- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

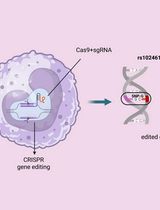
CRISPR/Cas9 + AAV-mediated Intra-embryonic Gene Knocking in Mice

Published: Vol 9, Iss 13, Jul 5, 2019 DOI: 10.21769/BioProtoc.3295 Views: 7387
Reviewed by: David PaulSylvain Baron Anonymous reviewer(s)
Original research article
The authors used this protocol in:

Nov 2018
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Intra-embryo genome editing by CRISPR/Cas9 has enabled rapid generation of gene knockout animals. However, large fragment knock-in directly into embryos’ genome is still difficult, especially without microinjection of donor DNA. Viral vectors are good transporters of knock-in donor DNA for cell lines, but seemed unsuitable for pre-implantation embryos with zona pellucida, glycoprotein membrane surrounding early embryos. We found adeno-associated virus (AAV) can infect zygotes of various mammals through intact zona pellucida. AAV-mediated donor DNA delivery following Cas9 ribonucleoprotein electroporation enables large fragment knock-in without micromanipulation.
Keywords: CRISPR/Cas9Background
Genome editing techniques, particularly the clustered regularly interspaced short palindromic repeats (CRISPR)-associated protein 9 (CRISPR/Cas9) technology, have significantly simplified the generation of transgenic animals. Conventionally, site-directed transgenesis of mice and rats was developed by founder chimeras with genetically modified pluripotent stem cells, which potentially contribute to the germline. However, this conventional strategy is labor-intensive and does not guarantee the germline transmission. Recently, genome editing has enabled site-directed mutagenesis by allowing the direct introduction of required components into embryos at a 1-cell stage. This alternative strategy eliminates the need for any transgenic donor cells, thus resulting in the rapid generation of founder rodents. The genome editing components Cas9 and guide RNA (gRNA) in the CRISPR/Cas9 system, are introduced by microinjection into the zygote pronucleus. Although an intra-embryonic microinjection requires advanced training and expensive micromanipulation equipment, it has enabled the gene-knockout as well as knock-in of large-fragments (Yoshimi et al., 2016; Miura et al., 2018). Alternatively, CRISPR/Cas9 ribonucleoprotein-mediated editing in zygotes can be performed by electroporation; however, large fragment knock-in cannot be efficiently performed (Chen et al., 2016; Hashimoto et al., 2016). The difference between Cas9 RNP-mediated gene knock-in and knockout is the presence of knock-in donor DNA. The donor DNA must be transported inside the cell nucleus; however, this is challenging because of the requirement of a driving force for transport from the cytosol to nucleus as opposed to Cas9 RNP with a nuclear localization signal. Unlike direct microinjection into the pronucleus, electroporation may not introduce a sufficient amount of the donor DNA into the pronucleus. Theoretically, DNA viral vectors are efficient transporters of knock-in donor DNA because they can transfer their genomic DNA to host cells’ nucleus where the in-transit nuclease-mediated degradation of DNA is prevented. However, this strategy seemed unsuitable for pre-implantation embryos possessing a zona pellucida, which inhibits viral infection. In recent studies, we and other groups found AAV can infect mice zygotes with intact zona pellucida (Mizuno et al., 2018; Yoon et al., 2018). We have successfully performed knock-in of a large fragment directly into mice embryos via CRISPR/Cas9 RNP electroporation following a trans-zona pellucida delivery of donor DNA using AAV vectors. Here, we demonstrate a detailed protocol for AAV-mediated intra-embryo knock-in in laboratory mice. This technique does not require either micromanipulation or germline competent pluripotent stem cells, which are successfully established only for rodents to date. AAV can infect various mammalian zygotes, including rats and cows. Thus, this simple genome editing method could be potentially applied to several species.
