- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Tandem Affinity Purification of SBP-CBP-tagged Type Three Secretion System Effectors
Published: Vol 9, Iss 12, Jun 20, 2019 DOI: 10.21769/BioProtoc.3277 Views: 5785
Reviewed by: Ralph Thomas BoettcherYing FengAnonymous reviewer(s)
Original research article
The authors used this protocol in:
Jun 2016

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Identification of protein-protein interactions of bacterial effectors and cellular targets during infection is at the core to understand how bacteria manipulate the infected host to overcome the immune response. Potential interacting proteins might be identified by genetic methods, i.e., two hybrid screens and could be verified by co-immunoprecipitation. The tandem affinity purification (TAP) method allows an unbiased screen of potential interaction partners of bacterial effectors in a physiological approach: target cells can be infected with a bacterial strain harboring the TAP-tagged bacterial effector protein which is translocated in the host similar as under physiological infection conditions. No transfection and overexpression of the bacterial protein in the eukaryotic host are needed. Therefore, also host target cells not easy to transfect can be analyzed by this method. Moreover, the two consecutive affinity tags Calmodulin-Binding-Peptide (CBP) and Streptavidin-Binding-Peptide (SBP) fused to the translocated bacterial protein allow an outstanding clear purification of protein complexes formed between the bacterial protein of interest and host cell proteins with less occurrence of contaminants. Mass spectrometry allows an unbiased identification of interacting eukaryotic proteins.
Keywords: YersiniaBackground
The interaction of bacterial effectors with eukaryotic proteins during infection is fundamental to understand the processes initiated by the bacteria to manipulate the host cell responses in order to establish infection. The type three secretion system (TTSS) is a needle-like structure found in several gram-negative bacteria that facilities direct secretion of bacterial effector proteins into the host cell. The tandem affinity purification (TAP) of TAP-tagged bacterial effector proteins secreted directly into the host cell via the TTSS provides the invaluable possibility to identify eukaryotic proteins interacting with bacterial effector proteins in a physiological approach. No transfection and overexpression of the bacterial protein in the eukaryotic host is needed once the tandem affinity tag labeled bacterial protein is translocated in the host cell cytoplasm during infection with the manipulated bacteria. The use of two affinity tags increases the specificity of the protein-protein interaction and ensures a low level background of nonspecific interactions.
This method can be adapted to a wide range of gram-positive or gram-negative bacterial species and various host target cell types. We have utilized this assay to identify new interaction partners of the Yersinia effector protein YopM. In an approach with TAP-tagged YopM being translocated into the macrophage cell line J774A.1 via the natural type three secretion system of Yersinia we verified the interaction of YopM with host kinases PKN2 and RSK1. Moreover, we detected association with additional PKN and RSK isoforms and identified a new interaction partner of YopM, the DEAD box Helicase DDX3. This assay can be adapted to other bacterial species and host cell types.
Materials and Reagents
- Pipette tips (sarstedt, catalog numbers: 70762010 [1,000 μl]; 70762012 [100 μl] and 701116 [10 μl])
- Cell culture dishes 100 mm (sarstedt, catalog number: 83.3902)
- Falcon tubes 15 ml (Sarstedt, catalog number: 62.544.502)
- Micro tubes 1.5 ml (Sarstedt, catalog number: 72.690.001)
- Cell scraper (Sarstedt, catalog number: 83.1830)
- J774A.1 cells (ATCC# TIB-67)
- Yersinia enterocolitica strain deltaYopM (Yersinia enterocolitica Serotype O:8 strain WA-314 with the YopM gene replaced by a kanamycin resistance cassette Trulzsch et al., 2004)
- Yersinia enterocolitica strain deltaYopM(pYopM-CBP-SBP) (deltaYopM strain complemented with the DNA construct YopM-CBP-SBP in pACYC184; Hentschke et al., 2010)
