- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Visualizing Filamentous Actin in Chlamydomonas reinhardtii

Published: Vol 9, Iss 12, Jun 20, 2019 DOI: 10.21769/BioProtoc.3274 Views: 6202
Reviewed by: Samantha E. R. DundonKae-Jiun ChangAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2019
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








Abstract
This protocol aims to visualize the filamentous actin network in Chlamydomonas reinhardtii. We improved fixed-cell labeling conditions using the F-actin probe, phalloidin. We created a Chlamydomonas-optimized protocol by halving the phalloidin incubation time, electing for optimal fixation conditions, and selecting for a healthy cell population. This phalloidin protocol is quick, effective, and is the only labeling method to date that allows for reliable actin filament detection in fixed vegetative Chlamydomonas cells. This method reveals previously unidentified actin structures in Chlamydomonas and novel insights into cytoskeletal dynamics.
Keywords: Chlamydomonas reinhardtiiBackground
Chlamydomonas reinhardtii is a single-celled green alga widely used in studying photosynthesis, cilia, chloroplast biology, and the synthesis of biofuels. It is a leading model system in these fields due to the organism’s well-characterized two apical flagella, inexpensive and simple culture conditions, and a fully sequenced haploid genome allowing for excellent genetic studies. Understanding the Chlamydomonas cytoskeleton will play an important role in advancing these areas of research. However, the filamentous actin network of Chlamydomonas reinhardtii remains uncharacterized, partly due to the difficulties in visualizing filaments in this eukaryotic alga.
Recently, there has been some success in detecting filamentous actin in Chlamydomonas. Through the expression of the fluorescently-tagged actin binding peptide, LifeAct, live-cell imaging revealed an actin-based perinuclear structure (Avasthi et al., 2014; Onishi et al., 2016). However, there is increasing evidence that LifeAct alters cell morphology, actin organization, actin-dependent functions, and preferentially labels a subset of actin structures (Courtemanche et al., 2016, Flores et al., 2019). Further, achieving stable expression is difficult in Chlamydomonas due to epigenetic silencing of exogenous genes (Cerutti et al., 1997) and the need to colocalize actin probes with fixed cell organelle markers is essential for understanding the revitalized field of Chlamydomonas actin biology. Thus, an efficient and specific fixed-cell method of actin visualization would lead to significant advancement in the field and offer novel insights into basic actin biology.
Available actin antibodies do not discriminate between monomeric and filamentous actin. It was previously shown that the widely used fluorescent actin probe phalloidin was ineffective in labeling filamentous actin in vegetative Chlamydomonas cells (Harper et al., 1992). However, phalloidin does label an actin-dense structure in gametic cells known as the fertilization tubule (Detmers et al., 1985). By optimizing phalloidin staining conditions, we are now able to visualize the Chlamydomonas actin network with exquisite detail in vegetative cells (Figure 1). Specifically, reducing the staining incubation time from 30 min to 16 min was essential in achieving optimal signal to noise which allowed for detection above Chlamydomonas’s auto-fluorescent chloroplast. Usage of a brighter and more photostable fluorophore, Atto 488, was also critical in obtaining a high signal to noise ratio and uniform labeling. This protocol allows us to understand how cell body actin filaments are distributed in gametic and vegetative cells for the first time. The previously unidentified filamentous actin structures will be crucial in exploring actin-dependent behaviors and how co-expressed actin genes in Chlamydomonas function to regulate cytoskeletal dynamics. Development of these strategies may prove useful for the study of actin filaments in other protists for which actin visualization has also been challenging.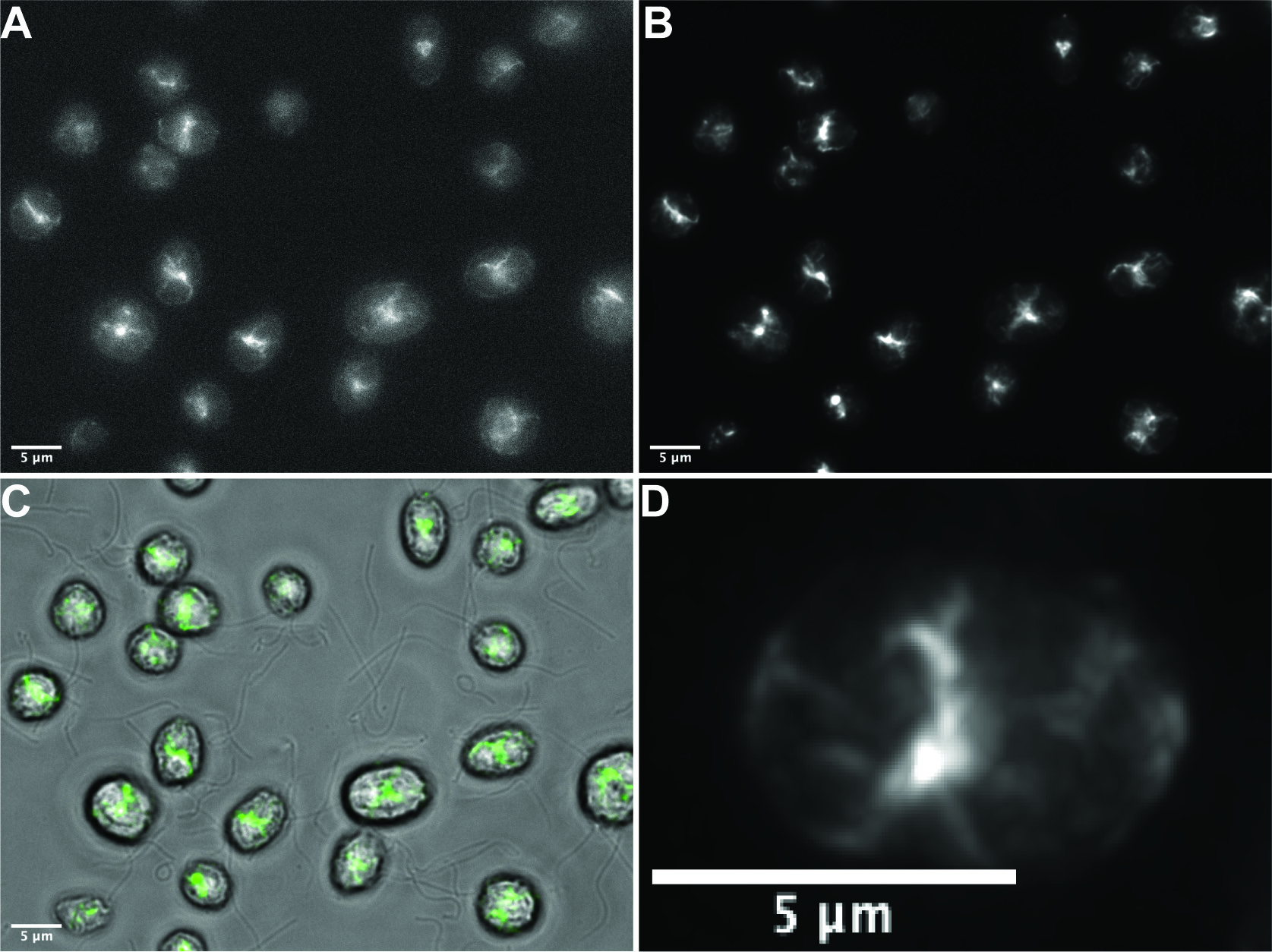
Figure 1. Phalloidin staining in wild-type vegetative Chlamydomonas reinhardtii. A. Representative raw single widefield image of specific filamentous actin staining using our optimized protocol. B. The deconvolved maximum intensity projection of a Z-stack (0.3 μm steps) of the field visualized in A. C. Overlay of brightfield and fluorescence channels with phalloidin signal indicated in green. D. Deconvolved maximum intensity projection of a Z-stack (0.3 μm steps) of a single cell demonstrating effective phalloidin staining. Scale bars = 5 μm.
