- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Imaging Gene Expression Dynamics in Pseudomonas fluorescens In5 during Interactions with the Fungus Fusarium graminearum PH-1


Published: Vol 9, Iss 12, Jun 20, 2019 DOI: 10.21769/BioProtoc.3264 Views: 5357
Reviewed by: Juan Facundo Rodriguez AyalaFernando A Gonzales-ZubiateJose Antonio Reyes-Darias
Original research article
The authors used this protocol in:

Mar 2017

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Genomics, transcriptomics and metabolomics are powerful technologies for studying microbial interactions. The main drawback of these methods is the requirement for destructive sampling. We have established an alternative but complementary technique based on a microplate system combined with promoter fusions for visualizing gene expression in space and time. Here we provide a protocol for measuring spatial and temporal gene expression of a bacterial reporter strain interacting with a fungus on a solid surface.
Keywords: Microbial interactionsBackground
Microbial interactions underpin many important biotechnological applications spanning medicine, food development and processing, bioremediation and biocontrol. In order to study microbial interactions, a broad range of technologies exist including genomics, transcriptomics and mass spectrometry imaging to study gene expression and metabolites exchanged during interactions. While these methods advance our understanding of microbial interactions, they also have limitations namely the requirement for destructive sampling. We have developed a microplate reader-based system for visualizing gene expression dynamics in living bacterial cells in response to a fungus in space and real-time. Pseudomonas fluorescens In5 is a Gram-negative soil bacterium and potent producer of secondary metabolites with antifungal activity (Michelsen et al., 2015; Hennessy et al., 2015). We previously identified the LuxR-type regulator NunF as a key regulator for synthesis of the antifungal compounds nunamycin and nunapeptin (Hennessy et al., 2017a). In this protocol, we detail how in vivo monitoring of target gene expression in living bacterial cells interacting with living fungal cells can be performed using a microplate-reader based technique (Hennessy et al., 2017b). P. fluorescens In5 expressing the red fluorescent protein mCherry fused to the promoter region of a regulator gene nunF indicating activation of an antifungal secondary metabolite gene cluster was used as a reporter system. Time-lapse image recordings of the reporter red signal and a green signal from fluorescent metabolites naturally produced by the bacterium combined with microbial growth measurements showed that nunF-regulated gene transcription is switched on when the bacterium enters the deceleration growth phase and upon physical encounter with fungal hyphae. The established non-destructive method has many advantages notably the ability to provide detailed space and time information on the transcription of target genes in living organisms. Importantly, the technique is a fast and simple alternative and complementary tool to the many technologies already used for studying microbial interactions.
Here we present a detailed protocol for imaging microbial interactions. In this example, we describe the construction of a reporter strain of the antimicrobial isolate Pseudomonas fluorescens In5 coupled with imaging analysis of gene expression of the reporter strain during an interaction with the fungus Fusarium graminearum PH-1 on an agar surface.
Materials and Reagents
- Eclipse pipette refilling system (Labcon, catalog numbers: 1045-260-000, 1093-260-000, 1036-260-000)
- NuncTM OmniTrayTM Single-Well Plate (Fischer Scientific, catalog number: 140156)
- Gene Pulser® Cuvettes 0.2 cm gap (Bio-Rad, catalog number: 1652086)
- 2 ml Microcentrifuge tubes (Fischer Scientific, catalog number: 21-402-905)