- RPMI1640 (Gibco, catalog number: 61870-010)
- Fetal bovine serum (FCS, Gibco, catalog number: 26140-079)
- Penicillin-Streptomycin (Gibco, catalog number: 15140122)
- L-Glutamine (Gibco, catalog number: A2916801)
- pACYC184 vector (NEB; Hentschke et al., 2010)
- InterPlay Mammalian TAP System (Agilent, catalog number: 240103)
- Bromphenol Blue (Serva, catalog number: 15375.02)
- Coomassie brilliant blue (Merck, catalog number: 1154440025)
- Calmodulin Sepharose 4B (GE Healthcare, Life, catalog number: 17052901)
- Streptavidin Sepharose High Performance (GE Healthcare, Life, catalog number: 17511301)
- Complete Protease inhibitor (Roche Diagnostics)
- Methanol (Roth, catalog number: 8388.1)
- Trypsin, sequencing grade modified (Promega, catalog number: V5111)
- a-SBP antibody (Millipore, clone 20, catalog number: MAB10764)
- Luria-Bertani broth (Roth, catalog number: X986.2)
- Acetic acid (Roth, catalog number: T179)
- SDS (Roth, catalog number: CN30.3)
- Molecular weight marker (Molecular weight range 10-180 kDa; PageRuler Prestained Protein Ladder, Thermo Scientific, catalog number 26616)
- Glycerol (Calbiochem, catalog number: 356350)
- β-Mercaptoethanol (Sigma, catalog number: M3148-25ml)
- Tris (Sigma, catalog number: T1503-1KG)
- EDTA (Roth, catalog number: 80432)
- Dithiothreitol (DTT; Roth, catalog number: 6908.2 259)
- Iodacetamid (Sigma, catalog number: I6125-25g)
- NH4HCO3 (Sigma, catalog number: 09830)
- Acetonitrile (Thermo, catalog number: 51101)
- Formic acid (Fluka, catalog number: 94318)
- Trifluor acetic acid (TFA; Roth, catalog number: CN30.3)
- Coomassie Brilliant Blue R-250 (Serva, catalog number: 17524.01)
- Bromphenol blue (Merck, catalog number: 8122)
- NaCl (Roth, catalog number: 9265.1)
- KCl (Merck, catalog number: 1049361000)
- Coomassie staining solution (see Recipes)
- Coomassie destain solution (see Recipes)
- SDS-PAGE sample buffer (2x) (see Recipes)
- PBS (10x) (see Recipes)
Equipment
- Pipettes (Eppendorf, Eppendorf Research Plus, catalog number: 3123000900)
- Power supplies (Bio-Rad, Bio-Rad power PC 200)
- SDS-PAGE electrophoresis cell (Bio-Rad Mini-Protean II)
- Cell counting chamber (Hartenstein, Neubauer-cell counting chamber)
- Mammalian cell culture incubator (Binder, CB Series)
- Shaking incubator (Sartorius, model Certomat BS-1)
- Rotary mixer (Hartenstein, catalog number LD26/L263)
- Centrifuge (Eppendorf, model: 5417R)
- Vacuum dryer (Speed Vac SC110, Savant)
- MALDI-TOF mass spectrometer (Bruker Daltonics, REFLEX IV)
Procedure
- Cloning of the CBP-SBP-tagged bacterial effector protein for bacterial expression
The tandem-affinity-tag containing the Calmodulin-Binding-Peptide (CBP; Stofko-Hahn et al., 1992) and the Streptavidin-Binding-Peptide (SBP; Keefe et al., 2001) can be derived from the pCTAP-vector commercially available from the InterplayTM Tandem Affinity Purification system (Agilent). The cloning is carried out with standard molecular biology techniques. Our cloning strategy to create YopM-CBP-SBP in pACYC184 is described in detail in the original publication (Hentschke et al., 2010). Briefly, the cloning can be performed as follows:- Remove the stop codon of your bacterial effector of interest by PCR amplification for insertion of a C-terminal CBP-SBP-tag. The resulting PCR product should contain the putative endogenous promoter region of the effector protein of interest to ensure physiological expression levels.
- Digest the PCR product and the pCTAP with appropriate restriction enzymes.
- Ligate the effector protein into pCTAP.
- Digest the resulting pCTAP effector protein construct with the appropriate enzymes to ligate it in the next step into a plasmid.
- Ligate the pCTAP effector protein construct from Step A4 into a low copy vector. In our case, we used pACYC184 (Hentschke et al., 2010).
Note: Use a low copy plasmid to ensure physiological expression levels of the bacterial effector protein. - This results in a vector harboring the bacterial effector protein C-terminally tagged with the two consecutive affinity tags CBP and SBP from the pCTAP vector (Figure 1).