Materials and Reagents
- Kimwipes
- Parafilm
- Hydrophobic Marker (Elite PAP Pen, Diagnostic Biosystems Ref: K039)
- 1.5 ml Microcentrifuge Tubes
- Columbia Jars (DWK Life Sciences WheatonTM Columbia Jars for Coverslips, catalog number: 02-912-637)
- 10 μl inoculating loop
- Culture tubes (Borosilicate Glass Tubes 13 x 100 mm item) (Globe Scientific, catalog number: 1510)
- Micropipette tips
- Microscope slides
- Coverglass (22 x 22 mm square, 0.13 mm to 0.16 mm thick) (Corning, catalog number: 2845-22)
- Disposable Petri Dishes 60 x 15 mm (VWR®, catalog number: 25384-164)
- Whatman® Qualitative Filter Paper, Grade 1 (Whatman, catalog number: 1001 125)
- Chlamydomonas Strains
Note: The wild-type strain CC-125 mating type + was obtained from the Chlamydomonas Resource Center (University of Minnesota). - Atto 488 Phalloidin (Sigma, catalog number: 49409), store at -20 °C, shield from light
- Poly-L-lysine solution (Sigma, catalog number: P8920)
- PBS Tablet (Research Products International PBS 100 ml Tablets SKU P32080-100T)
- PFA aqueous solution 16% (Electron Microscopy Sciences, catalog number: 15710)
- HEPES (Corning, catalog number: 61-034-RM)
- NaOH pellets (J.T. Baker, catalog number: 3722)
- HCl (Fischer Chemical, catalog number: A481-212)
- Acetone (Histological) (Fisher Chemical, catalog number: A16P-4)
- Fluoromount-GTM (Invitrogen, Ref: 00-4958-02)
- DI Water
- BD Biosciences DifcoTM Agar, Granulated (Ref: 214530)
- Hunter’s Trace Elements (Can be ordered from the Chlamydomonas Resource Center)
- Acetic Acid, Glacial (Certified ACS) (Fisher Chemical, catalog number: A35S-500)
- Methanol
- Tris Base (C4H11NO3), Ultra Pure
- Ammonium Chloride (NH4Cl)
- Magnesium Sulfate Heptahydrate (MgSO4·7H2O)
- Calcium Chloride Dihydrate (CaCl2·2H2O)
- Potassium Phosphate Dibasic Anhydrous (K2HPO4)
- Potassium Phosphate Monobasic (KH2PO4)
- 1x PBS (see Recipes)
- 4% PFA in 7.5 mM HEPES Buffer (see Recipes)
- Phalloidin Stock Preparation (see Recipes)
- TAP liquid media (Tris Acetate Phosphate Media) (see Recipes)
- 100x Tris buffer (see Recipes)
- 100x TAP Salts (see Recipes)
- 1000x Phosphate Solution (see Recipes)
- 1.5% TAP Agar Plates (see Recipes)
Equipment
- Chemical spatula
- Graduated cylinder
- Beaker
- Stir plate
- Stir rod
- pH meter
- Scale
- Tweezers
- Pipettes
- Gusto® High-Speed Mini Centrifuge (Heathrow Scientific, catalog number: HEA10050)
- Empty glove boxes, cover to shield samples from light
- Microscope for image acquisition (We use a Nikon Eclipse Ti-S equipped with a QiIMAGING QICAM)
- Roller Drum (Cel Gro Tissue Culture Rotator) (Thermo Scientific, catalog number: 1640Q)
- -20 °C freezer
Software
- Huygens Essential (Scientific Volume Imaging)
- ImageJ (Schneider et al., 2012)
Procedure
- Cell culture and experimental setup
- Grow two inoculating loops of CC-125 mating type + cells, taken from a Tris Acetate Phosphate (TAP) agar plate, in 2 ml of tris acetate phosphate (TAP) liquid media on a roller drum (40 rpm) overnight (~16 h) in growth lighting at room temperature.
