- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Immunoprecipitation and Sequencing of Acetylated RNA

Published: Vol 9, Iss 12, Jun 20, 2019 DOI: 10.21769/BioProtoc.3278 Views: 8059
Reviewed by: Gal HaimovichVaibhav B. ShahAnca Flavia Savulescu
Original research article
The authors used this protocol in:

Dec 2018
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Generation of the epitranscriptome through chemical modifications of protein-coding messenger RNAs (mRNAs) has emerged as a new mechanism of post-transcriptional gene regulation. While most mRNA modifications are methylation events, a single acetylated ribonucleoside has been described in eukaryotes, occurring at the N4-position of cytidine (N4-acetylcytidine or ac4C). Using a combination of antibody-based enrichment of acetylated regions and deep sequencing, we recently reported ac4C as a novel mRNA modification that is catalyzed by the N-acetyltransferase enzyme NAT10. In this protocol, we describe in detail the procedures to identify acetylated mRNA regions transcriptome-wide using acetylated RNA immunoprecipitation and sequencing (acRIP-seq).
Keywords: N4-acetylcytidineBackground
Chemical modifications of RNA have emerged as a new mechanism for the post-transcriptional regulation of gene expression (Zhao et al., 2017). In contrast to a handful of well-studied modified bases that constitute the DNA epigenome, over a hundred modified ribonucleosides are present in all types of RNA. This diversity offers new possibilities to modulate gene expression, significantly expanding the metabolic and regulatory functions of RNA (Zhao et al., 2017). Mainly studied in abundant transfer RNA (tRNA) and ribosomal RNA (rRNA), ribonucleoside modifications have more recently been described in messenger RNAs (mRNAs), where they form the basis of the epitranscriptome (Zhao et al., 2017). These epitranscriptomic modifications have the potential to control all steps of mRNA metabolism including structure, stability, location and translation.
Several modifications, including N6-methyladenosine (m6A), N6,2’-O-dimethyladenosine (m6Am), N1-methyladenosine (m1A), pseudouridine (Ψ), inosine (I), 5-methylcytidine (m5C), 5-hydroxylmethylcytidine (hm5C), 7-methylguanosine (m7G) and 2’-O-methylnucleosides (Nm) are found in eukaryotic mRNA and can influence the metabolism and function of mRNA (Li et al., 2016). While the majority of mRNA modifications are methylation events, only a single acetylated ribonucleoside has been described in eukaryotes, occurring at the N4-position of cytidine (N4-acetylcytidine, ac4C, Figure 1A). ac4C is one of the universally conserved RNA modifications present in all domains of life and had previously been observed in rRNA and tRNA (Ito et al., 2014a and 2014b; Sharma et al., 2015). More recently, ac4C was observed in RNA viruses and in polyadenylated RNA (poly(A)-RNA) of yeast and human cancer cells (Dong et al., 2016; McIntyre et al., 2018; Tardu et al., 2018), raising the possibility that ac4C is present in mRNA. In all cases, ac4C production was attributed to the enzyme NAT10 (Figure 1A), or its homologs (Ito et al., 2014a and 2014b; Sharma et al., 2015), suggesting a non-redundant activity.
To investigate the occurrence of ac4C in human mRNA, we generated tools for antibody-based enrichment and transcriptome-wide mapping, a method we named acRIP-seq (Arango et al., 2018). Additionally, to assess ac4C regulation and function, we deleted NAT10 in HeLa cells. The sum of these studies confirmed NAT10-dependent acetylation of mRNAs (Arango et al., 2018). Overall, acetylation was enriched within coding sequences and loss of ac4C through NAT10 ablation led to a specific decrease in the expression of defined ac4C targets. More specifically, we found that ac4C increases mRNA stability and enhances translation efficiency of mRNAs when the modification is present in coding sequences (Arango et al., 2018).
Here, we report a detailed protocol for acetylated RNA immunoprecipitation and sequencing, acRIP-seq (Figures 1B-1C). This method is used to identify acetylated mRNA regions transcriptome-wide.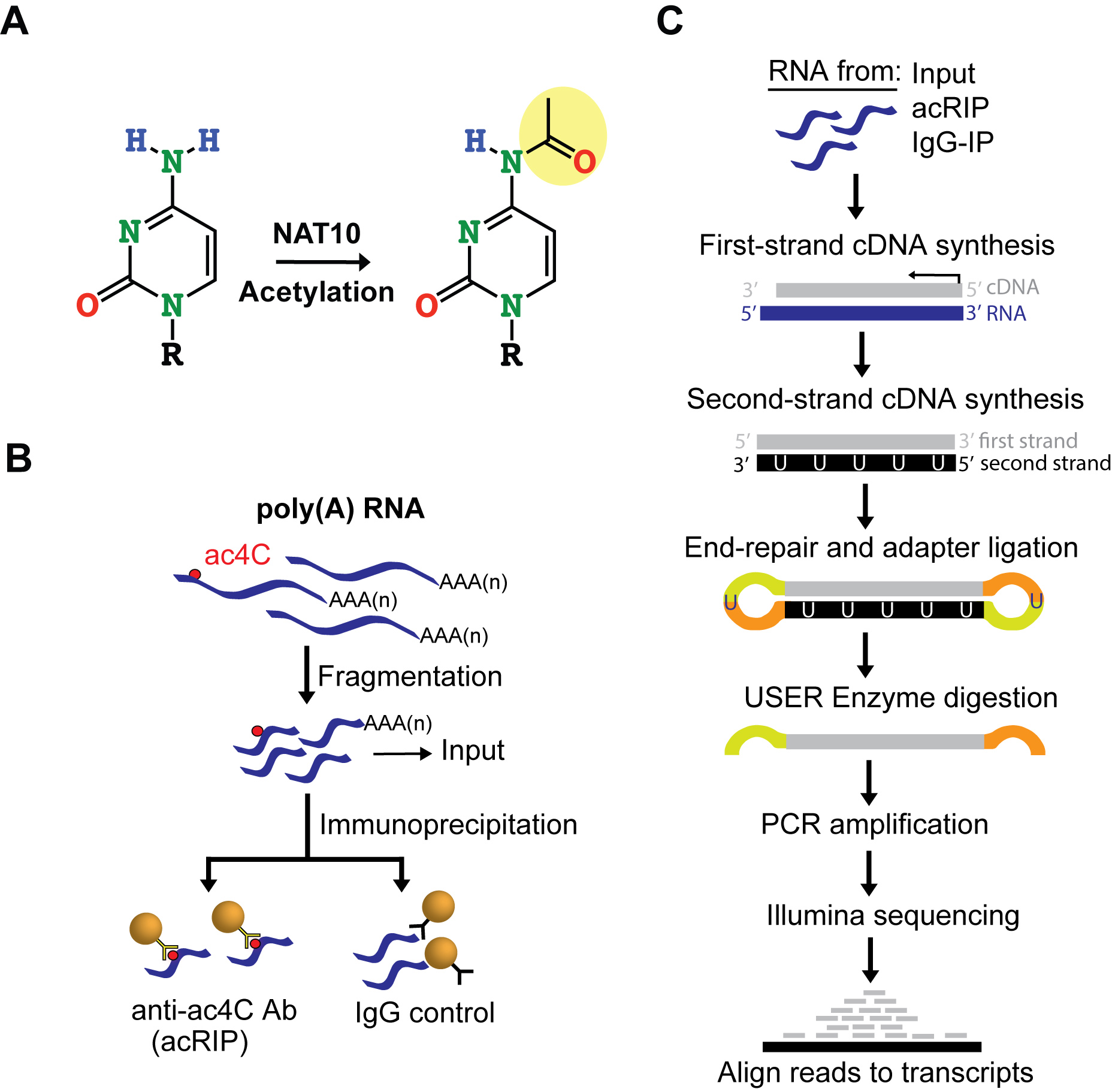