Note: TAP-tag can be cloned to the N-terminus using the pNTAP vector provided in the kit.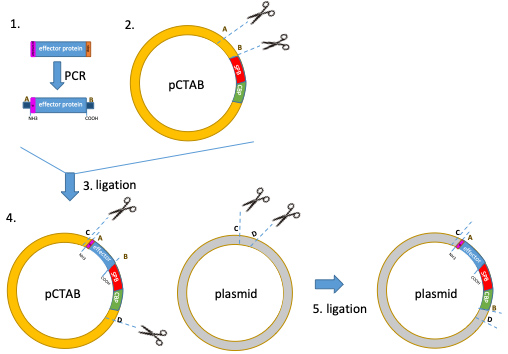
Figure 1. Schematic illustration of the cloning procedure described in the protocol. (1) Remove the stop codon of your bacterial effector of interest by PCR amplification. Digest the PCR product and the pCTAP with appropriate restriction enzymes (A and B). (3) Ligate the effector protein into pCTAP. (4) To express the CBP-SBP-tagged effector protein of interest in the (investigated) bacterium, digest pCTAP construct “effector-CBP-SBP” (left) and a low copy cloning plasmid of your choice (suitable for the investigated organism) with appropriate restriction enzymes (C and D). (5) Ligate the construct “effector-CBP-SBP” from step 4 into a low copy cloning plasmid.
- Construction of the bacterial strain harboring CBP-SBP-tagged effector protein
- Remove the endogenous effector protein from the bacterial strain you desire to investigate. In our case the YopM protein on the Yersinia virulence plasmid pYV had been replaced by a kanamycin resistance cassette (as described in more detail in the original publication; Trulzsch et al., 2004) resulting in YopM deletion strain deltaYopM.
- The effector deletion strain can be complemented with the plasmid harboring the TAP-tagged effector protein. In our case the Yersinia enterocolitica Serotype 0:8 strain WA-314 (Heesemann and Laufs, 1983), lacking the YopM was complemented with the pACYC184 vector expressing the YopM-SBP-CBP resulting in a strain named WA-314ΔYopM (pYopM-CBP-SBP) (Hentschke et al., 2010).
- Infection of target cells
Infect the cells with the strain harboring the CBP-SBP-tagged effector protein for an appropriate time period.- Prepare ten 100 mm culture dishes of cells (in total 108 cells).
- Grow J774A.1 cells overnight at 37 °C and 5% CO2 in RPMI1640 supplemented with 10% FCS, 100 IU/ml Penicillin and 100 μg/ml Streptomycin and additional 2 mmol Glutamine.
- Prepare overnight bacterial pre-cultures one day prior to the experiment. Briefly, streak out a colony from a plate and suspend the colony in 2 ml Luria-Bertani broth (LB) supplemented with appropriate antibiotics. Shake Yersinia strains overnight at 27 °C.
- Dilute overnight culture 1:20 in fresh LB (40 ml in total). Grow Yersinia for another 2 h at 37 °C in order to induce activation of the Yersinia type III secretion machinery and expression of Yops.
- Centrifuge the bacteria culture (4 °C, 5,000 x g, 10 min) and resuspend in ice-cold PBS.
- Adjust bacterial suspension to OD600 = 0.36.
- Upon infection, remove the medium of J774A.1 cells and change to antibiotic-free RPMI1640.
- Infect eukaryotic cells by adding 1 ml of adjusted bacterial suspension prepared in the previous Step E6 per 100 mm culture dish. 1 ml per 100 mm culture dish equals a multiplicity-of-infection of about 50:1 (50 bacteria per cells, Hentschke et al., 2010).
- Culture cells at 37 °C and 5% CO2 during the infection time.
Note: The J774A.1 cells were infected with Yersinia for 90 min. Time of infection might differ from cell type to cell type.
- Preparing the protein extracts
All protein purification steps should be performed at 4 °C to prevent the interacting proteins from dissociating. All amounts are indicated for a lysate volume of 10 ml.- Wash cells after infection three times with ice-cold PBS supplemented with 1x complete Protease inhibitor (Roche).
- Harvest cells by scraping off the cells with a cell scraper (Sarstedt) in 10 ml lysis buffer (1 ml per plate) supplied with the InterplayTM Tandem Affinity Purification kit (Agilent) supplemented with 1x complete Protease inhibitor (Roche) and transfer them to a 15 ml Falcon tube. Combine lysates of all 10 dishes in one 15 ml Falcon tube.
- Lyse the cells by three successive freeze-thaw rounds in a -80 °C freezer for 20 min and 10 min incubation in cold water.
- Clear the cells by centrifugation at 16,000 x g for 10 min at 4 °C.
- Collect the supernatant in a new 15 ml Falcon tube.
- Store an aliquot of 5 μl Lysate at -20 °C for further analysis by Western blot.
