- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Tryptophan Fluorescence Quenching Assays for Measuring Protein-ligand Binding Affinities: Principles and a Practical Guide

Published: Vol 9, Iss 11, Jun 5, 2019 DOI: 10.21769/BioProtoc.3253 Views: 22194
Reviewed by: Andrea PuharAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Oct 2018
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Tryptophan fluorescence quenching is a type of fluorescence spectroscopy used for binding assays. The assay relies on the ability to quench the intrinsic fluorescence of tryptophan residues within a protein that results from changes in the local environment polarity experienced by the tryptophan(s) upon the addition of a binding partner or ligand. The quenching can arise from local changes near the interaction site or from binding-induced conformational changes. In cases where the titrant absorbs at or near the excitation or emission wavelengths of tryptophan, significant quenching can occur even without an interaction. This is known as the inner filter effect. This protocol describes how to use tryptophan fluorescence quenching to investigate the binding affinity of a protein for its partner/ligand and how to check and correct for the inner filter effect. As an example, we measured the binding affinity of the haem-binding protein, HusA, from Porphyromonas gingivalis for haem, and showed how we accounted for the inner filter effect.
Keywords: Tryptophan fluorescence quenchingBackground
Fluorescence is a form of luminescence used in many fields ranging from fluorescent labeling in biology and medical diagnostics to fluorescence spectroscopy in chemistry. In fluorescence spectroscopy, incident radiation can promote a molecule to adopt one of many higher vibrational energy states, including those in an excited state. Upon collision with other molecules, some of the vibrational energy is lost until it returns to the lowest vibrational level of an excited state. Upon transition from the excited to the ground state, photons are then emitted at wavelengths that depend on the vibrational state the electron re-occupies in the ground state. Therefore, typically the intensity of photon emission varies with wavelengths and the emitted light has a longer wavelength than the incident radiation (Lakowicz, 1999).
In protein biochemistry, fluorescence spectroscopy is routinely used for monitoring protein (un)folding, and investigating protein conformational changes, binding and interactions. One common fluorescence assay relies on the intrinsic fluorescence of proteins that arises from excitation of aromatic amino acids, mainly tryptophan (Teale and Weber, 1957). Phenylalanine, despite being excitable, has a low quantum yield while tyrosine, despite having high quantum yield, is often quenched naturally (Möller and Denicola, 2002). Tryptophan, being a large hydrophobic amino acid is usually partially or fully buried in hydrophobic sites within proteins, or bound to ligands through hydrophobic interactions such as π-π stacking. Tryptophan can be selectively excited at 295 nm as there is little absorption by other residues at this wavelength. Upon excitation, tryptophan gives rise to an emission spectrum that peaks at 355 nm. Structural changes in the vicinity of tryptophan residues induced by ligand/partner interactions, protein conformational changes, self-association or protein folding/denaturation, can alter the intensity of fluorescence as well as introduce a wavelength shift in the emission spectrum (Möller and Denicola, 2002). In most binding assays, a decrease in fluorescence and a blue shift to lower wavelengths are observed upon binding which is attributed to the increase in hydrophobicity around the tryptophan sites. This is known as tryptophan fluorescence quenching and under the right conditions, can be used to measure the equilibrium binding constant, also known as the association constant Ka.
If P is the protein being studied and L is the ligand (we take this to mean any binding partner which can include protein, peptide and small molecule ligands) that P binds, then at equilibrium: 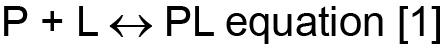
and the association constant Ka is defined as (![]() ) while its inverse (i.e.,
) while its inverse (i.e., ![]() ) is the dissociation constant, Kd.
) is the dissociation constant, Kd.
To assess whether tryptophan fluorescence quenching may be a suitable technique for measuring the binding constant of a protein for its partner or ligand, several questions that should be considered are listed below. “No’s” in Questions 1 and 2 would suggest tryptophan fluorescence quenching is unlikely to be the method of choice and other techniques should be explored for measuring the binding affinity of interest. A “Yes” to Question 3 would indicate further investigations are required to check the feasibility of the experiment and in favorable cases, the effect may be corrected for during data analysis, as shown in the HusA:haem example. Note that in the case where the interaction of interest is a protein:protein or protein:peptide interaction, then either component can be considered the “protein” and the “ligand”.
- Does the protein have one or more tryptophan(s) that are likely to be in different environments when the protein is free versus bound?
- Is there an absence of tryptophan(s) in the ligand?
- Does the ligand absorb at or near the excitation or emission wavelengths of tryptophan at the concentrations required for the titration study? If so, is the inner filter effect significant? In some cases, the inner filter effect can be minimized by using lower ligand concentrations or its contribution accounted for with “control” titrations (see later sections).