- The next day, set up a parafilm workspace (Figure 2). Use a hydrophobic marker to draw a circle on the coverslip where you will eventually pipette the cells and all future media used in the experiment. Make sure to label coverslips appropriately for each trial, we find it helpful to write “F” in the corner of each coverslip to help denote front from back. Add 200 μl of Poly-L-Lysine at room temperature to the hydrophobic circle and wash after 10 min by quickly dipping in distilled water. The Poly-L-Lysine will immediately fall off the coverslip once it is submerged in water, quickly place coverslip back onto parafilm and use a Kimwipe to remove any excess moisture around the edges of the coverslip. Cover your workspace to shield from debris that might adhere to the Poly-L-Lysine coated coverslips. Unused Poly-L-Lysine coverslips can be stored at room temperature for future use.

Figure 2. Parafilm Workspace - It is essential to select healthy cells by centrifuging 1 ml of cell culture at 1,800 rpm (203 x g) for 1.5 min. Discard the supernatant and resuspend cells in 600 μl fresh TAP media. Slowly pipette media up and down a few times then let culture sit. Healthy, swimming cells should migrate to the top of the culture after about 10 min. Then pipette (from the top of the culture) 200 μl of resuspended cells to Poly-L-Lysine-coated coverslips for 5 min and shield samples from light. The cells are shielded from the light because they phototax and swim upward towards the light, reducing the number of cells that adhere to the coverslip.
- Cell fixation and permeabilization
- After 5 min, tilt off liquid from the coverslips and replace with 200 μl 4% fresh paraformaldehyde (PFA) in 7.5 mM HEPES pH 7.4 to the middle of the hydrophobic circle. Allow cells to incubate in fixative for 15 min at room temperature.
Note: If non-specific chloroplast or bright pyrenoid signal is present (Figures 3A and 3B), dilute PFA solution fresh. We do not make up PFA from powder, but find by simply diluting fixative to 4% from an unopened ampule of 16% PFA solution improves labeling. We find after 2-3 weeks of PFA use, labeling efficiency decreases and makes pyrenoid signal very prominent. Diluting fixative from a fresh ampule alleviates this issue.
Figure 3. Phalloidin staining using non-optimal conditions. A. If cells incubate too long in staining solution or PFA has expired, cells will show strong pyrenoid signal (yellow arrow) and non-specific cytoplasmic staining. This type of signal can be easily distinguished from Atto 488 signal as there will be no filamentous perinuclear or apical staining, instead a dim hazy signal found throughout the entire cell body as pictured in A and B above. Scale bars = 5 μm. C. A general diagram of a Chlamydomonas cell highlighting filamentous actin in pink, the pyrenoid in blue, and the chloroplast in green. - Tilt of PFA solution into a Kimwipe and place coverslips in a Columbia jar containing 1x PBS to wash for 3 min. Use enough PBS so that the coverslip is completely submerged in liquid.
- For cell permeabilization, submerge coverslips in a Columbia jar containing 80% pre-cooled acetone (diluted in water and stored at -20 °C) and then incubate for 5 min at -20 °C.
- Quickly transfer coverslips into a second Columbia jar containing 100% pre-cooled acetone and incubate for another 5 min at -20 °C.
- Place coverslips back on parafilm and allow them to air dry for a minimum of 2 min or longer if needed.
- After 5 min, tilt off liquid from the coverslips and replace with 200 μl 4% fresh paraformaldehyde (PFA) in 7.5 mM HEPES pH 7.4 to the middle of the hydrophobic circle. Allow cells to incubate in fixative for 15 min at room temperature.
- Phalloidin Stain
- Rehydrate cells by transferring coverslips to a Columbia jar containing 1x PBS and incubate for 5 min.
- Place coverslips back on parafilm. Stain coverslips with Atto 488 Phalloidin (Sigma) for 16 min in the dark. This shorter than recommended staining time significantly reduces background and increases the signal to noise ratio.