Figure 1. acRIP-seq identifies acetylated regions within mRNA transcriptome-wide. A. NAT10 catalyzes cytidine acetylation. B-C. Schematics of acRIP-seq. Fragmented poly(A) RNA is immunoprecipitated (IP) with anti-ac4C antibodies or Isotypic IgG control (B). RNA from acRIP, IgG-IP and Inputs are used to construct Illumina sequences using the NEBNext® UltraTM II Directional RNA Library protocol followed by high-throughput sequencing and bioinformatical analysis to identify putative acetylated regions transcriptome-wide (C). USER: Uracil-Specific Excision Reagent.
Materials and Reagents
- Pipette tips (RNase-free)
- 0.2 ml PCR tubes (RNase-free)
- 1.5 ml microcentrifuge tubes (RNase-free)
- 2.0 ml microcentrifuge tubes (RNase-free)
- NEBNext® Magnesium RNA Fragmentation Module (NEB, catalog number: E6150)
- NEBNext® UltraTM II Directional RNA Library Prep Kit (NEB, catalog number: E7760S)
- NEBNext® Multiplex Oligos for Illumina® (Index Primers Set 1, NEB, catalog number: E7335S)
- RNase inhibitor, murine (NEB, catalog number: M0314S)
- Agilent RNA 6000 kit (Agilent, catalog number: 5067-1511)
- Agencourt AMPure XP Beads (Beckman Coulter, catalog number: A63881)
- Agencourt RNAclean XP Beads (Beckman Coulter, catalog number: A63987)
- MAXIscript T7 Transcription Kit (Thermo Fisher Scientific, catalog number: AM1312)
- Protein G-Magnetic Dynabeads (Thermo Fisher Scientific, catalog number:10003D)
- DynabeadsTM oligo(dT)25 (Thermo Fisher Scientific catalog number: 61002)
- Linear acrylamide (Thermo Fisher Scientific, catalog number: AM9520)
- Acid-Phenol:Chloroform, pH 4.5 (Thermo Fisher Scientific, catalog number: AM9722)
- Proteinase K, RNase-free (Thermo Fisher Scientific, catalog number: AM2546)
- EDTA, RNase-free (Thermo Fisher Scientific, catalog number: AM9260G)
- Sodium Acetate, RNase-free (Thermo Fisher Scientific, catalog number: AM9740)
- Nuclease-free H2O (Thermo Fisher Scientific, catalog number: AM9938)
- Tris-HCl, RNase-free (Thermo Fisher Scientific, catalog number: 15567027)
- NaCl, RNase-free (Thermo Fisher Scientific, catalog number: AM9759)
- SDS, RNase-free (Thermo Fisher Scientific, catalog number: AM9823)
- Bovine Serum Albumin (BSA, Thermo Fisher Scientific, catalog number: AM2616)
- Ethanol (Fisher Scientific, catalog number: BP2818100)
- Chloroform (Fisher Scientific, catalog number: BP1145-1)
- Sodium Phosphate monobasic (Sigma-Aldrich, catalog number: S0751)
- Sodium Phosphate dibasic (Sigma-Aldrich, catalog number: S3264)
- Triton X-100 (Sigma-Aldrich, catalog number: 93443)
- Rabbit monoclonal IgG isotype control (Cell Signaling Technologies, catalog number: 3900S)
- Rabbit monoclonal anti-N4-acetylcytidine (Abcam, catalog number: ab252215)
- ac4CTP (Trilink, catalog number: C-0001)
- Purified poly(A) RNA
- Acetylated RNA (see section C: In vitro transcription of acetylated RNA)
- LiCl (Sigma-Aldrich, catalog number: L7026)
- 10x acRIP buffer (see Recipes)
- Proteinase K buffer (see Recipes)
- Poly(A) RNA binding buffer (see Recipes)
- Poly(A) RNA washing buffer (see Recipes)
Equipment
- Pipette 0.2 to 2 μl (Thermo Fisher Scientific, catalog number: 4642010)
- Pipette 2 to 20 μl (Thermo Fisher Scientific, catalog number: 4642060)
- Pipette 20 to 200 μl (Thermo Fisher Scientific, catalog number: 4642080)
- Pipette 100 to 1000 μl (Thermo Fisher Scientific, catalog number: 4642090)
- -20 °C freezer (General Electric, model FUM 21SVCRWW)
- -80 °C freezer (Thermo Fisher Scientific, model Ultima Plus)
- Thermal Cycler (Thermo Fisher Scientific, catalog number: 4484073)
- ThermoMixer (Eppendorf, catalog number: 2231000574)
- Refrigerated Microcentrifuge (Eppendorf, catalog number: 5404000138)
- Rotator RotoFlex (Denville Scientific, catalog number: H5700)
- Magnetic rack for 1.5 ml tubes DynaMagTM-2 Magnet (Thermo Fisher Scientific, catalog number: 12321D)
- Magnetic rack for 0.2 ml tubes DiaMag02 (Diagenode, catalog number: B04000001)
- Nanodrop Spectrophotometer (Nanodrop, catalog number: ND-1000)
- Agilent 2100 Bioanalyzer (Agilent, catalog number: G293BA)
- HiSeq 2500 Illumina sequencer (Illumina, model HiSeq 2500)
Procedure
Notes:
- The acRIP-seq protocol starts with purified RNA. We used poly(A) RNA to specifically map acetylated regions in mRNA. However, this procedure can be extended to any RNA class of interest.