- NEB® 5-alpha Competent E. coli (Subcloning Efficiency) (NEB, catalog number: C2988J)
- Restriction enzymes: BamHI (NEB, catalog number: R3136S ), EcoR1 (NEB, catalog number: R0101S )
- Gentra Puregene Yeast/Bact. kit (QIAGEN, catalog number: 158567)
- Reporter plasmid pSEVA237R expressing the red fluorescent protein mCherry (Hennessy et al., 2018)
- Phusion high-fidelity polymerase (Fisher Scientific, catalog number: F530S)
- Wizard® SV Gel and PCR Clean-Up System (Promega, catalog number: A9281)
- Plasmid Mini Kit QIAprep Spin Miniprep Kit (QIAGEN, catalog number: 27104)
- Gibson Assembly® Cloning Kit (NEB, catalog number: E5510S)
- BactoTM agar (Difco, catalog number: 214050)
- Luria-Bertani broth (LB) (Difco, catalog number: 244610)
- LB agar (LBA) (Difco, catalog number: 244520)
- SOC outgrowth medium (NEB, catalog number: B9020S)
- Defined Fusarium Medium (DFM) (Hennessy et al., 2017b)
- Kanamycin sulfate monohydrate (Duchefa Biochemie, catalog number: K0126)
- Sucrose (Sigma-Aldrich, catalog number: S0389)
- Bacto-tryptone (VWR Chemicals, catalog number: 84610)
- Yeast extract (VWR Chemicals, catalog number: 84601)
- NaCl (Millipore, catalog number: 1.06404.1000)
- Glucose (Sigma-Aldrich, catalog number: 49159)
- Asparagine (Difco, catalog number: 214410)
- MgSO4 (VWR Chemicals, catalog number: 25164.265)
- KH2PO4 (Sigma-Aldrich, catalog number: NIST200B)
- KCl (Sigma-Aldrich, catalog number: P9333)
- Na2B4O7·10H2O (Sigma-Aldrich, catalog number: S9640)
- CuSO4·5H2O (Sigma-Aldrich, catalog number: C8027)
- FeSO4·7H2O (Sigma-Aldrich, catalog number: 1270355)
- MnSO4·H2O (Sigma-Aldrich, catalog number: M7634)
- NaMoO2·2H2O (Sigma-Aldrich, catalog number: 331058)
- ZnSO4·7H2O (Sigma-Aldrich, catalog number: Z0251)
- 300 mM sucrose (see Recipes)
- LB medium (see Recipes)
- DFM agar (see Recipes)
- 1,000x Trace solution (see Recipes)
Equipment
- Pipettes (VWR, catalog numbers: 613-2696E, 613-2702E, 613-2704E)
- Microplate reader (BMG LABTECH, catalog number: FLUO-star Omega)
- Eppendorf® Mastercycler® (Sigma-Aldrich, catalog number: EP6313000018-1EA)
- Gene Pulser® II (Bio-Rad, catalog number: 165-2105)
- Sterile inoculation loops (10 μl) (VWR, catalog number: 12000-812)
- Cork borer (6 mm Ø) for making fungal plugs
- Water bath or a heat block
Software
- BMG Omega Mars Software (Omega V5.11; BMG LABTECH)
- ImageJ analysis software (https://imagej.nih.gov/ij/)
- "Text image to stack” macro (ImageJ, http://rsbweb.nih.gov/ij/macros/ImportTextImageSequence.txt)
- Bulk Rename Utility, (https://www.bulkrenameutility.co.uk)
- Batch converter software converting BMG Omega Mars exported well scan text files to image text files for a rectangle of adjacent wells (MofM.exe)
Procedure
- Selection of target gene and construction of reporter strains
The first step of this protocol is to select a gene of interest (GOI) for gene expression analysis. Once selected, the promoter region located upstream of the GOI is PCR-amplified and cloned in front of a fluorescent protein-encoding gene. The promoter region (400 bp) upstream of the start codon is PCR-amplified using a high-fidelity polymerase and primers introducing overhangs to enable Gibson Assembly® cloning (Gibson et al., 2009) into the target plasmid. - PCR to amplify promoter region located upstream of target gene
- Thaw the Phusion kit reagents and thoroughly mix after thawing.
- Make a 2x Phusion High-Fidelity (HF) DNA Polymerase PCR master mix using the following recipe:
20 μl Phusion HF Buffer
2 μl 10 mM dNTPs
1 μl DMSO
2 μl forward primer (10 μM) (5′-CACAGGAGGCCGCCTAGGCCGCGGCCGCGCGAATTCGCCGACCGTTGGTCGGCTTTGTC-3′)
2 μl reverse primer (10 μM) (5′-GCTTGCATGCCTGCAGGTCGACTCTAGAGGATCCAAGCCTGCATACCAAAATCGCTG-3′)
1 μl Phusion HF Taq Polymerase
70 μl Nuclease-free water - Dispense 49 μl of 2x Phusion PCR master mix into sterile 0.5 μl PCR tubes and add 1 μl genomic DNA (50 ng μl-1) purified from P. fluorescens In5 as previously described (Dichmann et al., 2018).