Materials and Reagents
- 15 ml tube (TPP, catalog number: 91015)
- 50 ml tube (Hitachi, catalog number: S307904A)
- 1.5 ml tube (Simport, catalog number: T332-5S)
- Millex-GV Syringe Filter Unit, 0.22 µm, PVDF, 33 mm (Merck, catalog number: SLGV033RB)
- Millex-HV Syringe Filter Unit, 0.45 µm, PVDF, 33 mm (Merck, catalog number: SLHV033RS)
- 10 ml syringe (Terumo, catalog number: SS-10SZ)
- P200 Tip (QSP, catalog number: 110RL-NEW)
- P1000 Tip (Rikaken, catalog number: RST-4810BR)
- 5 ml pipette (Nunc, catalog number: 170355N)
- 10 ml pipette (Nunc, catalog number: 170356N)
- Aspirating pipette (Corning, catalog number: 357558)
- 10 cm culture dish (TPP, catalog number: 93100)
- PCR tube (FastGene, catalog number: FG-0028DC/SE)
- 35 mm culture dish, non-treated (Iwaki, catalog number: 1000-035)
- Fine glass pipette (Prepared in the laboratory, made of glass tube, ERMA, catalog number: 05-791-0)
- Amicon Ultra-4, 100 kDa (Merck, catalog number: UFC810024)
- C57BL/6N male mice (Japan SLC)
- B6D2F1 female mice (Japan SLC)
- ICR female mice (Japan SLC)
- 293T cell (Clontech, catalog number: 632180)
- Recombinant AAV vector plasmid (for your knock-in cassette flanked by homology arms toward your gene of interest)
- pAAM-MCS2 (recombinant AAV vector backbone, a gift from Steve Jackson, Addgene, Addgene plasmid number: 46954)
- pDGM6 (a gift from David Russell, Addgene, Addgene plasmid number: 110660)
- Cas9 protein (IDT, catalog number: 1074182)
- crRNA (IDT, AltR grade, for your gRNA sequence)
- tracrRNA (IDT, catalog number: 1072533)
- DMEM (SIGMA, catalog number: D5796-500ML)
- FBS (Gibco, catalog number: 10437-028)
- L-Glutamine-Penicillin-Streptomycin solution (SIGMA, catalog number: G1146-100ML)
- Trypsin-EDTA (0.05%), phenol red (Thermo, catalog number: 25300-120)
- Distilled water (Wako, catalog number: 043-16785)
- BES (SIGMA, catalog number: B4554)
- NaCl (SIGMA, catalog number: 28-2270-5)
- Na2HPO4 (Wako, catalog number: 197-02865)
- NaOH (Wako, catalog number: 198-13765)
- CaCl2·2H2O (Wako, catalog number: 031-00435)
- AAVpro Purification Kit (All Serotypes) (Takara, catalog number: 6666) (This kit contains AAV Extraction Solution A plus, AAV Extraction Solution B, Cryonase Cold-active Nuclease, Precipitator A, and Precipitator B)
- EDTA-2Na·2H2O (SIGMA, catalog number: 09-1420-5)
- 10x D-PBS(-) (KCl 0.2% w/v, KH2PO4 0.2% w/v, NaCl 8.0% w/v, Na2HPO4 1.15% w/v, pH 7.1-7.7) (Wako, catalog number: 048-29805)
- M2 medium (Merck Millipore, catalog number: MR-015P-5D)
- KSOM (Merck Millipore, catalog number: MR-020P-5D)
- Mineral oil (SIGMA, catalog number: M8410-1L)
- MEM-EAA (Gibco, catalog number: 11130-051)
- MEM-NEAA (Gibco, catalog number: 11140-050)
- Hyaluronidase (SIGMA, catalog number: H4272-30MG)
- Opti-MEM I medium (Gibco, catalog number: 31985-062)
- Pregnant mare serum gonadotropin (PMSG) (Aska Pharmacies)
- Human chorionic gonadotropin (hCG) (Aska Pharmacies)
- 1x PBS (see Recipes)
- DMEM supplemented with 10% FBS (see Recipes)
- 2x BBS (see Recipes)
- 150 mM Na2HPO4 (see Recipes)
- 2.5 M CaCl2 (see Recipes)
- 0.5 M EDTA (pH 8.0) (see Recipes)
- KSOM-AA medium (see Recipes)
Equipment
- P20 pipette (GILSON, catalog number: F123600)
