
- Place the sample in a beaker of H2O and into an ultrasonic cleaner.
- Stop the wheel and mount the MD-Dac polishing pad and apply a small amount of 3 μm diamond suspension.
- Turn the polishing wheel to 200 rpm and spread the suspension across the polishing pad.
- Press the sample face down with even pressure and gently polish then lift from the wheel.
- Place the sample in a beaker of H2O and into an ultrasonic cleaner.
- Stop the wheel and mount the MD-Nap polishing pad and apply a small amount of 1 μm diamond suspension.
- Turn the polishing wheel to 200 rpm and spread the suspension across the polishing pad.
- Press the sample face down with even pressure and gently polish then lift from the wheel.
- Place the sample in a beaker of H2O and into an ultrasonic cleaner.
- Gently clean the sample with absolute ethanol and a lint free wipe.
- Ensure the sample is thoroughly dry and free of dust by placing it under a steady stream of inert gas (nitrogen).
- Precision CNC-milling of bulk embedded samples
- Chemical etching
- Mount the sample onto the specimen holder, fixing it in place using silver paint (alternately carbon or copper tape may be used).
- Gently clean the sample with absolute ethanol and a lint free wipe.
- Ensure the sample is thoroughly dry and free of dust by placing it under a steady stream of inert gas (nitrogen).
- Define edges of etched zone and protect areas unetched surfaces by covering with masking medium, leaving a large margin, i.e., electrical tape and/or finger nail polish.
- Pipette off 2 ml of HCl solution and place on the surface of the sample that is to be etched.
- Gently expose entire surface of etch area to solution using a cotton swab for 60 s.
- Rinse with copious ultrapure H2O and blot dry with dust-free wipes.
- Pipette off 2 ml of HCl solution and place on the surface of the sample that is to be etched.
- Gently expose entire surface of etch area to solution using a cotton swab for 25 s.
- Rinse with copious ultrapure H2O and blot dry with dust-free wipes.
- To remove flaked off organic material, pipette off 2 ml of NaClO and place on the surface of the sample that is to be etched.
- Gently expose entire surface of etch area to solution using a cotton swab for 60 s.
- Rinse with copious ultrapure H2O and blot dry with absorbent, lint free wipes.
- Gently clean the sample with absolute ethanol and a lint free wipe and if electrical tape has been removed, making sure to remove any adhesive residue.
- Ensure the sample is thoroughly dry and free of dust by placing it under a steady stream of inert gas (nitrogen).
- Place in a protective specimen storage box to prevent exposure to elements or dust prior to carbon sputtering and SEM imaging.
Note: Samples can remain in storage indefinitely before proceeding to the following methods.
- Preparation of sample for imaging
- Gently clean the sample with absolute ethanol and a dust-free wipe.
- Ensure the sample is thoroughly dry and free of dust by placing it under a steady stream of inert gas (nitrogen).
- Carbon-coat the sample with a 20 nm thick layer of film.
- Mount the specimen to the stage or metal stub using silver paint (alternately, carbon or copper tape can be used), ensuring an unbroken and robust connection is made between the sample surface and sample holder.
- Place the sample under vacuum in preparation for imaging in accordance to your electron microscopy system.
Recipes
- 16% (w/v) paraformaldehyde solution.
- Add 8 g of paraformaldehyde into 50 ml of ultra-pure H2O
- Stir the mixture in a fume hood at 60 °C until fully dissolved
Note: If solution does not become clear, add a single crystal of sodium hydroxide. - Allow to cool in fume hood until room temperature (25 °C) has been reached then place in fridge (4 °C).