The two following questions which apply equally to other techniques used for measuring binding affinity also need to be considered. To a large degree, “Yes’s” are required but the answer may depend on the techniques and set up used which can alter detection limits and dynamic range, as well as the solution conditions required.
- Are the protein, ligand and the complex soluble at the concentrations required in the solution condition used for the titration? This will depend on the sensitivity of the instrument, Kd and solution properties of the components. Point 5 below further explains concentration considerations.
- Can you work with a protein concentration that is not too high relative to the expected Kd? As a rough rule of thumb, a concentration that ranges from as low as practical to 10x that of the estimated Kd is a good starting point. Can you titrate in the ligand such that its final concentrations in the sample can span 0.1x to at least 5x (and preferably 10x) that of the estimated Kd. A titration may need to be repeated with additional titration points if the measured Ka is significantly different from the range estimated initially to get an accurate measurement of Ka.
The relationship between protein:ligand complex, protein and ligand concentrations, Ka and appearance of the binding curve.
To illustrate the importance of Question 5, an interactive Excel spreadsheet (binding.xls) is available to download. The following section explains why binding affinity measurements are only meaningful when “sensible” protein and ligand concentrations are chosen relative to the binding affinity. In the spreadsheet, only the yellow boxes (Ka, Ptotal–total protein concentration, Ltotal–total ligand concentration at the end of the titration) are modifiable. The graphs on the left-hand side in the Excel spreadsheet are obtained by solving the quadratic equation as derived from equation [1] while the graph on the right-hand side shows the comparison between the quadratic solution and using the Lfree (Ligand free) can be approximated by Ltotal assumption. This assumption is valid when the protein concentrations used and/or the affinity (or Ka) is relatively low. This is often the case for enzyme-linked immunosorbent assay (ELISA) type reactions unless the binding affinity is very high (Ka > ~109 M-1) and hence this model is often included as the default for 1:1 binding in many curve fitting software. However, this does not always apply especially in biophysical studies where the protein concentrations need to be high due to detection limits.
The following set of graphs (Figures 1A and 1B) illustrates the case when the ligand concentration chosen is too low relative to the protein concentration in an example titration where L is being titrated into P resulting in a straight line for the concentration of PL which has a very similar appearance despite a 10-fold difference in the Kd.
Figure 1. Calculated binding curves showing the relationship between [PL] and [Ltotal] at a constant [Ptotoal]. Here [Ptotal] is chosen to be 10 times that of the final [Ltot]. A. Kd = 0.1 μM, Ptotal = 10 μM, Ltotal =1 μM. B. Kd = 1 μM, Ptotal = 10 μM, Ltotal =1 μM.
If this titration has continued to a ligand concentration that is 10x that of the protein concentration, the two PL concentration profiles can now be easily distinguished (Figures 2A and 2B).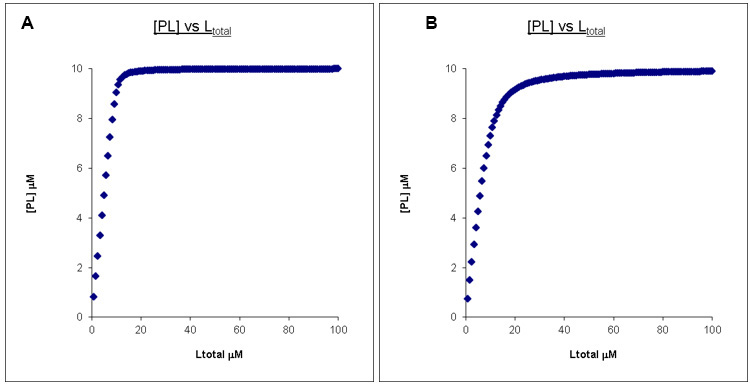
Figure 2. Calculated binding curves showing the relationship between [PL] and [Ltot] at a constant [Ptot]. Here [Ptot] is chosen to be 10 times less than that of the final [Ltot]. A. Kd = 0.1 μM, Ptotal = 10 μM, Ltotal =100 μM. B. Kd = 1 μM, Ptotal = 10 μM, Ltotal =100 μM.