- Add 4 μl 0.5 M EDTA and 0.7 μl 14.4 M β-mercaptoethanol provided in the InterplayTM Tandem Affinity Purification kit (Agilent) per ml lysate.
- Purification of the protein complexes using Streptavidin sepharose beads
- Prepare the Streptavidin binding buffer (SBB) by adding 7 μl of 14.4 M β-mercaptoethanol (provided in the kit) and 1x complete Protease inhibitor (Roche) to 10 ml of SBB. Mix the buffer by inverting the tube and keep it on ice. The prepared buffer may be stored at 4 °C for up to two weeks.
- Wash the Strepavidin resin slurry: centrifugate 500 μl of the provided streptavidin sepharose at 1,500 x g for 10 min at 4 °C. (Five-hundred microliters of provided sepharose has 250 μl resin plus 250 μl ethanol storage buffer resulting in 500 μl 50% slurry.)
- Remove the supernatant with a pipette.
- Discard the supernatant to remove the ethanol storage buffer.
- Resuspend the resin in 1 ml SBB.
- Wash the resin twice each with 1 ml SBB buffer by centrifugation and resuspention as described in Steps E2-E5. Use a new 1.5 ml tube for each washing step.
- Collect the resin in 250 μl SBB in a new 1.5 ml tube resulting in 500 μl 50% slurry.
- Add 500 μl of the washed streptavidin 50% slurry to the lysate (Figure 2-1).
- Incubate for 2 h at 4 °C under constant rotation at 10 rpm on a rotary mixer (Hartenstein).
- Collect the sepharose by centrifugation at 1,500 x g for 5 min and remove and store the supernatant for further Western blot analysis in a 1.5 ml tube at -20 °C.
- Wash the sepharose twice each with 1 ml SBB (Figure 2-2).
- Use a new 1.5 ml tube for each washing step.
- Collect the washed sepharose by centrifugation at 1,500 x g for 5 min and discard the supernatant.
- Supplement the Streptavidin elution buffer (SEB) with 7 μl of 14.4 M β-mercaptoethanol (provided in the kit) and 1x complete Protease inhibitor (Roche) to 10 ml of SEB. The SEB buffer must be kept at 4 °C and should be protected from light.
- Add 500 μl Streptavidin elution buffer (SEB) to the sepharose beads.
- For Elution of the protein complex incubate for 30 min at 4 °C rotating.
- Collect the sepharose by centrifugation at 1,500 x g for 5 min at 4 °C.
- Transfer the supernatant in a new 1.5 ml tube.
- Store 10 μl of the supernatant for later Western blot analysis.
- Prepare the Calmodulin binding buffer (CBB) by adding 7 μl of 14.4 M β-mercaptoethanol (provided in the InterPlay Mammalian TAP System kit) and 1x complete Protease inhibitor (Roche) to 10 ml of CBB. Mix the buffer by inverting the tube. Keep the prepared CBB on ice.
- Add 10 μl streptavidin supernatant supplement and then 4 ml CBB to the supernatant.
- Second purification step using Calmodulin sepharose beads
- Wash the Calmodulin sepharose as described in Steps E2-E6 with CBB supplemented with β-mercaptoethanol (InterPlay Mammalian TAP System kit) and 1x complete Protease inhibitor (Roche) (Figure 2-3).
- Add the washed calmodulin sepharose to the eluate (from Step E17).
- Incubate overnight at 4 °C under constant rotation.
- Wash the calmodulin sepharose twice with CBB.
- Store the supernatant for further Western blot analysis.
- Boil the washed beads for 10 min in 60-100 μl 2x SDS-loading buffer (Figure 2-3).

Figure 2. Schematic illustration of the Tandem affinity purification AP system. This figure is modified from InterPlay Mammalian TAP System protocol (https://www.agilent.com/cs/library/usermanuals/public/240101.pdf.) (1) Add the streptavidin sepharose to the cell lysate of infected cells containing the SBP-CBP-tagged effector protein with interacting partners. (2) Wash contaminants with Streptavidin binding buffer and add calmodulin sepharose. (3) Wash contaminants with Calmodulin binding buffer and elute your effector protein with potential interacting partners by boiling the Calmodulin sepharose beads in 2x SDS loading buffer.
- Identification of protein complexes
The eluate can be subjected to SDS-PAGE followed by Coomassie staining to determine purification efficiency and allow mass-spectrometry analysis of visible bands.- Load the lysate control, the streptavidin sepharose supernatant (Eluate-SBP) and the boiled calmodulin sepharose supernatant (Eluate-CBP) on a 10% SDS-PAGE (Figure 3).