- The steps described below are for one biological replicate. However, acRIP-seq should be performed in at least two biological replicates. A negative control consisting of ac4C-depleted RNA should be included. This can be accomplished by using poly(A) RNA from NAT10-/- cells (Arango et al., 2018).
- Poly(A) RNA purification
- For poly(A) RNA purification, we used two rounds of poly(A) selection using DynabeadsTM oligo(dT)25. Precautions should be taken to avoid ribonuclease contamination during the isolation procedure.
- Resuspend 1.8 mg of total RNA, obtained using the TRizol method (Rio et al., 2010), in 2.4 ml of Nuclease-free H2O at a concentration of 750 μg/ml. This amount of starting material is enough to obtain ~20 μg of poly(A) RNA.
- Resuspend the Dynabeads® Oligo (dT)25 and transfer 1.2 ml (6 mg) of beads to a 2.0 ml microcentrifuge tube.
- Magnetically separate the Dynabeads using a magnetic stand.
- Wash once with 1.2 ml poly(A) RNA washing buffer.
- Resuspend beads in 1.2 ml poly(A) RNA binding buffer.
- Pipette 600 μl of total RNA from Step A2 into a 1.5 ml microcentrifuge tube and heat at 75 °C for 2 min in a thermomixer set at 1,200 rpm.
- Immediately add the entire 600 μl of heated RNA to 1.2 ml beads from Step A6 to achieve a total volume of 1.8 ml.
- Mix thoroughly and anneal through continuous rotation for 5 min at room temperature.
- Magnetically separate the Dynabeads using a magnetic stand and carefully remove the supernatant.
- Remove the tube from the magnet and add 1.2 ml poly(A) RNA washing buffer.
- Repeat Steps A10-A11 for a total of three washes.
- Add 600 μl of 10 mM Tris-HCl [pH 7.5] and heat at 75 °C for 2 min in a thermomixer set at 1,200 rpm.
- Add 1.2 ml poly(A) RNA binding buffer.
- Mix thoroughly and anneal through continuous rotation for 5 min at room temperature.
- Magnetically separate the Dynabeads using a magnetic stand and carefully remove the supernatant.
- Remove the tube from the magnet and add 1.2 ml poly(A) RNA washing buffer.
- Repeat Steps A16-A17 for a total of three washes.
- Add 50 μl of 10 mM Tris-HCl [pH 7.5] and heat at 75 °C for 2 min in a thermomixer set at 1,200 rpm.
- Quickly place the tube in a magnetic stand and transfer the supernatant to a new 1.5 ml microcentrifuge tube.
- Add another 50 μl of 10 mM Tris-HCl [pH 7.5] to beads and heat at 75 °C for 2 min in a thermomixer set at 1,200 rpm.
- Quickly place the tube in a magnetic stand and transfer the supernatant to the same tube from Step A20.
- Repeat Steps A6 to A22 for a total of four times using the same set of beads. This procedure will process 2.4 ml of total RNA prepared in Step A2. After this step, the volume of poly(A) RNA will be 400 μl.
- Add 40 μl of 3 M sodium acetate, pH.5.5, 3 μl linear acrylamide, a coprecipitant to aid recovery of RNA during alcohol precipitation, and 1.1 ml of 100% ethanol.
- Incubate at -20 °C overnight.
- Centrifuge at 20,000 x g for 20 min at 4 °C in a microcentrifuge.
- Carefully remove supernatant.
- Wash pellet with 750 μl of 70% ethanol.
- Centrifuge at 20,000 x g for 5 min at 4 °C in a microcentrifuge.
- Carefully remove ethanol.
- Air dry pellet for 5 min at room temperature to remove residual ethanol.
- Resuspend in 50 μl Nuclease-free H2O.
- Evaluate poly(A) RNA quality using the Agilent RNA 6000 kit and Bioanalyzer (Figure 2A, top).
- RNA fragmentation
To narrow the mapping of acetylated regions, input poly(A) RNA is fragmented prior to immunoprecipitation. The optimal fragment size range of RNA is between 100-200 nucleotides. This range of RNA sizes is achieved by divalent metal cations such as Mg2+ or Zn2+ at high temperatures. Divalent cations attack the 2’ hydroxyl group of ribose ultimately cleaving the RNA strand with no sequence specificity (Shelton and Morrow, 1991; AbouHaidar and Ivanov, 1999).- Mix 20 μg of poly(A) RNA in 180 μl of Nuclease-free H2O in a sterile PCR tube. This amount of poly(A) RNA will be split between IgG and anti-ac4C immunoprecipitations.
- Add 20 μl of 10x RNA Fragmentation Buffer from the NEBNext® Fragmentation Module.
- For better performance of the fragmentation reaction, divide the solution into four PCR tubes (50 μl each).
- Incubate at 94 °C for 5 min in a pre-heated thermal cycler with lid set at 105 °C.
- Immediately place PCR tubes on ice.
- Add 5 μl of 10x RNA Fragmentation Stop Solution from the NEBNext® Fragmentation Module to each PCR tube.