However, no matter how high the final Ltotal concentration is, the PL concentration profiles remain very similar once the affinity is “too” high. Shown in Figure 3 is the comparison between a Kd of 0.01 μM and 0.001 μM with Ptotal and Ltotal otherwise unchanged.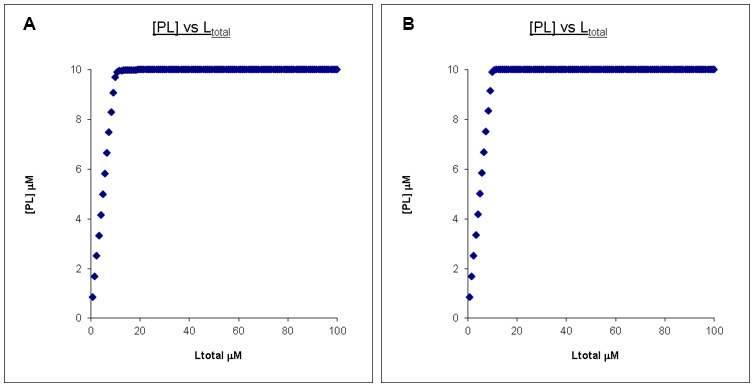
Figure 3. Calculated binding curves showing the relationship between [PL] and [Ltot] at a constant [Ptot]. Here [Ptot] is chosen to be far higher than the Kd’s. A. Kd = 0.01 μM, Ptotal = 10 μM, Ltotal =100 μM. B. Kd = 0.001 μM, Ptotal = 10 μM, Ltotal =100 μM.
The only way to “fix” this is we can drop the protein concentration. As seen in Figure 4, we can recover the difference in PL concentration profiles by dropping the protein concentration (and corresponding ligand concentration) to 0.1 μM so it is comparable to the Kd’s. However, in the case of tryptophan fluorescent quenching or other techniques that require protein signals to be measured, this may not be feasible given the detection limit of the technique. 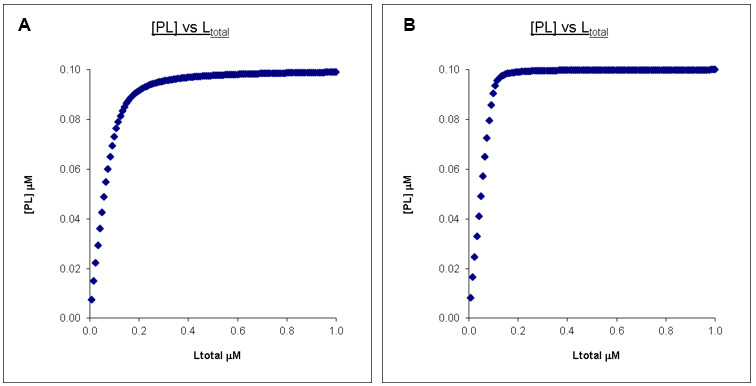
Figure 4. Calculated binding curves showing the relationship between [PL] and [Ltot] at a constant [Ptot]. Here [Ptot] is chosen to be higher than the Kd’s but still in a reasonable range. A. Kd = 0.01 μM, Ptotal = 0.1 μM, Ltotal = 1 μM. B. Kd = 0.001 μM, Ptotal = 0.1 μM, Ltotal =1 μM.
As such, it may be necessary to use more than one technique to “nail” a binding affinity. It is also advisable to predict what the binding curve looks like given what one knows about the system before choosing a technique and the concentrations of protein and ligand to use.
What other techniques are commonly used for measuring binding affinity?
In addition to the tryptophan fluorescent quenching assay described, several other assays can be used to measure protein binding affinities. Generally, techniques with lower sensitivities (e.g., NMR spectroscopy) are more suited for measuring weaker binding and vice versa. That is, the typical concentrations need for signal detection should match the expected Kd’s. Common assays include labeled ligand-binding assays, label-free binding assays and thermodynamic binding assays. An example of a labeled ligand-binding assay is a fluorescent ligand assay in which the binding partner is fluorescently labeled and monitored for a change in fluorescent intensity or anisotropy once the complex forms (Breen et al., 2016). This assay is useful as it offers a range of wavelengths depending on the fluorophore, however attachment of the fluorophore to the ligand may lead to conformational changes that can interfere with the binding reaction. An example of a label-free binding assay is UV-visible (UV-vis) absorption spectroscopy. It is a powerful technique to study the binding reaction of a chromophore that is sensitive to changes induced by ligation (Nienhaus and Nienhaus, 2005). The binding affinity can be measured by observing absorption profiles of a ligand upon titration of its binding partner. Despite being useful for haem and other “colored” ligands, this assay is not suitable for most ligands. This technique also fails to explore the binding reaction from the protein’s point of view. Thermodynamic binding assay, such as Isothermal Titration Calorimetry (ITC) uses enthalpy changes in the ligand binding reaction to measure the binding affinity and thermodynamics of an interaction (Krainer and Keller, 2015). It is very useful for binding reactions that produce significant enthalpy changes but not otherwise. Other commonly used biophysical techniques for measuring protein:ligand binding affinities include surface plasmon resonance (SPR), microscale thermophoresis (MST), and Bio-Layer Interferometry (BLI) with SPR and BLI having the additional capability to follow binding kinetics.