- PCR program:
a.98 °C 30 s
b.98 °C 10 s
c.62 °C 15 s
d.72 °C 15 s
e.Repeat steps b-d, 30x
f.72 °C 10 min
PAUSE POINT: PCR products can be stored at 4 °C or -20 °C. - Preparation of reporter plasmid for Gibson assembly® cloning
- Digest plasmid DNA of the reporter plasmid pSEVA237R (Hennessy et al., 2017a) harboring a gene encoding the red fluorescent marker mCherry using the restriction enzymes BamHI-EcoR1. Once digested, purify the digested plasmid DNA using the Promega Gel extraction kit according to the manufacturer’s instructions.
- Set up a Gibson assembly® reaction on ice according to the manufacturer’s instructions. Incubated the Gibson assembly® reaction for 1 h at 50 °C without shaking.
- Thaw 50 μl of E. coli DH5α chemically competent cells on ice for 30 min and add 5 μl of Gibson assembly® mix and incubate cells for further 30 min on ice.
- Heat-shock cells using either a water bath for 45 s at 42 °C or a heat block warmed to 42 °C for 90 s.
- Add 250 μl of SOC outgrowth medium to the heat-shocked cells and incubate shaking 200 x g at 37 °C for 1 h.
- Plate 100 μl and 200 μl on LBA supplemented with kanamycin (50 μg/ml).
- Isolate antibiotic resistant colonies into 10 ml LB supplemented with kanamycin (50 μg/ml) and incubate shaking 200 x g overnight at 37 °C.
- Purify plasmid DNA and digest using BamHI-EcoR1 to identify positive clones.
- Perform DNA sequencing to confirm the integrity of the DNA. Once the plasmid DNA is confirmed by sequencing, it can be transformed into P. fluorescens In5.
- Transformation of P. fluorescens In5 with the reporter plasmid pSEVA237R::PGOI
- Inoculate P. fluorescens In5 in 10 ml LB broth shaking 200 x g overnight at 28 °C.
- Centrifuge 6 ml of overnight culture at 4,000 x g for 5 min at 20 °C.
- Wash cells three times each with 4 ml 300 mM sterile filtered sucrose.
- Resuspend the washed bacterial pellet in 400 μl of 300 mM sterile filtered sucrose.
- Add 300-500 ng plasmid DNA from each construct: tester plasmid (pSEVA237R::PGOI) and empty vector control (pSEVA237R) to 100 μl competent cells.
- Transfer mixture to 2 mm gap width electroporation cuvette.
- Apply a pulse (25 μF, 200 Ω, 2.5 kV) in the electroporation apparatus.
- Add 1 ml LB at room temperature.
- Transfer mixture to a 2 ml Eppendorf tube.
- Shake mixture at 200 x g 2 h at 28 °C.
- Isolate antibiotic resistant colonies into 10 ml LB supplemented with kanamycin (50 μg/ml) and incubate shaking 200 x g overnight at 28 °C.
- Purify plasmid DNA and digest using BamHI-EcoR1 to identify positive clones.
- Pre-culturing of F. graminearum PH-1 and P. fluorescens In5
- Using a sterile cork borer, transfer a 6 mm Ø sized plug of F. graminearum PH-1 onto DFM media prepared as previously described (Hennessy et al., 2017b) and incubate for 5 days at 28 °C.
- Inoculate P. fluorescens In5 harboring either pSEVA237R::PGOI or pSEVA237R in 10 ml LB broth supplemented with 50 μg ml-1 kanamycin (antibiotic selectable marker on pSEVA237R backbone) and incubate shaking 200 x g overnight at 28 °C.
- Establishment of bacterial-fungal interaction on solid surface
- Add 35 ml of DFM agar to a sterile NuncTM OmniTrayTM. Place 10 plugs of F. graminearum PH-1 (6 mm Ø) down the center of the plate in a straight line.
- Using a 10 μl sterile loop streak the bacterial reporter strains 3 cm away from the fungal plugs. Allow bacterial streaks to dry on the surface.
- Experimental set-up
- The bacterial reporter strains and fungus are then co-cultured in the microplate reader, programmed to simultaneously measure red and green fluorescence respectively in addition to cell density (Figures 1 and 2).
- For step-by-step instructions for defining plates and time-series recording of images using the plate reader software BMG Omega Mars Software see Hennessy et al. (2017b).