- Transfer the fragmentation RNA reactions into one 1.5 ml microcentrifuge tube and add the following components:
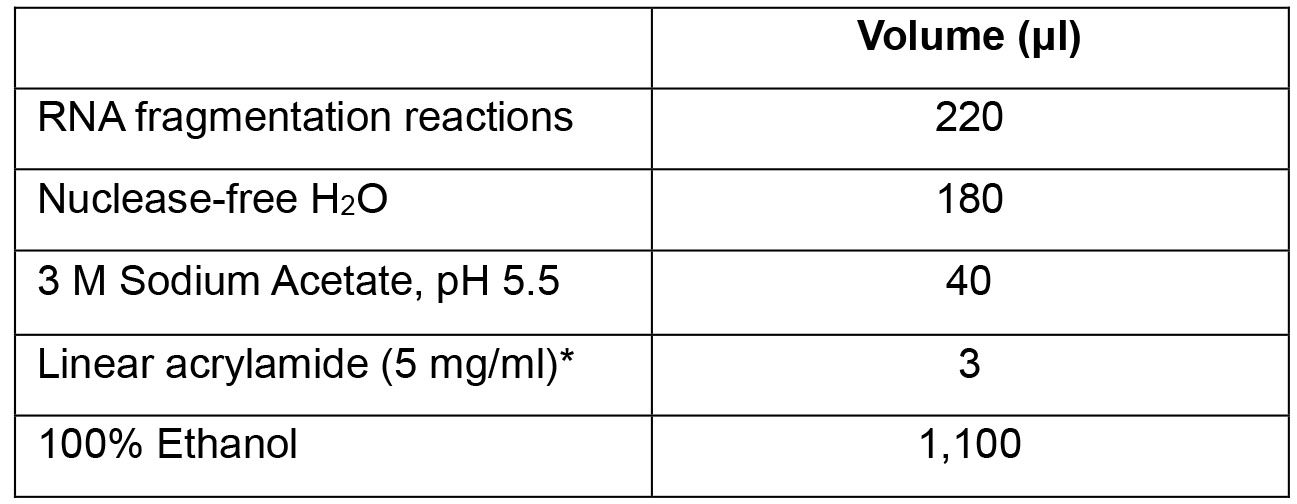
*We recommend the use of linear acrylamide rather than other co-precipitants such as glycogen. We have observed that glycogen can bind non-specifically to anti-ac4C antibodies. - Incubate at -20 °C overnight.
- Centrifuge at 20,000 x g for 20 min at 4 °C in a microcentrifuge.
- Carefully remove supernatant.
- Wash pellet with 750 μl of 70% ethanol.
- Centrifuge at 20,000 x g for 5 min at 4 °C in a microcentrifuge.
- Carefully remove ethanol.
- Air dry pellet for 5 min at room temperature to remove residual ethanol.
- Resuspend in 50 μl Nuclease-free H2O.
- Evaluate fragmentation efficiency and concentration using the Agilent RNA 6000 kit and Bioanalyzer (Figure 2A, bottom).
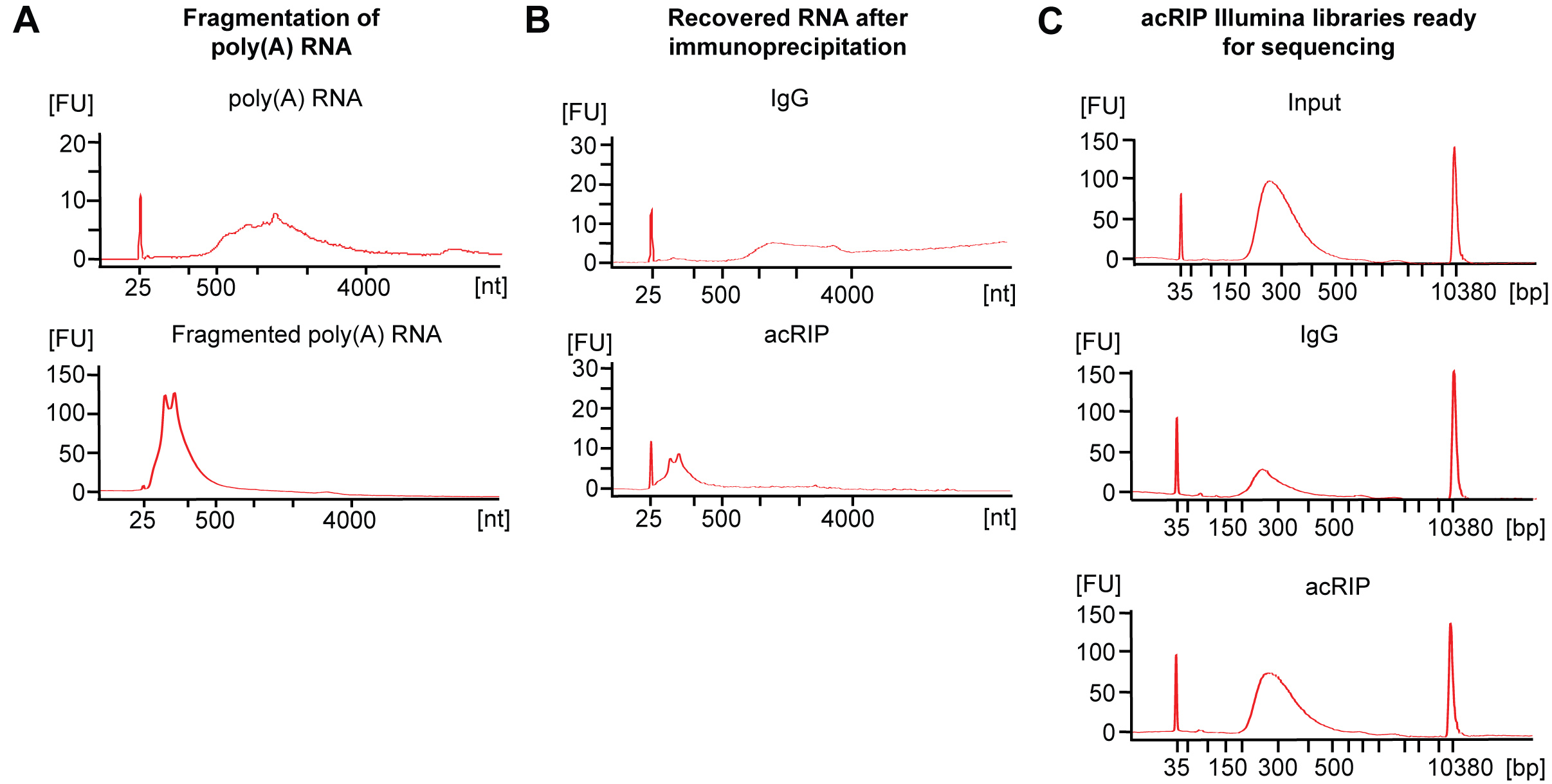
Figure 2. Bioanalyzer profiles. A. Representative profiles of unfragmented (top) and fragmented (bottom) poly(A) RNA. B. Representative profiles of RNA isolated from immunoprecipitation with IgG control (top) or anti-ac4C antibodies (bottom). C. Representative profiles of DNA libraries from Input (top), IgG (middle) or acRIP (bottom).