HusA:haem titration as an example
Our previous work demonstrated that the tryptophan fluorescence of a haem-binding protein from Porphyromonas gingivalis, termed Haem uptake system protein A (HusA), can be quenched by its ligand haem (Gao et al., 2010). HusA has three tryptophan residues however only one is located at the haem-binding site (Protein Data Bank accession code 6BQS; Figure 5A) and binds a range of porphyrins including haem (Figure 5B). UV-visible (UV-vis) spectroscopy was also used to explore haem binding by HusA and produced a similar Ka estimate. ITC was also attempted, however, the heat changes were too small even at the highest concentrations of protein and haem used so was not pursued.
Figure 5. Structure of HusA (A) and haem (B). The structure of HusA is displayed in ribbon with its three tryptophan residues shown as sticks. The tryptophan (W130) at the haem binding pocket is highlighted in cyan.
Despite being an efficient static fluorescent quencher of HusA, haem also displays background quenching known as the inner filter effect. This inner filter effect occurs when haem absorbs excitation light or fluorescence emitted by tryptophan, known as the primary or secondary inner filter effects, respectively (Ghisaidoobe and Chung, 2014). This creates an apparent continual quenching and hence binding. To correct for this, control titrations were carried out by titrating haem into N-Acetyl-L-tryptophanamide (NATA) (Zelent et al., 1998; Fonin et al., 2014), a fluorophore that does not bind haem. This data was used to correct for the inner filter effect.
Materials and Reagents
- Pipette tips
- Protein with tryptophan residues affected by binding (e.g., HusA)
- Ligand (e.g., Hematin/haem, Sigma-Aldrich, catalog number: H3505)
- N-Acetyl-L-tryptophanamide, analytical grade (NATA, Sigma-Aldrich, St Louis, MO)
- Tris(hydroxymethyl)aminomethane, analytical grade (chem-supply, https://www.chemsupply.com.au/)
- NaCl, analytical grade (chem-supply, https://www.chemsupply.com.au/)
- NaOH, analytical grade (chem-supply, https://www.chemsupply.com.au/)
- Binding buffer (see Recipes)
- Buffer for dissolving ligand (see Recipes)
Equipment
- Quartz 5-mm cuvette (Starna Pty LTD, Baulkham Hills, Aus)
- Pipettes
- Carey Eclipse Fluorescence Spectrophotometer (Agilent)
Software
- OriginLab for data analysis
- Microsoft Excel
Procedure
- Experimental Rationale
The binding assay should have at least ten titration points that span from ~0.1x to 5-10x the final Kd (Figure 6). We made Haem stock solutions at four concentrations (Table 1) to avoid diluting the target by more than 5% during the titration to simplify data analysis. For the HusA experiment, previous work had suggested that haem was bound with a Kd of ~10 μM. However, due to the significant inner filter effect, the final titration point was chosen to be ~5x the final Kd. The 11 titration points were designed using stock solutions (Table 1), and these are outlined in Table 2.
Table 1. Haem stock solutions prepared. Haem powder is dissolved in 0.1 M NaOH, pH 12, by gentle pipetting and inversion for 5 min, and incubation at room temp for 10 min before use.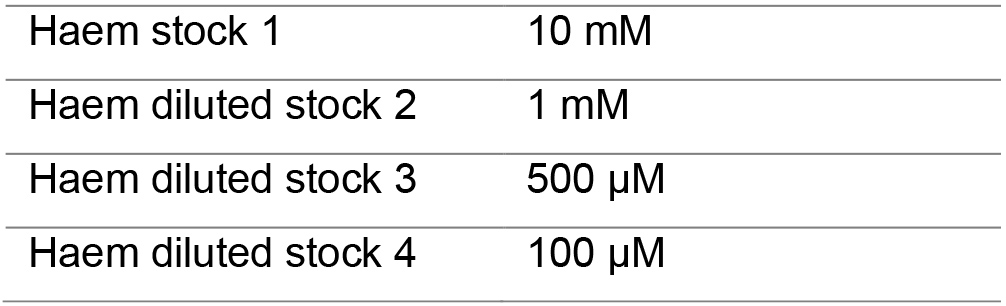
Table 2. Titration protocol for haem into HusA using four stock solutions. 11 titration points chosen are listed below. Target concentration is diluted by less than 5%, and ligand concentration should span up to at least 5x estimated Kd.