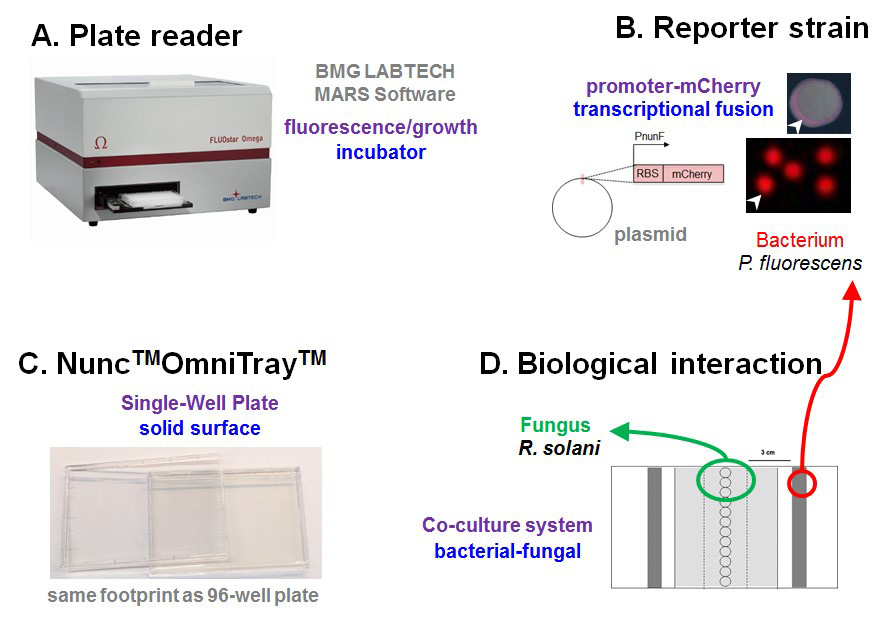
Figure 1. Schematic of experimental setup. A. Plate reader programmed to measure fluorescence and cell density and serves as an incubator to co-cultivate microbes at selected temperatures. B. 96-well microplates exchanged for OmniTray enabling the addition of agar medium providing a solid surface for interactions. C. Bacterial reporter strains are constructed whereby the promoter region located upstream of a gene of interest is cloned in front of a gene encoding a fluorescent protein (e.g., mCherry) to measure gene transcription in living cells. D. Biological interaction is established where fungal plugs are placed down the center of the OmniTray and bacterial reporter strains streaked either side of the fungal plugs.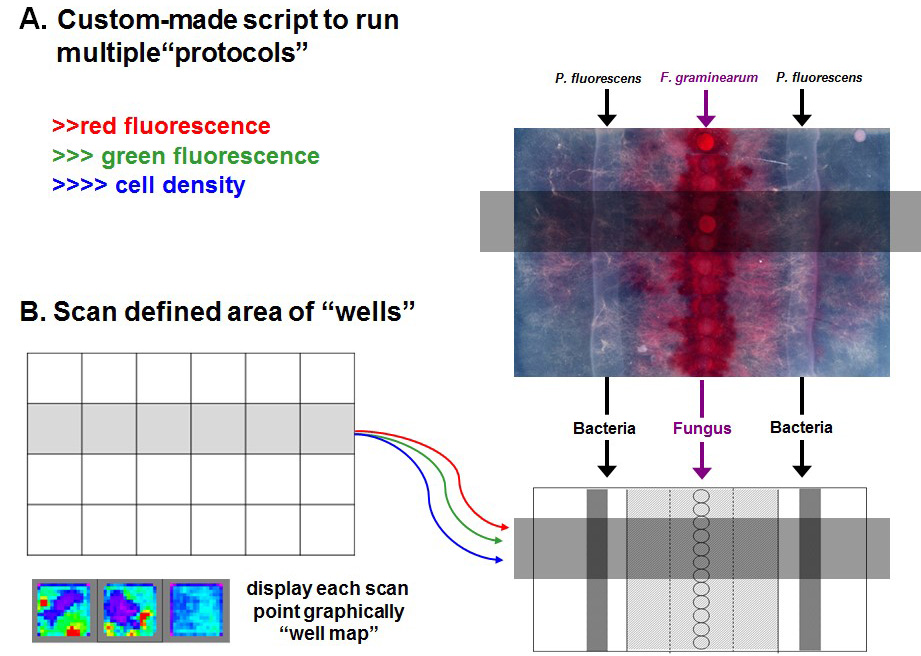
Figure 2. Schematic of scanning function. A script can be written to instruct the plate reader to measure multiple parameters simultaneously (A) and perform a scanning function “imaging” a specific area of the plate measuring gene expression and growth. Bacteria streaked on the left-hand side of the fungal strains is the reporter strain (pSEVA237R::PGOI) and the bacterial streak on the right-hand side of the fungal plugs is the control strain (pSEVA237R). “Well map” is shown using a rainbow (blue-green-yellow-red) representation of intensities of signals for one of the protocols from low to high (B).