- In vitro transcription of acetylated RNA
To evaluate acRIP efficiencies between different conditions, an in vitro transcribed acetylated mRNA is spiked into input RNA prior to immunoprecipitation. In vitro transcription is performed using the MAXIscript T7 Transcription Kit. We use a 177 nt probe derived from the mouse β-globin gene with a nucleotide length within the range of poly(A) RNA after fragmentation (100-200 nt). The instructions described below are for the 177 nt probe derived from the mouse β-globin gene. However, any DNA sequence can be used for the same purpose.
The sequence of probe: Probe can be purchased as Ultramer from Integrated DNA Technologies (IDT)
TAATACGACTCACTATAGGGGAGGAAGTAGTGAAGAGTGTTAGAGGATGCTTGTCATCACCGAAGCCTGATTCCGTAGAGCCA CACCCTGGTAAGGGCCAATCTGCTCACACAGGATAGAGAGGGCAGGAGCCAGGGCAGAGCATATAAGGTGAGGTAGGATCAGTTGGATGTGGGAGTTGTAAGGTAGAATGTG
Grey: T7 promoter
Red: cytidines- Mix the following components:
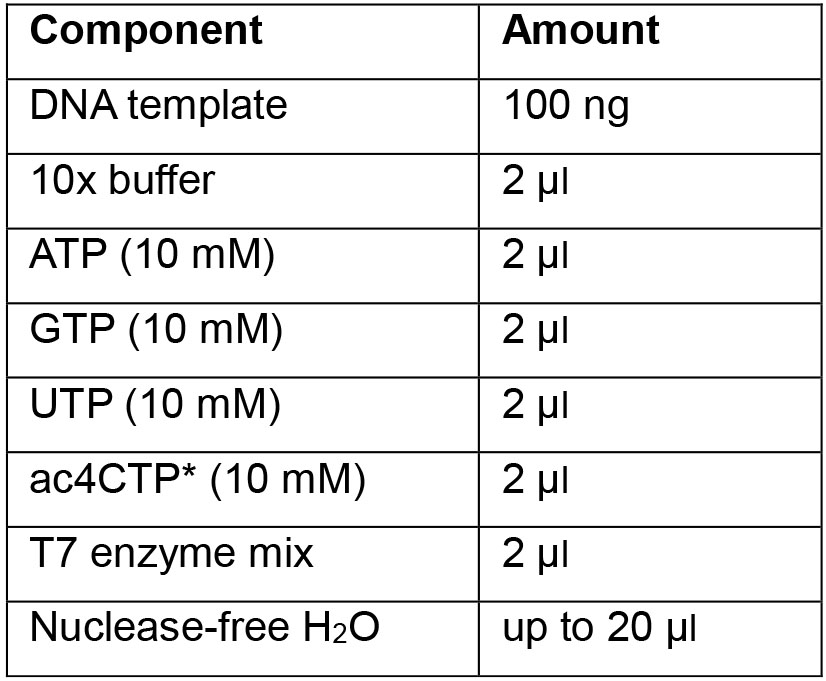
*ac4CTP can be purchased from Trilink. - Incubate at 37 °C for 1 h.
- Add 1 μl of DNase I (provided in the MAXIscript T7 Transcription Kit, see Materials and Reagents) and incubate at 37 °C for 20 min.
- Add 44 μl of Agencourt RNAclean XP beads and mix well.
- Incubate for 10 min at room temperature.
- Spin and place the tube on a magnetic rack for 5 min. Discard the supernatant.
- Wash twice each with 200 μl of freshly prepared 80% ethanol while the tubes are on the magnetic rack.
- Briefly spin the tube, and replace in the magnetic rack.
- Completely remove the residual ethanol, and air dry beads for 5 min while the tube is on the magnetic rack with the lid open.
- Elute RNA with 21 μl Nuclease-free H2O. Mix well by pipetting up and down, incubate for 2 min at room temperature. Put the tube in the magnetic rack until the solution is clear.
- Without disturbing the bead pellet, transfer 21 μl of the supernatant to a clean PCR tube.
- Assess the quality of the RNA fragment in a denaturing agarose gel.
- Measure concentration using the Nanodrop.
- Mix 16.2 μg fragmented poly(A) RNA from Step B15 with 16.2 pg of In vitro transcribed acetylated RNA from Step C13.
- Save 200 ng (~1.2%) of material from Step C14 as Input.
- Mix the following components:
- Coupling antibodies to protein G Dynabeads
acRIP is achieved with a rabbit monoclonal antibody against ac4C (Sinclair et al., 2017) and isotypic rabbit monoclonal IgG as control.- Magnetically separate 20 μl (10 μl for IgG and 10 μl for acRIP) of Protein G-Magnetic Dynabeads using a magnetic stand.
- Wash twice with 200 μl 1x acRIP Buffer each.
- Resuspend Dynabeads in 200 μl 1x acRIP Buffer.
- Divide Dynabeads into two sterile 1.5 ml microcentrifuge tubes:
Tube 1: rabbit monoclonal IgG; 100 μl of bead solution.
Tube 2: rabbit monoclonal anti-ac4C antibody; 100 μl bead solution. - Add 1 μg of monoclonal IgG to Tube 1. Add 1 μg of rabbit monoclonal anti-ac4C antibody to Tube 2.
- Incubate for 1 h at room temperature in a rotator.
- Magnetically separate the Dynabeads using a magnetic stand.
- Wash twice with 200 μl 1x acRIP Buffer.
- Resuspend Dynabeads in 200 μl 1x acRIP Buffer.
- RNA Immunoprecipitation
- Mix the following components:

- Magnetically separate Dynabeads from Step D9, discard the supernatant and add 100 μl of RNA solution from Step E1 into each tube.
- Incubate at 4 °C for 4 h in a rotator.
- Magnetically separate Dynabeads using a magnetic stand.
- Collect supernatants in a sterile microcentrifuge tube and save at -80 °C (optional).
- Add 500 μl of cold 1x acRIP buffer. Quickly flick tubes to resuspend beads.
- Magnetically separate Dynabeads on ice using a magnetic stand and discard the supernatant.
- Repeat Steps E6-E7 for a total of three washes.
- After last wash, resuspend Dynabeads in 100 μl 1x proteinase K buffer.
- Incubate at 37 °C for 30 min in a Thermomixer. Shake at 1,200 rpm.