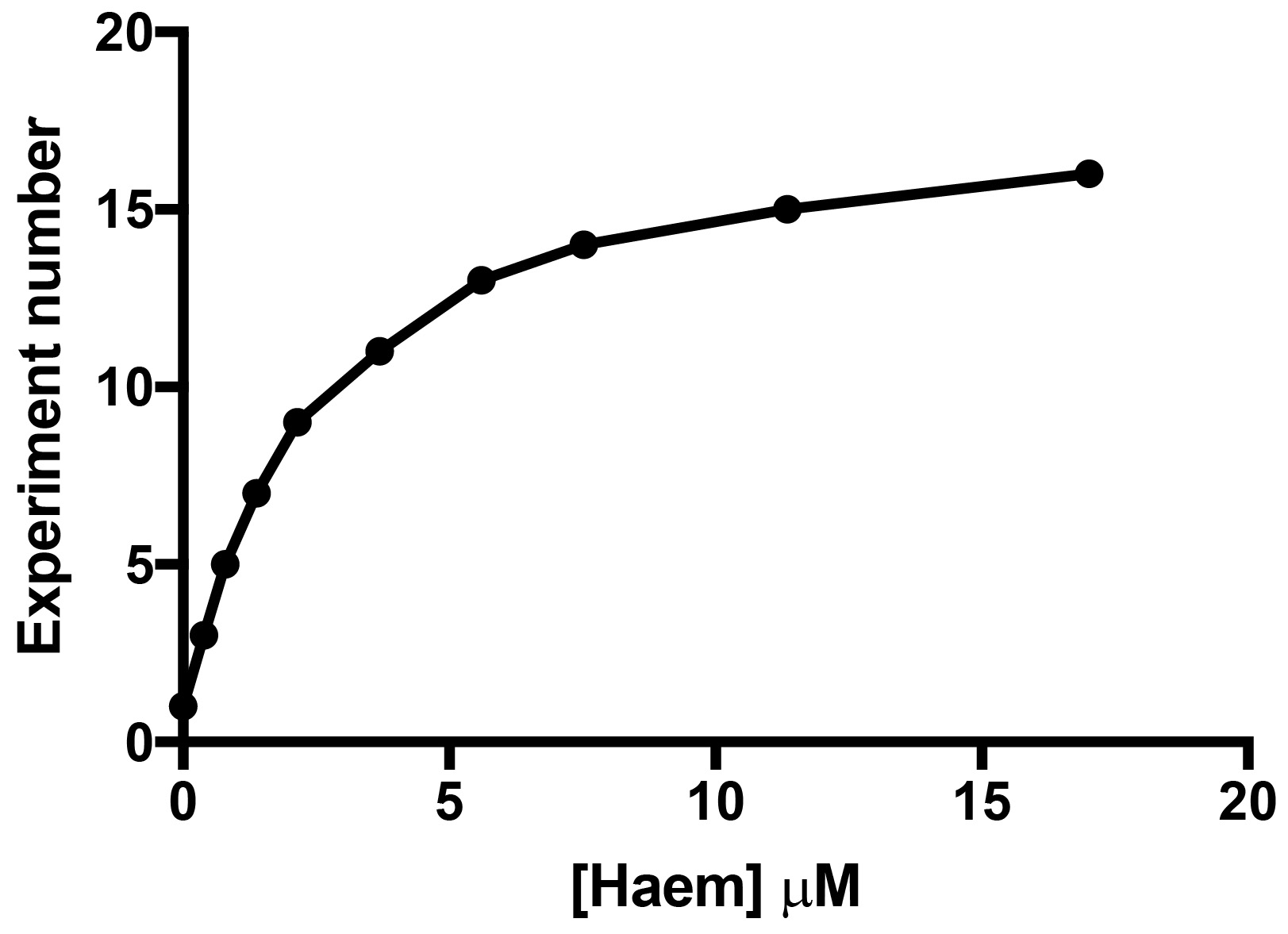
Figure 6. Experimental rationale where at least ten titration points were chosen to allow the titrant to span the estimated Kd by a factor of five - Control titrations and accounting for the inner filter effect
- Fluorimeter settings used:
- Selective excitation: 295 nm
- Photon emission: 310 nm to 500 nm
- Path length: 5 mm
- Photomultiplier effect (PME): 850
- Blank the spectrophotometer with 0.5 ml binding buffer in a 5 mm Quartz cuvette.
- Prepare a 1 μM solution of NATA in 0.5 ml binding buffer in a cuvette.
- Excite and record emissions on NATA.
- Titrate ligand into the NATA and mix by gentle pipetting for 1 min.
- After 10 min of incubation following mixing, excite and record emissions.
- Continue according to your titration protocol, with pipetting and incubating for 10 min after each ligand addition.