- 2.5% (v/v) glutaraldehyde and 4% (w/v) paraformaldehyde in 0.1 M cacodylate buffer (200 ml)
Mix 50 ml of 10% (v/v) glutaraldehyde with 50 ml of 16% (w/v) paraformaldehyde in 100 ml of 0.2 M cacodylate buffer
Note: Prepare fresh prior to use. - 0.02 M Hydrochloric Acid Solution (200 ml)
- Add 800 μl of 5 M hydrochloric acid
- Make up the volume to 200 ml using ultra-pure H2O
- Sodium Hypochlorite Solution (200 ml)
Mix 160 ml of 12.5% sodium hypochlorite with 40 ml of ultra-pure H2O - Cleaned Monomer
The methyl methacrylate monomer contains inhibitor hydroquinone to prevent polymerization in transit. The hydroquinone must be removed prior to use. To do this it is ‘washed’ with 5% sodium hydroxide solution. Cleaned monomer is highly flammable and explosive and must be stored in an explosion proof refrigerator at 4 °C prior to use.- Mix 1,000 ml of 5% NaOH and 1,000 ml of methyl methacrylate (MMA)
- Allow the solutions to separate in a separatory funnel, mounted on a ring stand
- Drain off only the lower solution for disposal, repeat twice
- Add 1,000 ml dH2O to the remaining solution, allow it to separate and drain off the lower solution for disposal, repeat twice
- Drain the MMA into a flask through a Buchner funnel lined with filter paper and contains 100 g of calcium chloride pellets
- Repeat with new calcium chloride pellets
- Allow monomer to reach room temperature before using (25 °C)
- Polymer solution
- Add 3 g of dried benzoyl peroxide to 300 ml of clean monomer in the glass screw-top jar and stir until dissolved
- Add 120 g methyl methacrylate beads to the jar gradually over a period of 2-3 days, stirring constantly and allowing beads to dissolve completely between additions
Acknowledgments
This work has been supported in part by the Paul Trainor Foundation and the Alexander von Humboldt Foundation. We would like to acknowledge Gregor Dellemann of Zeiss for his support of the project since its inception in 2011, Professor Thomas Bauer (former affiliation: Orthopedic Pathology, Cleveland Clinic; current affiliation: Pathologist-in-Chief, Department of Pathology and Laboratory Medicine, Hospital for Special Surgery, New York) for his expert advice and assistance, as well as Dr. Judy Drazba and her team at the Core Research Services of the Lerner Research Institute (Cleveland Clinic Foundation), including Mei Yin and Diana Mahovic, for their assistance with embedding of the specimens in EPON® and PMMA. We would also like to acknowledge Dr. Nicholas Ariotti and Natasha Kapoor-Kaushik for technical assistance and use of facilities supported by AMMRF at the Electron Microscope Unit at UNSW. The contributions of Paul Feng, a cooperative student from Institut Polytechnique de Grenoble (Grenoble INP) who assisted with map generation, are also acknowledged with appreciation.
Competing interests
Zeiss Microscopy GmbH, a manufacturer of microscope, provided in-kind-support for the project, including use of their R&D prototyping and demonstration labs. Since 2011, M. Knothe Tate and her team have worked with Zeiss, first on multiSEM prototypes and then commercialized microscopes, in a collaboration based in fundamental discovery and translational R&D**, and without remuneration. Similarly, the Zeiss-affiliated authors have served as scientific R&D collaborators for the aforementioned collaboration (**).
Ethics
Femoral necks and heads, normally discarded after hip replacement surgery, were collected after sectioning into 1 mm thick sections by the Cleveland Clinic Pathology Department per IRB protocol (#12-335) guidelines. Informed consent was obtained from all subjects. Per the IRB protocol, all specimens were de-identified, by removing all identifying information such as name, medical patient numbers, etc. by the tissue procurement and/or pathology department of the clinic prior to release of the tissues for scientific research. Age and sex of the patient were retained on documentation. Together with date of acquisition, each specimen could be separately identified by researchers without having access to medical documentation.
References
- Anderson, E. J., Kreuzer, S. M., Small, O. and Knothe Tate, M. L. (2008) Pairing computational and scaled physical models to determine permeability as a measure of cellular communication in micro- and nano-scale pericellular spaces. Microfluid Nanofluid 4(3): 193-204.
- Chang, H., Docheva, D., Knothe, U. R. and Knothe Tate, M. L. (2014). Arthritic periosteal tissue from joint replacement surgery: a novel, autologous source of stem cells. Stem Cells Transl Med 3(3): 308-317.
- Eberle, A. L., Mikula, S., Schalek, R., Lichtman, J., Tate, M. L. K. and Zeidler, D. (2015). High-resolution, high-throughput imaging with a multibeam scanning electron microscope. J Microsc 259(2): 114-120.
- Hageman, D. et al. (2016). Cellular Epidemiology of Human Disease using Biogeographic and Google Maps Approaches - Towards Definition of Cell Network Indices for Rapid Diagnostics. In: Annals of Biomedical Engineering. 44: 3719.