- Separate magnetic Dynabeads and transfer the supernatant to a sterile 1.5 ml microcentrifuge tube.
- Add 300 μl of Nuclease-free H2O to Dynabeads. Flick tubes to resuspend beads.
- Magnetically separate Dynabeads and collect supernatant into the same microcentrifuge tube from Step E11.
- Add 400 μl Acid-Phenol:Chloroform, pH 4.5. Vortex.
- Centrifuge at 20,000 x g at 4 °C for 15 min in a microcentrifuge.
- Transfer the upper clear layer to a new 1.5 ml microcentrifuge tube.
- Add 400 μl chloroform.
- Centrifuge at 20,000 x g for 1 min.
- Transfer the upper layer to a new 1.5 ml microcentrifuge tube.
- Add the following components:

- Incubate at -20 °C overnight.
- Centrifuge at 20,000 x g for 20 min at 4 °C in a microcentrifuge.
- Carefully remove supernatant.
- Wash pellet with 750 μl of 70% ethanol.
- Centrifuge at 20,000 x g for 5 min at 4 °C in a microcentrifuge.
- Carefully remove ethanol.
- Air dry pellet for 5 min at room temperature to remove residual ethanol.
- Resuspend in 6 μl Nuclease-free H2O.
- Evaluate concentration using the Agilent RNA 6000 kit and Bioanalyzer (Figure 2). At least 1 ng of precipitated RNA should be obtained and used in library preparation.
- Mix the following components:
- Library preparation
Libraries are constructed using the NEBNext® UltraTM II Directional RNA Library Prep Kit for Illumina® and following the manufacturer’s suggestions (Figure 1C). All reagents are supplied with the kit.- Samples:
Input: Step C15
IgG: Step E28
acRIP: Step E28 - First strand cDNA synthesis
- Set up the following priming reaction in PCR tubes and mix by gentle pipetting. Use equivalent amounts of RNA (~1 ng) for all samples:
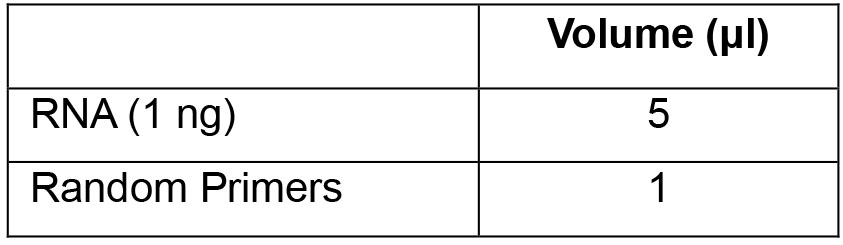
- Incubate the samples in a thermal cycler as follows:
5 min at 65 °C, with heated lid set at 105 °C.
Hold at 4 °C. - To the RNA from Step F2b (6 μl) add the following components and mix by gentle pipetting:

- Incubate samples in a preheated thermal cycler (with the heated lid set at 105 °C) as follows:
10 min at 25 °C
15 min at 42 °C
15 min at 70 °C
Hold at 4 °C.
- Set up the following priming reaction in PCR tubes and mix by gentle pipetting. Use equivalent amounts of RNA (~1 ng) for all samples:
- Second strand cDNA synthesis
- To the tubes from Step F2d (20 μl) add the following components and mix by gentle pipetting:

- Incubate samples in a thermal cycler (with the heated lid set at 37 °C) for 60 min at 16 °C.
- Add 144 μl (1.8x) of resuspended AMPure XP Beads to samples from Step F3b.
- Incubate for 5 min at room temperature.
- Spin, place the tube on the magnetic stand for 5 min. Carefully remove and discard the supernatant.
- Wash twice each with 200 μl of freshly prepared 80% ethanol while the tubes are on the magnetic rack.
- Air dry the beads for 5 min while the tube is on the magnetic rack with lid open.
- Elute into 53 μl 0.1x TE buffer. Mix well by pipetting up and down and incubate for 2 min at room temperature.
- Place the tube in the magnetic rack until the solution is clear.
- Remove 50 μl of the supernatant and transfer to a clean nuclease free PCR tube.
- To the tubes from Step F2d (20 μl) add the following components and mix by gentle pipetting:
- Perform end prep of cDNA library
- Mix the following components in a PCR tube:

- Mix well by pipetting up and down at least 10 times.
- Incubate samples in a thermal cycler (with the heated lid set at 75 °C) as follows:
30 min at 20 °C
30 min at 65 °C
Hold at 4 °C.
- Mix the following components in a PCR tube:
- Adaptor ligation
- Dilute the NEBNext Adaptor for Illumina 30-fold with Adapter Dilution Buffer prior to use.
- Add the following components directly to the End Prep Reaction.
Caution: Do not pre-mix the components to prevent adaptor-dimer formation.
*The NEBNext adaptor is provided in the NEBNext® Multiplex Oligos for Illumina®. - Mix well by pipetting up and down at least 10 times.
- Incubate the sample in a thermal cycler (with the heated lid set at 75 °C) for 15 min at 20 °C.
- Add 3 μl of USER (uracil-specific excision reagent) Enzyme to the ligation mixture resulting in a total volume of 96.5 μl. USER enzyme is provided in the NEBNext® UltraTM II Directional RNA Library Prep Kit.
- Mix well and incubate at 37 °C for 15 min with the heated lid set to ≥ 45 °C.
- Add 96.5 μl (1x) resuspended AMPure XP Beads and mix well.
- Incubate for 10 min at room temperature.
- Quickly spin, place on a magnetic rack for 5 min. Discard the supernatant.
- Wash twice with 200 μl of freshly prepared 80% ethanol while the tubes are on the magnetic rack.
- Briefly spin the tube, and put the tube back in the magnetic rack.
- Completely remove the residual ethanol, and air dry beads for 5 min while the tube is on the magnetic rack with the lid open.
- Elute DNA with 16 μl 0.1x TE buffer. Mix well, incubate for 2 min at room temperature. Put the tube in the magnetic rack until the solution is clear.
- Without disturbing the bead pellet, transfer 15 μl of the supernatant to a clean PCR tube.