- P200 pipette (GILSON, catalog number: F123601)
- P1000 pipette (GILSON, catalog number: F123602)
- 37.0 °C bath (lab armor, catalog number: M714)
- Centrifuge (TOMY, catalog number: AX-310)
- Centrifuge (HITACHI, catalog number: CR21GII)
- Thermal cycler (Takara, catalog number: TP350)
- Electroporator (BEX, catalog number: GEB15)
- 1 mm gap electrode (BEX, catalog number: LF501PT1-10)
- Stereoscopic microscope (Nikon, model: SMZ1000)
- Thermo plate for microscope (TOKAI HIT, model: TP-SMZSL)
- Anaesthetic vaporizer (Muromachi Kikai, model: MK-A110)
- CO2 incubator (Wakenyaku, model: 9300E)
- -80 °C freezer (Panasonic, model: MDF-U700VX-PJ)
Procedure
- AAV vector plasmid construction
- Design your AAV vector plasmid and construct it by conventional molecular biological techniques (Green et al., 2012; Behringer et al., 2003). Empty backbone of AAV vector, pAAV-MCS2, is deposited to Addgene.
- Insert your knock-in cassette flanked by 5′ and 3′ homology arms into your gene of interest between AAV inverted terminal repeats (ITRs). We recommend 300-1,000 bp homology arms for each side, but 100 bp homology arms are still sufficient for the knock-in.
- AAV vector production, extraction and purification
Day 0:- Seed 4.0-4.5 x 106 293T cells/10 cm dish with 10 ml DMEM supplemented with 10% FBS. Prepare 8 dishes.
- Incubate in a 37.0 °C, 10% CO2 incubator (or 5% incubator, alternatively).
Day 1:- Incubate sterile distilled water, 2.5 M CaCl2, 2x BBS in a 37.0 °C bath for 30-60 min. Vortex briefly.
- Add 80 μl AAV plasmid (1 mg/ml) and 200 μl pDGM6 (1 mg/ml) into 3,320 μl sterile distilled water in a 15 ml tube (10 μl AAV plasmid, 25 μl pDGM6, 415 μl sterile distilled water for one 10 cm dish transfection, respectively). Vortex briefly.
- Add 400 μl 2.5 M CaCl2 solution (50 μl for each 10 cm dish transfection) into the 15 ml tuble containting plasmids. Immediately vortex for 10 s.
- After vortexing, immediately add 4 ml 2x BBS (500 μl for one 10 cm dish transfection), and then shake the 15 ml tubes vigorously. Alternatively, add 2x BBS into the 15 ml tube under continuous vortexing.
- Incubate at RT for 20 min.
- Add 1 ml transfection solution (prepared on Procedure B Day 1: Steps 1 to 5) to each 10 cm dish.
- Incubate in a 37.0 °C, 3% CO2 incubator (or 5% incubator, alternatively).
- Aspirate medium containing transfection solution, and add 5 ml DMEM supplemented with 10% FBS with a 5 ml pipette.
- Incubate in a 37.0 °C, 10% CO2 incubator (or 5% incubator, alternatively) until Day 4.
- Add x1/80 volume of 0.5 M EDTA (pH 8.0) (62.5 μl into 5 ml medium per 10 cm dish).
- Incubate at RT for 10 min. The cells would be detached from the dish after incubation.
- Collect 293T cells and culture medium into a 50 ml tube with a 10 ml pipette.
- Centrifuge at 4 °C, 1,700-2,000 x g, 10 min.
- Aspirate supernatant.
- Centrifuge at 4 °C, 1,700-2,000 x g, 1 min to collect residual medium.
- Aspirate supernatant completely.
- Vortex the 50 ml tube and dissociate cell pellet.
- Add 4 ml AAV Extraction Solution A plus (0.5 ml for each 10 cm dish transfection).
- Vortex for 15 s.
- Incubate at RT for 5 min.
- Vortex again for 15 s.