- Separate the eluted proteins from Step F6 (50 μl) by SDS-PAGE.
- Stain the SDS gel in 10 ml Coomassie staining solution for 1 h.
- Destain the SDS gel in 10 ml Coomassie destain solution for 2 h or overnight.
- Cut out visible bands with a scalpel. Gel pieces size should be as small as possible.
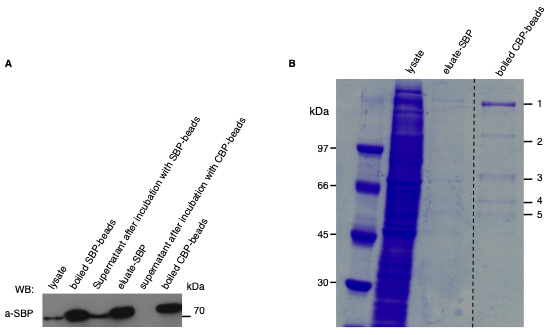
Figure 3. Identification of interacting proteins of the bacterial effector protein. A. Representative Western blot analysis to determine the purification efficiency of each step of the Tandem affinity purification. The presence of the tagged effector was detected with an antibody against the SBP (Millipore clone 20 #MAB10764). In this case, Tandem affinity purification was performed with cell lysate of WA-314ΔYopM (pYopM-CBP-SBP) infected J774 cells. B. Representative Coomassie stained SDS-PAGE including purification steps of the Tandem affinity purification. Visible bands (number 1 to 5) of the boiled CBP-beads need to be excised for further analysis by mass spectrometry.
- Analyze visible bands by mass spectrometry
The mass spectrometry was carried out with standard procedure techniques. Details of the identification of interacting proteins of the Yersinia effector protein YopM can be found in Hentschke et al. (2010).- Cut the gel bands into small pieces.
- Reduce the proteins with freshly prepared 10 mM DTT solved in a 100 mM ammonium bicarbonate stock solution at 56 °C for 30 min. Add enough DTT solution to cover the gel pieces completely.
- Modify the cysteine residues with 55 mM iodacetamid solved in a 100 mM ammonium bicarbonate stock solution at ambient temperature for 20 min in the dark. Gel pieces should be covered completely.
- Digest the protein in eluate with trypsin (5 ng trypsin/μl in 50 mM NH4HCO3, 37 °C, 16 h).
- Extract the peptides from the gel pieces by adding extraction solution containing 50% acetonitrile (ACN)/5% formic acid (v/v) in water. The volume of the extraction solution should be twice as much as the volume of the gel pieces. Incubate for 30 min on a mixer at 37 °C.
- Collect the supernatant.
- Add 100% ACN and incubate for 15 min on a mixer at 37 °C.
- Collect the supernatant and pool it with the supernatant collected in the previous Step H6.
- Add enough H2O to cover the gel pieces and incubate again for 15 min on a mixer at 37 °C.
- Pool the supernatant with the supernatant of previous Step H8.
- Add extraction solution to the gel pieces in a ratio of 2:1 and incubate for 30 min on a mixer at 37 °C. Unite the supernatant with the others collected.
- The volume of the extraction solution should be twice as much as the volume of the gel pieces.
- Dry down the extracts in a vacuum concentrator using medium to high drying rate at ambient temperature. Depending on the volume you have that step can take several hours.
- Dissolve the extracts in an appropriate volume of 5% methanol/5% formic acid.
Note: Extracts should be fully covered with solution. - Mix 0.5 μl of sample with an equal volume of matrix solution (saturated solution of cyano-4-hydroxycinnamic acid (HCCA) in 65% water/35% acetonitrile/0.1% trifluor acetic acid (TFA).
- Apply the mixture onto a MALDI target by the dried-droplet method.
- Determine peptide mass fingerprint data on a MALDI-TOF mass spectrometer (Bruker Daltonics) in reflector mode. Settings are dependent on the mass spectrometer available to you.
Data analysis
Data analysis of the obtained peptide mass fingerprints of MALDI-TOF analysis can be performed with the Mascot search algorithm version 2.2 (Matrix Sciences, London, UK).
- Use the following parameter: mass tolerance: 50 ppm, one missed tryptic cleavage allowed, fixed modification: carbamidomethyl cysteine, variable modification: monooxidized methionine.
- Limit searches to organism of infected cells, e.g., mus musculus.
- As a result, the expectation values can be matched to specific protein sequences to identify interaction partners of the effector protein.