- PCR Enrichment of Adaptor Ligated DNA
- Mix the following components:

*The Universal PCR Primer and index primers for Illumina are provided in the NEBNext® Multiplex Oligos for Illumina®. Use a different index primer for each sample. - PCR Cycling Conditions:
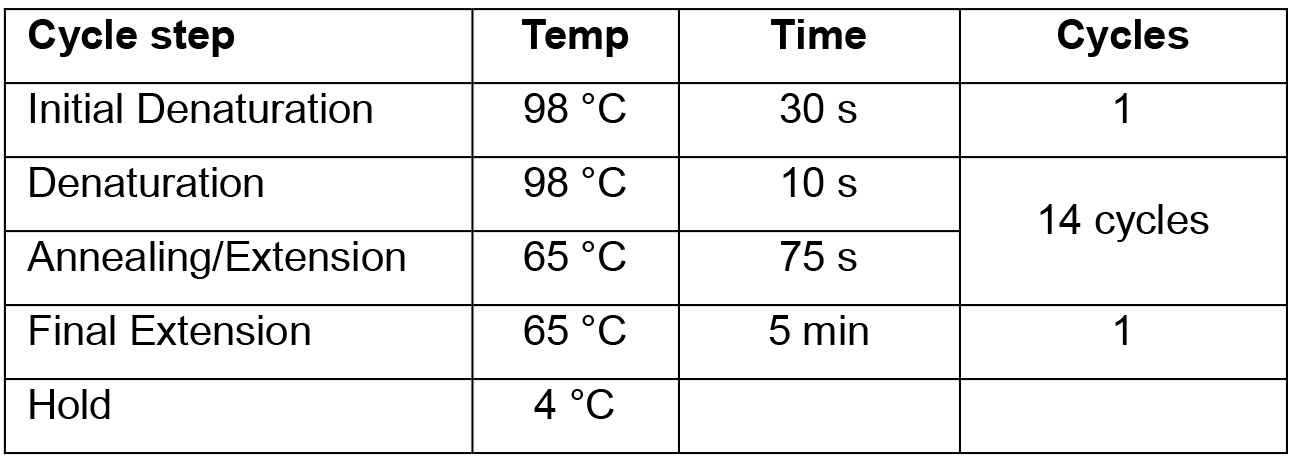
- Add 50 μl (1x) resuspended AMPure XP Beads and mix well.
- Incubate for 5 min at room temperature.
- Spin and place the tube on a magnetic rack for 5 min. Discard the supernatant.
- Wash twice each with 200 μl of freshly prepared 80% ethanol while the tubes are on the magnetic rack.
- Briefly spin the tube, and put the tube back in the magnetic rack.
- Completely remove the residual ethanol, and air dry beads for 5 min while the tube is on the magnetic rack with the lid open.
- Elute DNA with 23 μl of 10 mM Tris-HCl. Mix well by pipetting up and down, incubate for 2 min at room temperature. Put the tube in the magnetic rack until the solution is clear.
- Without disturbing the bead pellet, transfer 21 μl of the supernatant to a clean PCR tube.
- Assess library quality on an Agilent Bioanalyzer DNA Chip (Figure 2).
- Mix the following components:
- Sequence using an Illumina sequencer in paired-end mode read length 125 bp (2 x 125 bp).
- Samples:
Data analysis
- Pre-process raw reads to remove low-quality bases and adaptor sequences by dynamic trimming or fixed cropping using Trimmomatic (Bolger et al., 2014).
- Map reads to the human genome (hg19) with Tophat2 v.2.1.1 (Trapnell et al., 2009). Parameters used are: (i) reporting at most one alignment per read (-g 1); (ii) allowing maximally five mismatches per read (> 95% matching); (iii) supplying the Ensembl Release 75 gene annotation; and (iv) performing a post-alignment filter to remove alignments to mitochondrial DNA (chrM) and non-concordant mate pairs.
- Since sites of acetylation in transcripts will be non-contiguous in genomic coordinates, and difficult to assign to alternative isoforms, a representative transcript reference can be used for continuous peak calling. For example, canonical transcript sequences can be obtained from the UCSC genome browser (Rosenbloom et al., 2015) and used in the generation of a Bowtie2-based index (Langmead and Salzberg, 2012). Quantify the number of bases in this reference for use in step 5. Reads can then be aligned in local alignment mode with Bowtie2 (Langmead and Salzberg, 2012).
- Perform separate alignments to the spiked-in mouse β-globin probe sequence (see section C) and to the 43 kb human ribosomal DNA complete repeating unit (GenBank U13369.1) using Bowtie2 (Langmead and Salzberg, 2012), to specifically analyze reads originating from those features.
- Perform peak calling with MACS2 (Zhang et al., 2008), with parameters selected to optimize performance with transcript mapped reads [i.e., turning off the shifting model (-nomodel) and local lambda (-nolambda), and supplying transcript reference bases (with -gsize) as the genome size]. Input samples should be used as controls for peak calling. In addition to peak calling within replicates, a set of reference peaks can be generated on pooled data, on which to apply filtering criteria (step 7).
- Note that in the MACS2 peak calling output, each multi-base-pair peak includes ≥ 1 ‘‘summit,’’ or local peak maxima, defined at a single base position. Each of these summits is a putative ‘‘ac4C site.’’ The acRIP-seq approach is not a base-resolution method, so these ac4C sites are not required to be cytidines.
- Apply stringent filtering to enforce replicability and remove artifacts such as non-specific binding, with a routine like the following: (i) Compare peaks to select only those sites with a reduction in signal in NAT10-/- (non-acetylated control [Arango et al., 2018]) as compared to parental cells, following the expectation that non-artifactual ac4C sites would show diminished signal when the enzyme is reduced. To acquire an enrichment value for comparison, use the bedtools map function (Quinlan, 2014) to extract the value from the MACS2 pileup output at the position of the summit called in parental cells. Select peak summits with pileup values higher in parental cells than NAT10-/- cells to pass this filtering step. (ii) Remove peaks that result from non-specific IgG binding, by intersecting ac4C peaks with peaks called in IgG-IP, and keep only those with no coordinate overlap (Bedtools, [Quinlan, 2014]). (iii) Enforce detection in replicate experiments, by requiring peaks called in your reference set (pooled data) to overlap with peaks called in each individual replicate. (iv) Investigate the possibility of mapping errors, by spot-checking peak calls against genomically aligned reads to confirm concordance.