- After the final titration point, export data into Microsoft Excel for data processing.
- Fluorimeter settings used:
- Tryptophan fluorescence quenching of a protein by haem
- Repeat Steps B1-B2 from above.
- Prepare a solution of protein into 0.5 ml buffer in the cuvette where the starting fluorescence intensity is ~900 to ensure that the intensity is above 0 by the final titration point. For the HusA experiment, 1 μM HusA was dissolved into binding buffer in the cuvette.
- Repeat Steps B4-B8 from above.
Data analysis
- Data processing for inner filter effect
- Identify which wavelength records the highest intensity. Using five wavelengths around this point, take the average maximum intensity from each titration point. For the NATA control experiment, the highest intensity recorded was from averaging the intensity between 354-358 nm.
- Calculate the difference in intensity between subsequent titration points, and convert this to a percentage difference between subsequent titration points. The percentage decrease in fluorescence intensity between each titration point represents the inner filter effect proportion. Using the above, the data processing for the inner filter effect of haem is shown in Table 3, and the inner filter effect against Haem concentration is shown in Figure 7.
Table 3. Inner filter effect data processing for haem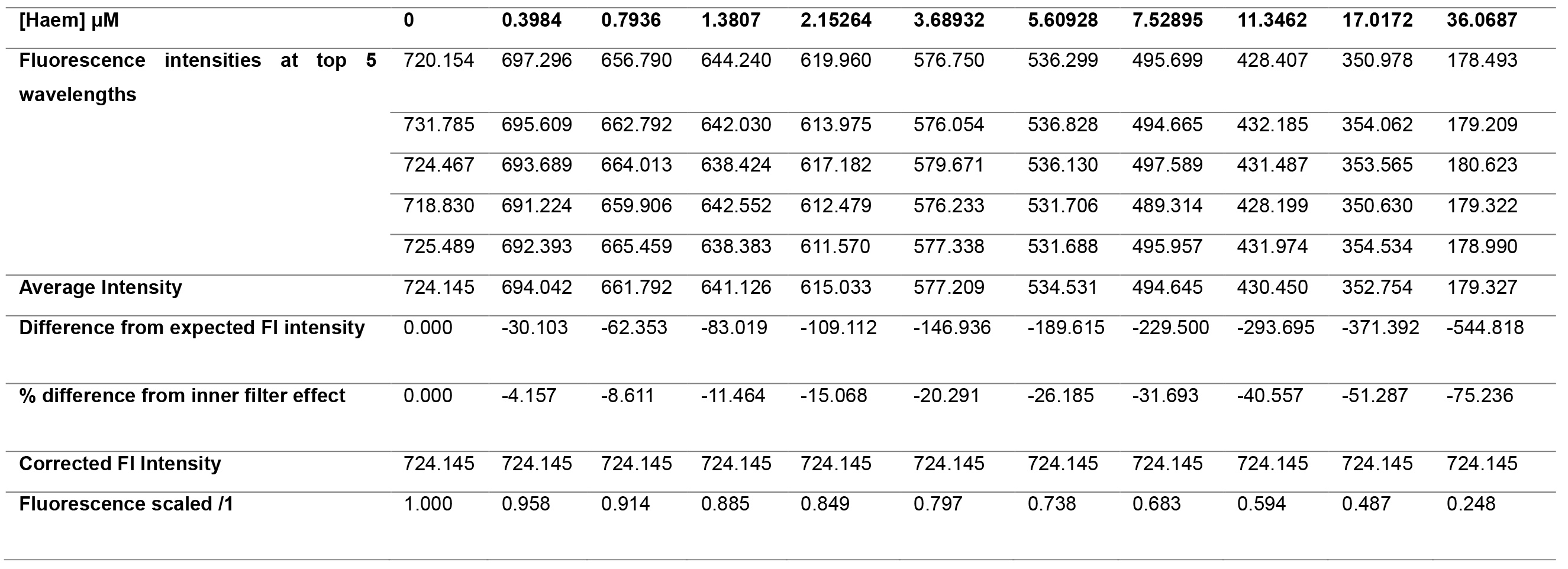
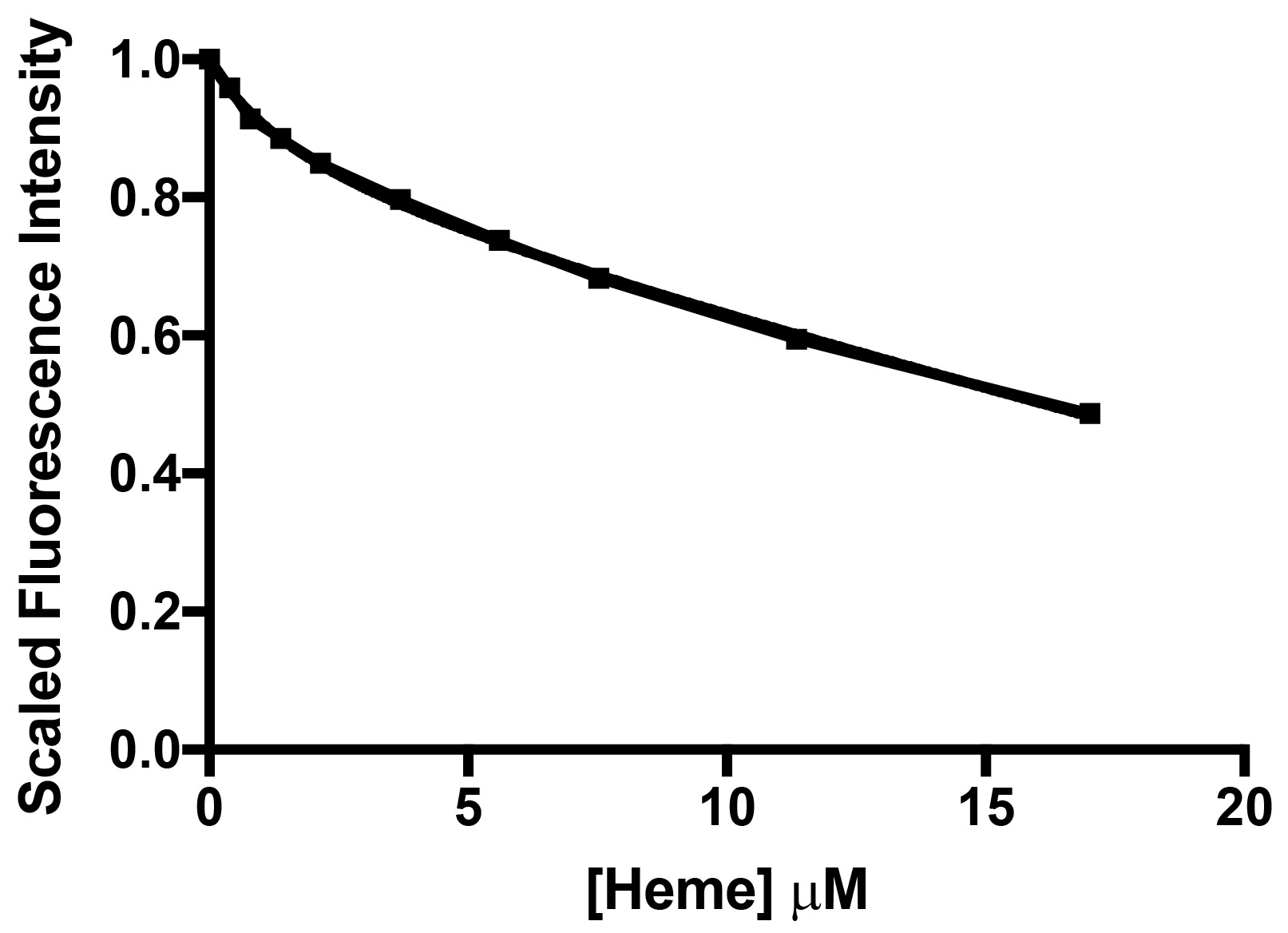
Figure 7. The quenching profile of NATA by haem illustrates the inner filter effect
- Data processing for tryptophan fluorescence quenching of protein by ligand
- Identify which wavelength records the highest intensity. Using five wavelengths around this point, take the average maximum intensity from each titration point. For the HusA experiment, the highest intensity recorded was from averaging the intensity between 333-337 nm.
- Correct for the inner filter effect for each titration point by adding the percent decrease in intensity due to the inner filter effect. Accounting for the inner filter effect of haem for the HusA experiment is shown in Table 4 and the corrected haem-binding curve of HusA is shown in Figure 8. It is important to check that the correction is appropriate–overcorrection usually manifests with the binding curve sloping upwards at high ligand concentrations and under-correction as sloping downwards when one would have expected saturation and therefore a plateau given the expected affinity.