Notes
- If antibody against your effector protein is not available, use the commercially available anti-SBP-tag-antibody (e.g., Millipore, clone 20, catalog number: MAB10764).
- If failed to get visible bands using Coomassie staining method, increase the number of cells and try silver staining of the SDS-PAGE.
- If you encounter problems with unspecific interactions or contamination, adding a negative control (i.e., pCTAP plasmid without the effector) to infect the target cells, would facilitate identifying unspecific binding of contaminants.
- We applied electroporation to transform the plasmid harboring the TAP-tagged effector protein in the Yersinia enterocolitica deletion strain. It is important that the bacteria are in the exponential growth phase and that they are washed carefully to ensure that they are salt-free. (Conchas and Carniel, 1990)
Recipes
- Coomassie staining solution
0.1% (w/v) Coomassie Brilliant Blue R-250 (Serva)
25% (w/v) Methanol (Roth)
10% (w/v) Acetic acid (Merck)
ddH2O - Coomassie destain solution
25% (w/v) Methanol (Roth)
10% (w/v) Acetic acid (Merck)
ddH2O - SDS-PAGE sample buffer (2x)
0.5 M Tris (Sigma)
125 mM SDS (Roth)
10% (w/v) Glycerol (Calbiochem)
2% β-Mercaptoethanol (Sigma)
0.1% Bromphenol blue (Merck)
ddH2O adjust to pH 6.8 - PBS (10x)
137 mM NaCl (Roth)
2.7 mM KCl (Roth)
Acknowledgments
This protocol is an extended version of the one described in the original papers (Hentschke et al., 2010; Berneking et al., 2016). Details of the cloning procedure and identification of interacting proteins of the Yersinia effector protein YopM can be found in the original papers.
We thank the UKE Core Facility Mass Spectrometry and Fritz Buck for help with experimental setup and data analysis.
This work was supported by grants from the Deutsche Forschungsgemeinschaft DFG (HE 5875/1-1) to MH and MS. LB was supported by the Deutsche Forschungsgemeinschaft (GRK-1459).
Competing interests
The authors declare that there is no conflict of interest.
References
- Berneking, L., Schnapp, M., Rumm, A., Trasak, C., Ruckdeschel, K., Alawi, M., Grundhoff, A., Kikhney, A. G., Koch-Nolte, F., Buck, F., Perbandt, M., Betzel, C., Svergun, D. I., Hentschke, M. and Aepfelbacher, M. (2016). Immunosuppressive Yersinia effector YopM binds DEAD box helicase DDX3 to control ribosomal S6 kinase in the nucleus of host cells. PLoS Pathog 12(6): e1005660.
- Heesemann, J. and Laufs, R. (1983). Construction of a mobilizable Yersinia enterocolitica virulence plasmid. J Bacteriol 155(2): 761-767.
- Hentschke, M., Berneking, L., Belmar Campos, C., Buck, F., Ruckdeschel, K. and Aepfelbacher, M. (2010). Yersinia virulence factor YopM induces sustained RSK activation by interfering with dephosphorylation. PLoS One 5(10): e13165.
- Keefe, A. D., Wilson, D. S., Seelig, B. and Szostak, J. W. (2001). One-step purification of recombinant proteins using a nanomolar-affinity streptavidin-binding peptide, the SBP-Tag. Protein Expr Purif 23(3): 440-446.
- Stofko-Hahn, R. E., Carr, D. W. and Scott, J. D. (1992). A single step purification for recombinant proteins. Characterization of a microtubule associated protein (MAP 2) fragment which associates with the type II cAMP-dependent protein kinase. FEBS Lett 302(3): 274-278.
- Trulzsch, K., Sporleder, T., Igwe, E. I., Russmann, H. and Heesemann, J. (2004). Contribution of the major secreted yops of Yersinia enterocolitica O:8 to pathogenicity in the mouse infection model. Infect Immun 72(9): 5227-5234.
- Conchas, R. F. and Carniel, E. (1990). A highly efficient electroporation system for transformation of Yersinia. Gene 87(1): 133-137.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Berneking, L., Schnapp, M., Nauth, T. and Hentschke, M. (2019). Tandem Affinity Purification of SBP-CBP-tagged Type Three Secretion System Effectors. Bio-protocol 9(12): e3277. DOI: 10.21769/BioProtoc.3277.
Category
Cell Biology > Cell signaling > Intracellular Signaling
Molecular Biology > Protein > Protein-protein interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.