- Incubate at RT for another 5 min.
- Vortex again for 15 s.
- Centrifuge the 50 ml tube at 4 °C, 9,000 x g, 10 min.
- Transfer the supernatant to a fresh 50 ml tube with a P1000 tip pipette.
- Add 400 μl AAV Extraction Solution B (10% volume of AAV Extraction Solution A plus), and mix it gently.
- Add 40 μl Cryonase Cold-active Nuclease (1% volume of AAV Extraction Solution A plus), and mix it gently.
- Incubate at 37.0 °C for 60 min.
- Add 400 μl Precipitator A (10% volume of AAV Extraction Solution A plus).
- Vortex for 10 s.
- Incubate at 37.0 °C for 30 min.
- Vortex for 10 s.
- Add 200 μl Precipitator B (5% volume of AAV Extraction Solution A plus).
- Vortex for 10 s.
- Centrifuge at 4 °C, 9,000 x g, 20 min.
- Transfer the supernatant to a fresh 50 ml tube with a P1000 tip pipette.
- Centrifuge again at 4 °C, 9,000 x g, 20 min.
- Transfer the supernatant to a 6-10 cm dish with a P1000 tip pipette (It might be difficult to aspirate the supernatant from the 50 ml tube directly with a 10 ml syringe).
- Aspirate the supernatant with a 10 ml syringe, and filtrate it with 0.45 μm PVDF filter into a 15 ml tube.
- Transfer the filtered supernatant into Amicon Ultra-4, 100 kDa.
- Centrifuge at 15 °C, 2,000 x g, 5 min.
- Discard flow-through, add 4 ml 1x PBS onto the filter-unit, and centrifuge at 15 °C, 2,000 x g, 5 min.
- Repeat the previous ultrafiltration step, 7 times.
- Collect AAV vector solution with a P200 pipette into a 1.5 ml tube. Approximately 100-120 μl of viral vector suspension would be recovered.
- Dilute AAV vector solution with 1x PBS up to 200 μl, and dispense 20 μl aliquots in 1.5 ml tubes.
- Store AAV vector solution at -80 °C.
- Estimation of AAV vector titer
Estimate the AAV vector titer by qPCR (Lock et al., 2014; Werling et al., 2015). - Preparation of pronuclear-stage mice embryos
Prepare pronuclear-stage mice embryos as described previously (Behringer et al., 2003). Transfer embryos with a fine glass pipette.- Administer 5 IU/mouse PMSG into B6D2F1 female mice by intraperitoneal injection at 15:00-18:00 PM.
- Forty-eight hours after PMSG administration, administer 5 IU/mouse hCG into B6D2F1 female mice by intraperitoneal injection.
- Immediately after hCG administration, mate female mice with C57BL/6N male mice.
- 14-16 h after hCG injection, retrieve zygotes from oviduct, and remove surrounding cumulus cells by short-term culture and pipetting in 0.1% hyaluronidase-containing M2 media.
- Incubate zygotes in KSOM-AA medium at 37.0 °C in 5% CO2 in air atmosphere for 1-4 h.
- Collect two-pronuclear zygotes.
- Preparation of Cas9 RNP solution
- Reconstitute lyophilized crRNA and tracerRNA with Opti-MEM I medium at 100 μM each.
- Mix 10 μl of crRNA and an equal amount of tracrRNA in PCR tube.
- Anneal crRNA with tracrRNA in a PCR thermal cycler. Heat it 95 °C for 5 min, and cool it down to 25 °C by 5 °C/min, then keep it at 4 °C.
- Dilute 100 ng/μl (0.61 μM) Cas9 protein, 200 ng/μl (2.94 μM) annealed gRNA complex (crRNA + tracrRNA) in Opti-MEM I medium. Prepare enough amount of the Cas9 RNP solution; 20-30 μl to wash embryos before electroporation, and 5 μl/run on electroporation.
- Keep Cas9 RNP solution on ice until the electroporation.