Data analysis
Once the runs are complete, a series of scans or images are obtained which must then be exported as text files prior to batch conversion of the files from Mars formatted well image text files into image text files for the whole plate. The text files can then be imported into the free image analysis software ImageJ. Once in ImageJ, the time-series image stack can be converted into an animation presentation and quantitative data can be extracted from specific areas of interest (Figures 3 and 4).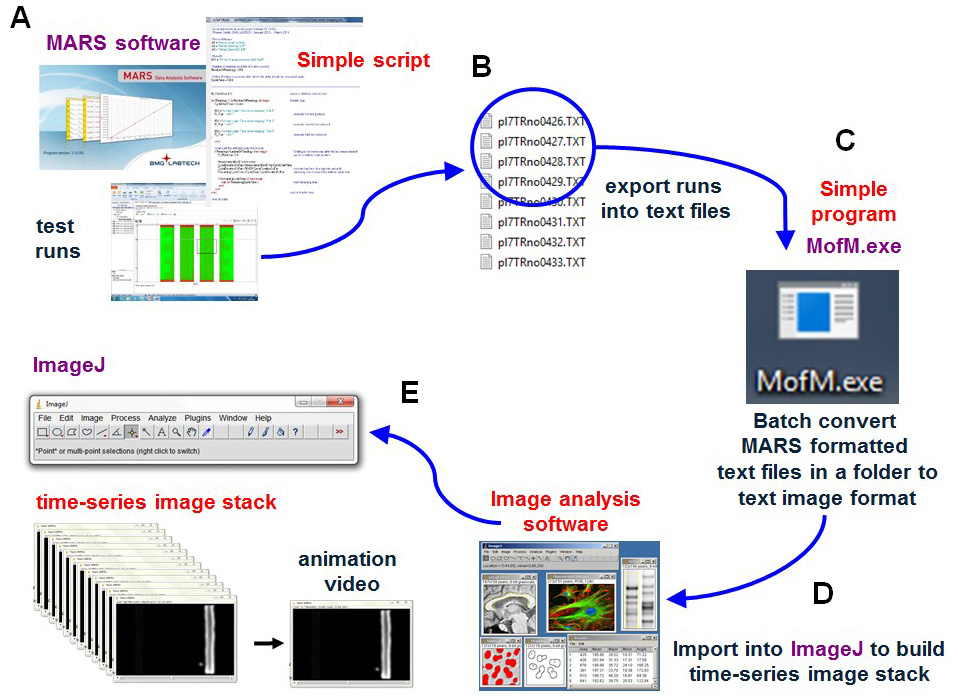
Figure 3. Schematic showing data processing. Upon completion of imaging, a time series of scans or images are (A) exported as MARS text files. (B) Batch converted into a time series of text image formatted files using the program MofM.exe. (C) The text image time series files are imported into the free image analysis software ImageJ (D) to make a TIF formatted time-series image stack retaining all the intensity levels of the original scans (32-bit TIF image stack used). Once in ImageJ, the time-series image stack can be converted into an animation presentation to visualize spatial and temporal gene expression or using the image analysis procedures to extract numerical data from the image stack (E).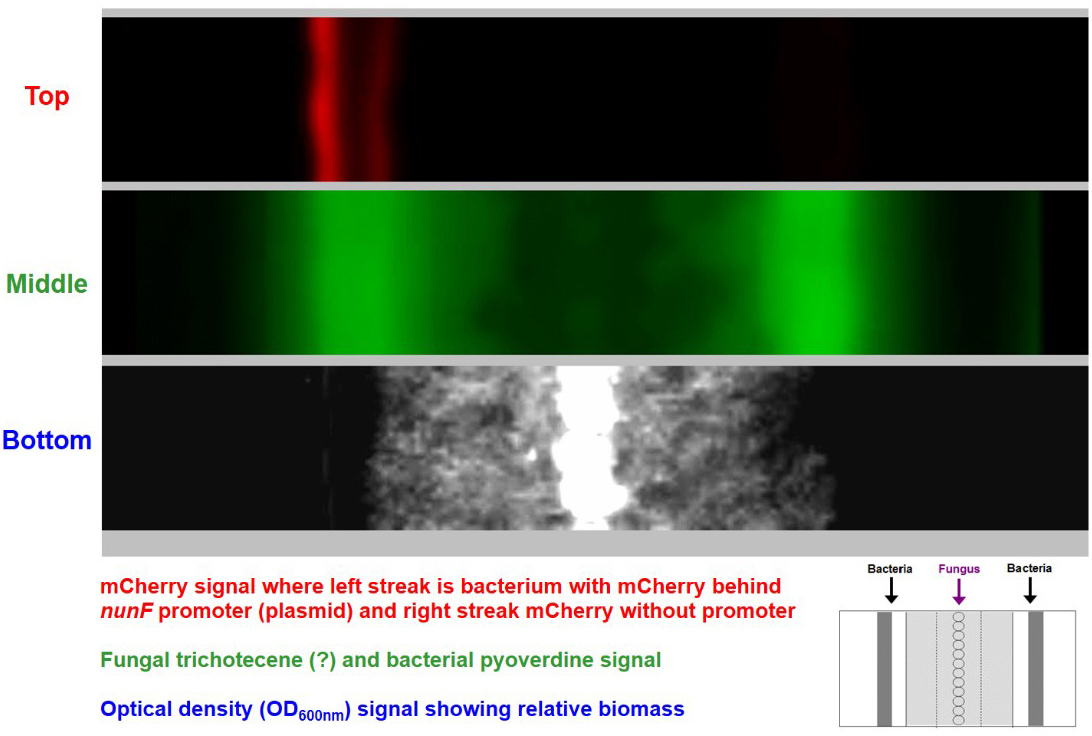
Figure 4. System enables real-time imaging of transcription. Example of time-series stack converted into an animation video (available at https://www.nature.com/articles/s41598-017-00296-4#Sec9). Top image shows a bacterial reporter strain emitting red fluorescence. Middle image shows fluorescent compounds produced by the bacterial reporter strains and a weak signal of fungal produced fluorescent compounds seen accompanying the expanding fungal mycelium growth. Bottom image shows fungal growth.
- Export of imaging runs into text files
- Open the MARS Omega Software.
- Import files (or scans) from the plate reader by selecting the .RUC files (note the plate reader files should reflect the number of parameters run e.g., 141 files divided by 3 measurements [cell density, red fluorescence, green fluorescence] should give 47 files per run).
- Once the files are imported select all the runs, select the set of stored test runs using the BMG Omega MARS program “Open function” (use the program version V3.20 R2 that includes the new “Export multiple Ascii files” function).