Note: The Atto 488 Phalloidin reagent greatly enhances photostability and brightness compared to Alexa-488, which allows for more uniform and reproducible filament labeling. Due to the photostability, Atto 488-labeled slides can be reimaged. - Quickly tilt of staining solution and wash cells by transferring coverslips to a Columbia jar containing 1x PBS. Wash once for 5 min.
- Remove excess liquid from the coverslip with a Kimwipe, but be careful to not disturb the inside of the hydrophobic circle (where the cells are adhered). Mount coverslips with self-sealing Fluoromount-GTM (Invitrogen) as quickly as possible. This sealant is designed for staining experiments where the final step is aqueous.
- Image acquisition
Capture 0.3 μm step Z-stacks in brightfield and widefield fluorescence channels (GFP filter set) using a Nikon Eclipse Ti-S equipped with a QiIMAGING QICAM. Deconvolve fluorescence images using Huygens Essential deconvolution software and format in ImageJ.
Data analysis
Hyugens Essential deconvolution software (Scientific Volume Imaging) was used for post-image analysis. Images were deconvolved using the following parameters:
- Algorithm: Classic MLE
- PSF
Mode: Theoretical
Max Iterations: 10 - Iteration Mode: Optimized
- Quality Change Thresh. (%): 0.001
- Signal to Noise Ratio: 20
- Background Mode: Auto
- Background Estimation Radius: 0.7
- Relative Background: 0.0
- Background Correction: If Possible
- Brick Mode: Auto
- PSFs per Brick Mode: Auto
- PSFs per Brick, Manual Mode: 1
Recipes
- 1x PBS
- Dissolve 1 tablet of PBS in 80 ml of purified water and add up to 100 ml for a 1x solution. Stir until dissolved
- 1 Tablet contains 137 mM Sodium Chloride, 2.7 mM Potassium Chloride, and 11.9 mM Phosphate Buffer
- 4% PFA in 7.5 mM HEPES buffer pH 7.4
- PFA aqueous solution 16% (Electron Microscopy Sciences)
- HEPES Stock Solution 10x (0.1 M, pH 7.4) (Farhat, 2013)
- For 100 ml of 0.1 M HEPES, pH 7.4, add 2.38 g of HEPES to an appropriate beaker (100-200 ml in this case)
- Add 80 ml of deionized water to the beaker
- Add a stir bar to the beaker and leave it on a stir plate until completely dissolved (~1 min)
- Add one NaOH pellet to raise pH towards 7.4. Adding one pellet will bring the pH to around 7
- Once the first pellet is fully dissolved, add a second NaOH pellet if necessary, to raise the pH to 7.4. Monitor carefully, and if the pH approaches 7.3/7.4 before the pellet is fully dissolved, carefully remove the NaOH pellet with a clean spatula
Note: In our experience, about 1.5 pellets are just the right amount to raise the pH to 7.4, so retrieving the second pellet is necessary for achieving the right pH. - If the pH goes too high, lower it back to a pH of 7.4 by carefully adding a little HCl, while monitoring the pH
Caution: Wear gloves, eye protection and exercise extreme caution with this acid solution. - Once the pH of the solution is 7.4, add enough deionized water to raise the volume to 100 ml
- Filter (if Whatman filter paper is used here), if possible, and store in the refrigerator for up to 4 months or aliquot and freeze at -20 °C for future use
- For 50 ml, add 37.5 ml of 1x HEPES solution to 12.5 ml fresh 16% PFA for final concentration of 4% PFA in 7.5 mM HEPES buffer. Vortex lightly and store at 4 °C
- Phalloidin stock preparation
- Follow the manufacturer’s instructions and resuspend lyophilized phalloidin reagent in 0.5 ml pre-cooled methanol and store at -20 °C. Shield from light
- To prepare working stock, add 5 μl of phalloidin stock to 200 μl of 1x PBS per coverslip. For example, if you need to stain 4 coverslips, add 20 μl of the stock to 800 μl of 1x PBS. Keep phalloidin stocks shielded from light at all times