- Preparation of embryo culture medium containing knock-in donor AAV vector
- Dilute knock-in donor AAV with KSOM-AA medium at 1 x 107-1 x 108 vg/ml. We recommend 3 x 107 vg/ml for the initial trial, but optimal AAV dose should be determined empirically in each laboratory.
- Prepare 100 μl drop of KSOM-AA medium containing AAV vector for embryo transduction, and three 100 μl drops of KSOM-AA medium without AAV for washing embryo after electroporation for every 40-60 embryos in a 35-mm non-treated culture dish. Cover it with mineral oil.
- Incubate the dish at 37.0 °C in 5% CO2 air atmosphere for more than 30 min.
- Cas9 RNP electroporation and donor AAV transduction
- Connect 1 mm gap electrode on stereoscopic microscope with the electroporator. DO NOT turn on the thermal plate of microscope during electroporation steps. Otherwise, electrode solution would evaporate immediately (Figure1A).
- Wash 1 mm gap electrode with 5 μl Opti-MEM I media, 3 times.
- Prepare 20-30 μl drop of Cas9 RNP solution on 35-mm non-treated culture dish for washing embryos.
- Wash 20-30 two-pronuclear zygotes with Cas9 RNP solution prepared on Step G3.
- Add 5 μl Cas9 RNP solution on 1 mm gap electrode.
- Immediately transfer 20-30 two-pronuclear zygotes on 1 mm gap electrode (Figure 1B).
- Perform electroporation within 1 min. Condition is as following: 25 V, 3 ms ON, 97 ms OFF, Pd Alt 3-4 times.
- Collect embryos on the electrode, and wash them with KSOM-AA medium three times.
- Transfer embryos into KSOM-AA medium containing AAV vector.
- Incubate embryos at 37.0 °C in 5% CO2 air atmosphere for 16-20 h.
- Repeat Steps G2-G10 for next run of electroporation. Cas9 RNP solution should be replaced with new 5 μl Cas9 RNP solution for the next 20-30 embryos.
- Prepare pseudopregnant female by mating proestrus stage ICR female mice with ICR male mice received vasectomy.
- Sixteen to twenty hours after electroporation, collect embryos at 2-cell stage.
- Wash embryos with M2 medium three times, and transfer to oviduct of E0.5 pseudopregnant females under anesthesia.

Figure 1. Cas9 RNP electroporation of mice zygotes. A. Required equipment for zygote electroporation. The 1 mm gap electrode on the microscope is connected to the electroporator. B. Two-pronuclear zygotes on 1 mm gap electrode. Scale bar, 100 μm.
Data analysis
The knock-in efficiency and the development rate of embryos were described in Table 2 of the original article (https://doi.org/10.1016/j.isci.2018.10.030). The knock-in efficiency blastocyst stage embryos cultured in vitro is similar to that of the offsprings. We recommend genotyping of blastocysts in preliminary experiments.
Notes
- All materials should be lot-checked for embryo culture.
- All mice were maintained in a 12 h:12 h light-dark cycle (08:00-20:00).
- In general, we prepare 100-200 zygotes for one experiment. This is enough to obtain a founder mouse in many cases.
- To exclude potential off-target insertion of transgenes, consider copy number estimation of the transgene on the offspring’s’ genome by digital PCR or conventional southern blotting.
Recipes
- 1x PBS
Dilute 500 ml 10x D-PBS(-) with 4.5 L distilled water
Sterilize by autoclave - DMEM supplemented with 10% FBS
DMEM
10% FBS (heat-inactivated)
1% L-Glutamine-Penicillin-Streptomycin solution - 2x BBS
50 mM BES
280 mM NaCl
1.5 mM Na2HPO4
Adjust the pH to 6.95-7.2 with NaOH
Optimal pH is different in each lot and laboratories
Sterilize with 0.22 μm filter, and store at -30 °C - 150 mM Na2HPO4
Dissolve 1.065 g Na2HPO4 in 50 ml distilled water - 2.5 M CaCl2
Dissolve 18.38 g CaCl2·2H2O in 50 ml distilled water
Sterilize with 0.22 μm filter, and store at -30 °C - 0.5 M EDTA (pH8.0)
- Dissolve EDTA-2Na·2H2O 93.06 g in 500 ml distilled water
- Adjust the pH to 8.0 with NaOH
- Sterilize with 0.22 μm filter
- KSOM-AA medium
1x KSOM media
1% MEM-EAA
0.5% MEM-NEAA
Acknowledgments
We thank F. Suchy, A. Ogawa, Dr. Toshiya Nishimura, Dr. Hideki Masaki, Dr. Sanae Hamanaka and A. Umino for helpful advice. We also thank Y. Yamamoto, J. Iwano, N. Sato, H. Tsukui for technical support, K. Okada for secretarial support, and M. Watanabe for advice in preparing the manuscript. This work was supported by grants from Leading Advanced Projects for medical innovation, Japan Agency for Medical Research and Development.