- Next sort the files in order (i.e., mCherry runs, GFP runs etc.).
- Click “sort according to name” (sort the test runs so that it is easy to select all test runs with the same protocol).
- Mark the set of test runs and right-click. Select “Export multiple ASCII files”. Check settings by clicking the “Settings” button: Untick boxes in the “File content options”. In the “File Name and Locations options” do the following:
- Select folder to store the set of output text files (there will be one file for each reading time).
- File name: use the < automatic filename creation > option.
- Select “File Extension”= TXT “Separator”=TAB and “If file exist”=”rename old file adding date/time”. In “Auto mode and Manage Test Runs File Export options” only tick the “Export microplate view” option. Then press the “OK” button.
- Next, replace the ‘o’ s with ‘0’ to ensure files are ordered correctly.
- Bulk rename utility tool for renaming files
- Download software Bulk Rename Utility tool (http://www.bulkrenameutility.co.uk/Main_Intro.php).
- Open Bulk Rename Utility.
- Open txt. Files.
- Highlight only those to change (only 2 digit files e.g., o98 not 100 or above).
- Go into documents folder where .txt files saved and check that they have been changed from noo98 to no098.
- Prepare destination folder for these files by removing all spaces e.g., Imaging Test Run to ImagingTestRun (necessary for converter program to function) and also find the location for this folder through my Computer: H:\Documents\ImagingTestRun. Then insert additional forward slashes for converter program to read: H:\\Documents\\ImagingTestRun.
- Double-click the converter program MofM.exe (in supplementary file S2) that can be placed in any folder and need no installation. In the dialogue enter full path name (note that use double backslash \\ in the path name) of the folder containing the text files saved in step 6 (and eventually renamed by giving a prefix for the converted files. The program is now run and there is created a subfolder containing the converted files that can be used for text image series import to stack using the ImageJ Macro in supplementary file S3).