Recipes
- 10x acRIP buffer
100 mM Na-Phosphate monobasic pH 7.0
100 mM Na-Phosphate dibasic
1.4 M NaCl
0.5% Triton X-100
1% BSA - 1x Proteinase K buffer
100 mM Tris-HCl, pH 7.5
150 mM NaCl
12.5 mM EDTA
2% SDS
5 mg/ml Proteinase K - Poly(A) RNA binding buffer
20 mM Tris-HCl, pH 7.5
1 M LiCl
2 mM EDTA - Poly(A) RNA washing buffer
10 mM Tris-HCl, pH 7.5
0.15 M LiCl
1 mM EDTA
Acknowledgments
This work was supported by the Intramural Research Program of the National Institutes of Health (NIH), NCI, Center for Cancer Research. We thank the members of the Center for Cancer Research Sequencing Facility at the National Cancer Institute (Frederick, MD) for providing Illumina sequencing services. This protocol utilized the Biowulf Linux cluster at the NIH, Bethesda, MD (https://hpc.nih.gov).
Competing interests
The authors declare no competing interests.
References
- AbouHaidar, M. G. and Ivanov, I. G. (1999). Non-enzymatic RNA hydrolysis promoted by the combined catalytic activity of buffers and magnesium ions. Z Naturforsch C 54(7-8): 542-548.
- Arango, D., Sturgill, D., Alhusaini, N., Dillman, A. A., Sweet, T. J., Hanson, G., Hosogane, M., Sinclair, W. R., Nanan, K. K., Mandler, M. D., Fox, S. D., Zengeya, T. T., Andresson, T., Meier, J. L., Coller, J. and Oberdoerffer, S. (2018). Acetylation of cytidine in mRNA promotes translation efficiency. Cell 175(7): 1872-1886 e1824.
- Bolger, A. M., Lohse, M. and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics (Oxford, England) 30: 2114-2120.
- Dong, C., Niu, L., Song, W., Xiong, X., Zhang, X., Zhang, Z., Yang, Y., Yi, F., Zhan, J., Zhang, H., Yang, Z., Zhang, L. H., Zhai, S., Li, H., Ye, M. and Du, Q. (2016). tRNA modification profiles of the fast-proliferating cancer cells. Biochem Biophys Res Commun 476(4): 340-345.
- Ito, S., Akamatsu, Y., Noma, A., Kimura, S., Miyauchi, K., Ikeuchi, Y., Suzuki, T. and Suzuki, T. (2014a). A single acetylation of 18 S rRNA is essential for biogenesis of the small ribosomal subunit in Saccharomyces cerevisiae. J Biol Chem 289(38): 26201-26212.
- Ito, S., Horikawa, S., Suzuki, T., Kawauchi, H., Tanaka, Y., Suzuki, T. and Suzuki, T. (2014b). Human NAT10 is an ATP-dependent RNA acetyltransferase responsible for N4-acetylcytidine formation in 18 S ribosomal RNA (rRNA). J Biol Chem 289(52): 35724-35730.
- Langmead, B. and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9(4): 357-359.
- Li, X., Xiong, X. and Yi, C. (2016). Epitranscriptome sequencing technologies: decoding RNA modifications. Nat Methods 14(1): 23-31.
- McIntyre, W., Netzband, R., Bonenfant, G., Biegel, J. M., Miller, C., Fuchs, G., Henderson, E., Arra, M., Canki, M., Fabris, D. and Pager, C. T. (2018). Positive-sense RNA viruses reveal the complexity and dynamics of the cellular and viral epitranscriptomes during infection. Nucleic Acids Res 46(11): 4776-5791.
- Quinlan, A. R. (2014). BEDTools: The Swiss-army tool for genome feature analysis. Curr Protoc Bioinformatics 47: 11.12.1-34.
- Rio, D. C., Ares, M., Jr., Hannon, G. J. and Nilsen, T. W. (2010). Purification of RNA using TRIzol (TRI reagent). Cold Spring Harb Protoc 2010(6): pdb prot5439.
- Rosenbloom, K. R., Armstrong, J., Barber, G. P., Casper, J., Clawson, H., Diekhans, M., Dreszer, T. R., Fujita, P. A., Guruvadoo, L., Haeussler, M., Harte, R. A., Heitner, S., Hickey, G., Hinrichs, A. S., Hubley, R., Karolchik, D., Learned, K., Lee, B. T., Li, C. H., Miga, K. H., Nguyen, N., Paten, B., Raney, B. J., Smit, A. F., Speir, M. L., Zweig, A. S., Haussler, D., Kuhn, R. M. and Kent, W. J. (2015). The UCSC Genome Browser database: 2015 update. Nucleic Acids Res 43(Database issue): D670-681.
- Sharma, S., Langhendries, J. L., Watzinger, P., Kotter, P., Entian, K. D. and Lafontaine, D. L. (2015). Yeast Kre33 and human NAT10 are conserved 18S rRNA cytosine acetyltransferases that modify tRNAs assisted by the adaptor Tan1/THUMPD1. Nucleic Acids Res 43(4): 2242-2258.
- Shelton, V. M. and Morrow, J. R. (1991). Catalytic transesterification and hydrolysis of RNA by zinc(II) complexes. Inorganic Chemistry 30(23): 4295-4299.
- Sinclair, W. R., Arango, D., Shrimp, J. H., Zengeya, T. T., Thomas, J. M., Montgomery, D. C., Fox, S. D., Andresson, T., Oberdoerffer, S. and Meier, J. L. (2017). Profiling cytidine acetylation with specific affinity and reactivity. ACS Chem Biol 12(12): 2922-2926.
- Tardu, M., Lin, L. and Koutmou, K. S. (2018). N4-acetylcytidine and 5-formylcytidine are present in Saccharomyces cerevisiae mRNAs. bioRxiv. doi.org/10.1101/327585.
- Trapnell, C., Pachter, L. and Salzberg, S. L. (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25(9): 1105-1111.
- Zhang, Y., Liu, T., Meyer, C. A., Eeckhoute, J., Johnson, D. S., Bernstein, B. E., Nusbaum, C., Myers, R. M., Brown, M., Li, W. and Liu, X. S. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9(9): R137.
- Zhao, B. S., Roundtree, I. A. and He, C. (2017). Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol 18(1): 31-42.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Arango, D., Sturgill, D. and Oberdoerffer, S. (2019). Immunoprecipitation and Sequencing of Acetylated RNA. Bio-protocol 9(12): e3278. DOI: 10.21769/BioProtoc.3278.
Category
Molecular Biology > RNA > RNA purification > Affinity purification
Molecular Biology > RNA > Epitranscriptome
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.