- TAP liquid media (1 L)
- Put ~800 ml deionized water into a beaker, flask, or bottle
- Add the following solutions:
- 10 ml 100x tris buffer
Dissolve 24.2 g tris (free base, not tris HCl) in a final volume of 100 ml H2O - 10 ml 100x TAP salts
Dissolve 18.75 g NH4Cl, 5 g MgSO4·7H2O (heptahydrate), and 2.5 g CaCl2·2H2O (dihydrate) in a final volume of 500 ml dH2O - 1 ml 1,000x Phosphate Solution
Dissolve 21.6 g K2HPO4 and 10.8 g KH2PO4 in a final volume of 200 ml dH2O - 1 ml Hunter trace elements
- 1 ml glacial acetic acid
- 10 ml 100x tris buffer
- Add deionized water to a total volume of 1 L
- Test the pH of the solution. It should be between 7 and 7.3. If required, adjust pH using acetic acid. If pH is below 7 initially, start the process over from scratch
- Autoclave and let cool before use
- 1.5% TAP Agar Plates
- Follow Recipe 4 a-d for preparing TAP liquid media
- For 1.5% TAP plates, add 15 g of DifcoTM Agar, granulated to the 1 L TAP solution
- Add a stir bar and autoclave
- Remove agar solution from autoclave and allow solution to cool. Stir the agar so it is evenly distributed throughout the container
- Pour 30-40 ml of agar into each Petri dish. 1 L of agar should make ~2 sleeves of 1.5% TAP agar plates which can be stored in a 4 °C refrigerator or at room temperature for short term use
Acknowledgments
Thank you to members of the Avasthi Lab for troubleshooting and manuscript feedback. This work was funded through P20GM104936 and P20GM103418 (to P.A.).
Competing interests
We have no conflict of interest or competing interests to declare.
References
- Avasthi, P., Onishi, M., Karpiak, J., Yamamoto, R., Mackinder, L., Jonikas, M. C., Sale, W. S., Shoichet, B., Pringle, J. R. and Marshall, W. F. (2014). Actin is required for IFT regulation in Chlamydomonas reinhardtii. Curr Biol 24(17): 2025-2032.
- Cerutti, H., Johnson, A. M., Gillham, N. W. and Boynton, J. E. (1997). Epigenetic silencing of a foreign gene in nuclear transformants of Chlamydomonas. Plant Cell 9(6): 925-945.
- Courtemanche, N., Pollard, T. D. and Chen, Q. (2016). Avoiding artefacts when counting polymerized actin in live cells with LifeAct fused to fluorescent proteins. Nat Cell Biol 18(6): 676-683.
- Detmers, P. A., Carboni, J. M. and Condeelis, J. (1985). Localization of actin in Chlamydomonas using antiactin and NBD-phallacidin. Cell Motil 5(5): 415-430.
- Farhat, Y. (2013). Reagent Preparation: HEPES Stock Solution (0.1 M, pH 7.4). Protocol Place Dec 2013.
- Flores, L. R., Keeling, M. C., Zhang, X., Sliogeryte, K. and Gavara, N. (2019). Lifeact-GFP alters F-actin organization, cellular morphology and biophysical behaviour. Sci Rep 9(1): 3241.
- Harper, J. D., McCurdy, D. W., Sanders, M. A., Salisbury, J. L. and John, P. C. (1992). Actin dynamics during the cell cycle in Chlamydomonas reinhardtii. Cell Motil Cytoskeleton 22(2): 117-126.
- Onishi, M., Pringle, J. R. and Cross, F. R. (2016). Evidence that an unconventional actin can provide essential F-Actin function and that a surveillance system monitors F-Actin integrity in Chlamydomonas. Genetics 202:977-996.
- Schneider, C. A., Rasband, W. S. and Eliceiri, K. W. (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9(7): 671-675.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Craig, E. W. and Avasthi, P. (2019). Visualizing Filamentous Actin in Chlamydomonas reinhardtii. Bio-protocol 9(12): e3274. DOI: 10.21769/BioProtoc.3274.
Category
Cell Biology > Cell staining > Protein
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.