Competing interests
The authors declare no competing interests.
Ethics
All animal experiments were approved by the Institutional Animal Care and Use Committee (PA17-59, November 22th 2017 March 31th 2022), and performed in accordance with the guidelines of the University of Tokyo and the National Institute for Physiological Sciences.
References
- Behringer, R., Gertsenstein, M., Nagy, K. V. and Nagy, A. (2003). Manipulating the mouse embryo a laboratory manual. Cold Spring Harbor Laboratory Press. New York.
- Green, M. R. and Sambrook, J. (2012). Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press. New York.
- Chen, S., Lee, B., Lee, A. Y., Modzelewski, A. J. and He, L. (2016). Highly efficient mouse genome editing by CRISPR ribonucleoprotein electroporation of zygotes. J Biol Chem 291(28): 14457-14467.
- Hashimoto, M., Yamashita, Y. and Takemoto, T. (2016). Electroporation of Cas9 protein/sgRNA into early pronuclear zygotes generates non-mosaic mutants in the mouse. Dev Biol 418(1): 1-9.
- Lock, M., Alvira, M. R., Chen, S. J. and Wilson, J. M. (2014). Absolute determination of single-stranded and self-complementary adeno-associated viral vector genome titers by droplet digital PCR. Hum Gene Ther Methods 25(2): 115-125.
- Miura, H., Quadros, R. M., Gurumurthy, C. B. and Ohtsuka, M. (2018). Easi-CRISPR for creating knock-in and conditional knockout mouse models using long ssDNA donors. Nat Protoc 13(1): 195-215.
- Mizuno, N., Mizutani, E., Sato, H., Kasai, M., Ogawa, A., Suchy, F., Yamaguchi, T. and Nakauchi, H. (2018). Intra-embryo gene cassette knockin by CRISPR/Cas9-Mediated genome editing with adeno-associated viral vector. iScience 9: 286-297.
- Werling, N. J., Satkunanathan, S., Thorpe, R. and Zhao, Y. (2015). Systematic comparison and validation of quantitative real-time PCR methods for the quantitation of adeno-associated viral products. Hum Gene Ther Methods 26(3): 82-92.
- Yoon, Y., Wang, D., Tai, P. W. L., Riley, J., Gao, G. and Rivera-Perez, J. A. (2018). Streamlined ex vivo and in vivo genome editing in mouse embryos using recombinant adeno-associated viruses. Nat Commun 9(1): 412.
- Yoshimi, K., Kunihiro, Y., Kaneko, T., Nagahora, H., Voigt, B. and Mashimo, T. (2016). ssODN-mediated knock-in with CRISPR-Cas for large genomic regions in zygotes. Nat Commun 7: 10431.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Mizuno, N., Mizutani, E., Sato, H., Kasai, M., Nakauchi, H. and Yamaguchi, T. (2019). CRISPR/Cas9 + AAV-mediated Intra-embryonic Gene Knocking in Mice. Bio-protocol 9(13): e3295. DOI: 10.21769/BioProtoc.3295.
Category
Stem Cell > Pluripotent stem cell > Regenerative medicine
Developmental Biology > Cell growth and fate > Regeneration
Cell Biology > Cell engineering > CRISPR-cas9
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.