- Copy location address by right-clicking in program > edit > paste then press enter and enter a prefix, e.g., pl (as the program is converting to a single well plate layout) > enter. You should find a new sub-folder under TestImagingRun called ‘plfolder’ with all the files.
- Installation of ImageJ analysis software and macro retrieval
- Download and install ImageJ.
- Go to the following link to retrieve the macro: http://rsbweb.nih.gov/ij/macros/ImportTextImageSequence.txt.
- Save the macro as a macro file (.ijm) in the macros folder of the ImageJ folder as “Text to images. ijm”.
- Open ImageJ software as follows:
> plugins > macros > install > select and open Text images to stack.ijm - The macro is now installed.
- ImageJ data analysis
- First open ImageJ then click plugins > macros > text image to stack > select plfolder.
- To reorder files, open image > stacks > tools > reverse.
- This results in a pixelated image so click image > adjust > brightness and contrast > auto.
- Once steps 1-3 are completed, the files are ready for analysis and the next step is extraction of data from Regions of Interest (ROI).
- Enlarge the image using the magnifying glass and click on the image (right click enlarges, left click reduces).
- Click on ROI and export data as a table at a specific slice (i.e., time-point slice or image 31/47).
- A picture shows the cumulative expression at a specific time over the whole area. To decide which time-point to investigate, scroll along e.g., 24/27 and draw on the image.
- To measure distance, the first step is to calibrate the image. For example, to measure the known distance between two streaks (e.g., 5 cm) first press analyze, set scale. Calculate the distance in pixels. Known distance 50 mm.
- To investigate expression over time on a plate reader, first mark the ROI, then select area > image > stacks > plot Z axis profile > save > save as excel (Figure 5).
- Click Live > File > save as > selection (save ROI file e.g., stack.roi).
- Go to “Files ready for analysis” and save as single TIFF file e.g., stack. tif.
- To open the file, click “File” > Open stack.tif > select a time-point e.g., 34/47 > File > save as >JPEG to get an image.
- For saving marked files (i.e., marking a specific area): File > save as selection > stack.roi. This step can be repeated to add a second ROI.
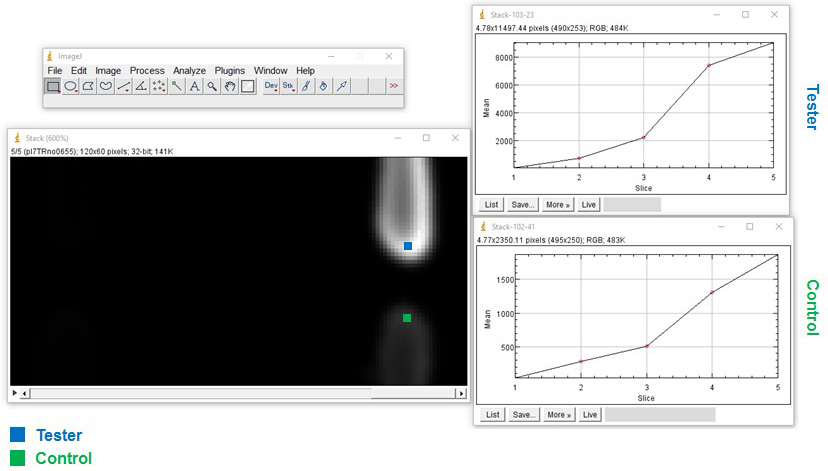
Figure 5. Extraction of quantitative data. Example of regions of interest (ROI) of two reporter strains: Tester (bacterium with mCherry behind nunF promoter) and the Control (bacterium with mCherry and no promoter). Using the free image analysis software ImageJ (https://imagej.nih.gov/ij/) ROIs are selected and a z-axis profile is plotted to obtain quantitative data.
Notes
- How to add color: work of the TIFF file created above then click image > look-up tables > red. Note that if the file is saved in TIFF format, the color will not be red so remember to save the file in PNG format.
- How to export a video: save image as an AVI file then compress as a JPEG and define the frame rate e.g., 7 fps (this can be manually decided based on the scrolling time through image).