- Knapp, H. F., Reilly, G. C., Stemmer, A., Niederer, P. and Knothe Tate, M. L. (2002). Development of preparation methods for and insights obtained from atomic force microscopy of fluid spaces in cortical bone. Scanning 24(1): 25-33.
- Knothe Tate, M. L. and Fath, T. (2016a). The only constant is change: next generation medical devices for physical and mental health. Adv Healthcare Mater 5(15): 1840-1843.
- Knothe Tate, M. L. (2017). Navigation of bee brains to human hips-microscopy and the modern Magellans. In: A new age in scanning electron microscopy: Applications in the life sciences. Science/AAAS 34-42.
- Knothe Tate, M. L., Adamson, J. R., Tami, A. E. and Bauer, T. W. (2004). The osteocyte. Int J Biochem Cell Biol 36(1): 1-8.
- Knothe Tate, M. L., Detamore, M., Capadona, J. R., Woolley, A. and Knothe, U. (2016b). Engineering and commercialization of human-device interfaces, from bone to brain. Biomaterials 95: 35-46.
- Knothe Tate, M. L., Falls, T., Mishra, S. and Atit, R. (2010). Engineering an ecosystem: taking cues from nature’s paradigm to build tissue in the lab and the body. In: New Perspectives in Mathematical Biology 57: 113-134. American Mathematical Society.
- Knothe Tate, M. L., Zeidler, D., Pereira, A. F., Hageman, D., Garbowski, T., Mishra, S., Gardner, L. and Knothe, U. R. (2016c). Organ-to-cell-scale health assessment using geographical information system approaches with multibeam scanning electron microscopy. Adv Healthc Mater 5(13): 1581-1587.
- Lichtman, J. W and Sanes, J. R. (2008). Ome sweet ome: what can the genome tell us about the connectome? Curr Opin Neurobiol 18(3): 346–353.
- Mikula, S. and Denk, W. (2015). High-resolution whole-brain staining for electron microscopic circuit reconstruction. Nat Methods 12(6): 541-546.
- Nathanson, A., Srikantha, A., Zeidler, D., Wojek, C. and Knothe Tate, M. L. (2019). Towards cellular epidemiology of degenerative diseases using multiSEM and machine learning approaches. Trans Orth Res Soc 65: 0085.
- National Institutes of Health. NIH to build a detailed map of cells within the human body. (2018). https://www.nih.gov/news-events/news-releases/nih-build-detailed-map-cells-within-human-body.
- Ngo, L., Knothe, L. E. and Knothe Tate, M. L. (2018). Knee joint tissues effectively separate mixed sized molecules delivered in a single bolus to the heart. Sci Rep 8(1): 10254.
- Pereira, A. F., Hageman, D. J., Garbowski, T., Riedesel, C., Knothe, U., Zeidler, D. and Knothe Tate, M. L. (2016). Creating high-resolution multiscale maps of human tissue using multi-beam SEM. PLoS Comput Biol 12(11): e1005217.
- Reilly, G. C., Knapp, H. F., Stemmer, A., Niederer, P. and Knothe Tate, M. L. (2001). Investigation of the morphology of the lacunocanalicular system of cortical bone using atomic force microscopy. Ann Biomed Eng 29(12): 1074-1081.
- Sporns, O., Tononi, G. and Kotter, R. (2005). The human connectome: A structural description of the human brain. PLoS Comput Biol 1(4): e42.
- Tapia, J. C., Kasthuri, N., Hayworth, K. J., Schalek, R., Lichtman, J. W., Smith, S. J. and Buchanan, J. (2012). High-contrast en bloc staining of neuronal tissue for field emission scanning electron microscopy. Nat Protoc 7(2): 193-206.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Ngo, L., Nathanson, A. D., Garbowski, T., Knothe, U., Zeidler, D. and Knothe Tate, M. L. (2019). Electron Microscopy Sample Preparation Protocol Enabling Nano-to-mesoscopic Mapping of Cellular Connectomes and Their Habitats in Human Tissues and Organs. Bio-protocol 9(14): e3298. DOI: 10.21769/BioProtoc.3298.
Category
Cell Biology > Cell imaging > Electron microscopy
Systems Biology > Connectomics > Cellular connectivity
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.