- How to overlay images: The above method describes the data processing required for analyzing one set of files from the plate reader (i.e., red fluorescence readings). Before overlaying, a TIFF file must be created for each parameter measured on the plate reader:
3x TIFF files for each “channel”- Channel 1: red fluorescence signal stack to TIFF (color red)
- Channel 2: blue fluorescence signal stack to TIFF (color blue)
- Channel 3: cell density signal stack to TIFF (color grey or black/white)
- Click image > colour > merge channels > create composite > ok
- Save the super-imposed file.
Note: For “create composite” step, tick “create composite” and leave the other parameters blank. Note this type of image can only be used for distance or visualization. Red fluorescence e.g., mCherry signal cannot be measured, for this the single TIFF file must be used. It is also important to measure the distance between strains before and after the runs and to take a scan or photo of the plate.
Recipes
- 300 mM sucrose (Filter sterilized)
Add 102.69 g sucrose to 800 ml sterilized MilliQ water and stir until the sucrose is dissolved, and make the final volume to 1 L - LB
- Add 10 g Bacto-tryptone, 5 g yeast extract, 10 g NaCl to 800 ml MilliQ water and stir until the reagents are dissolved
- Autoclave at 121 °C for 15 min
- To make LBA, add 15 g BactoTM agar
- DFM agar
- Add 20 g BactoTM agar to 716 ml MilliQ water and autoclave at 121 °C for 15 min
- Following autoclaving add the following:
20 ml 20% w/v glucose
1 ml of 1,000x Trace solution
50 mM asparagin
10 ml 0.21 M MgSO4
10 ml (1.12 M KH2PO4 + 0.7 M KCl), pH 6
- 1,000x Trace solution
Add 40 mg Na2B4O7·10H2O, 400 mg CuSO4·5H2O, 1.2 mg FeSO4·7H2O, 700 mg MnSO4·1H2O, 800 mg NaMoO2·2H2O, 10 g ZnSO4·7H2O to 800 ml sterilized MilliQ water and autoclave at 121 °C for 15 min
Acknowledgments
This work was supported by the Villum Foundation grant VKR7310 (Microbial Communication-A Key to the Development of Novel Sustainable Agri- and Aquaculture Practices Using Biological Control Bacteria) awarded to P. Stougaard.
Competing interests
None declared.
References
- Hennessy, R. C., Christiansen, L., Olsson, S. and Stougaard, P. (2018). A broad-host range dual-fluorescence reporter system for gene expression analysis in Gram-negative bacteria. J Microbiol Methods 144: 173-176.
- Dichmann, S. I., Park, B., Pathiraja, D., Choi, I. G., Stougaard, P. and Hennessy, R. C. (2018). Draft genome sequence of a novel Serratia sp. strain with antifungal activity. Microbiol Resour Announc 7(22) pii: e01340-18.
- Gibson, D. G., Young, L., Chuang, R. Y., Venter, J. C., Hutchison, C. A., 3rd and Smith, H. O. (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6(5): 343-345.
- Hennessy, R. C., Glaring, M. A., Michelsen, C. F., Olsson, S. and Stougaard, P. (2015). Draft genome sequence of Pseudomonas sp. strain in5 isolated from a greenlandic disease suppressive soil with potent antimicrobial activity. Genome Announc 3(6) pii: e01251-15.
- Hennessy, R. C., Phippen, C. B. W., Nielsen, K. F., Olsson, S. and Stougaard, P. (2017a). Biosynthesis of the antimicrobial cyclic lipopeptides nunamycin and nunapeptin by Pseudomonas fluorescens strain In5 is regulated by the LuxR-type transcriptional regulator nunF. Microbiologyopen 6(6).
- Hennessy, R. C., Stougaard, P. and Olsson, S. (2017b). A microplate reader-based system for visualizing transcriptional activity during in vivo microbial interactions in space and time. Sci Rep 7(1): 281.
- Michelsen, C. F., Jensen, H., Venditto, V. J., Hennessy, R. C. and Stougaard, P. (2015). Bioactivities by a crude extract from the Greenlandic Pseudomonas sp. In5 involves the nonribosomal peptides, nunamycin and nunapeptin. PeerJ 3: e1476.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Hennessy, R. C., Stougaard, P. and Olsson, S. (2019). Imaging Gene Expression Dynamics in Pseudomonas fluorescens In5 during Interactions with the Fungus Fusarium graminearum PH-1. Bio-protocol 9(12): e3264. DOI: 10.21769/BioProtoc.3264.
Category
Microbiology > Microbial genetics > Gene expression
Molecular Biology > Protein > Expression
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.