Table 4. Calculating the tryptophan fluorescence quenching of HusA by haem, accounting for the inner filter effect of haem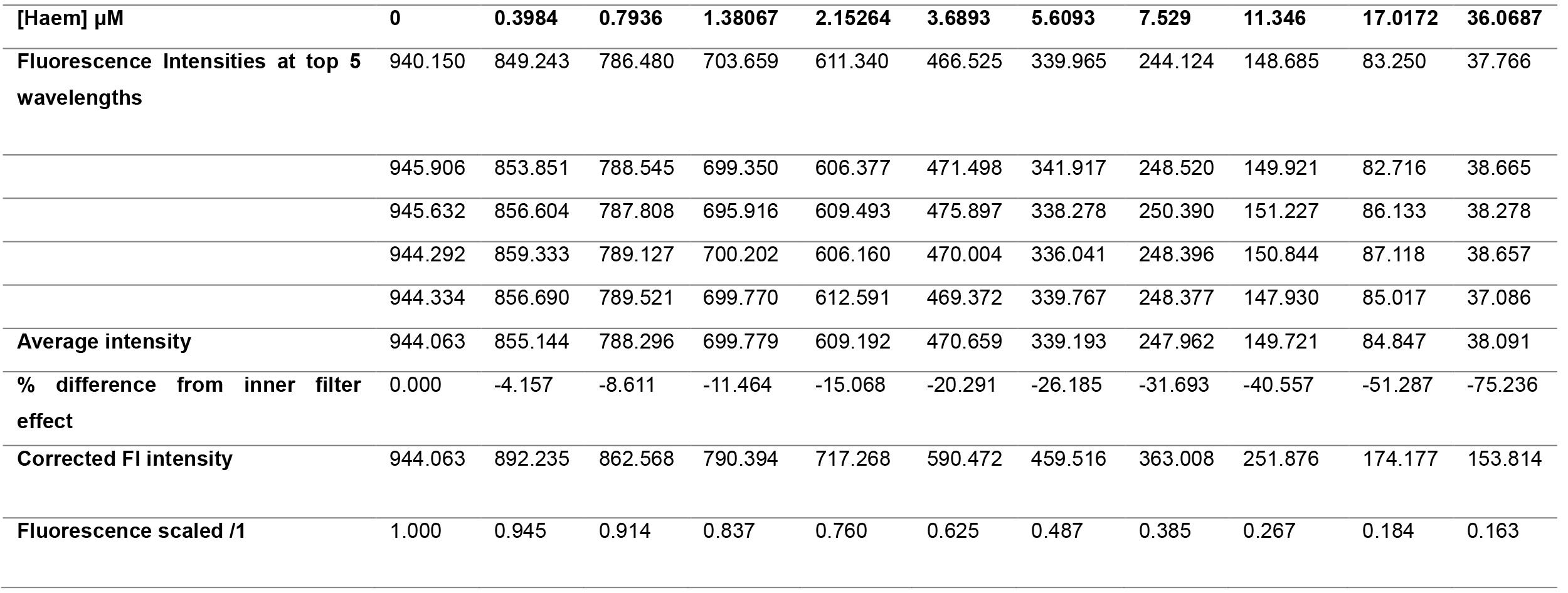
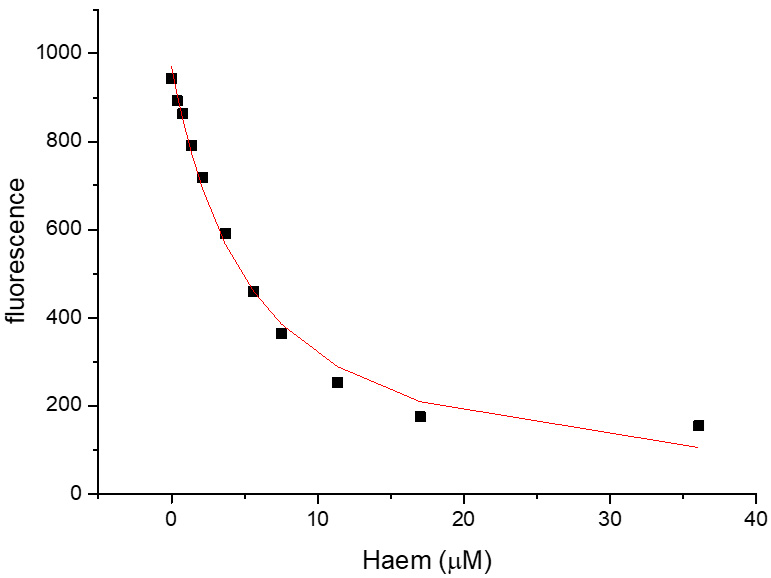
Figure 8. Haem-binding curves to 1 μM of HusA generated using the quenching of fluorescence at 355 nm by haem. Ka = 0.21 ± 0.034. R square = 0.9885.
- Binding affinity
Association constant, Ka, was fitted using the following equation on Origin2016, assuming a single binding site. The dissociation constant, Kd, can be calculated by taking the inverse of the Ka if desired.Xobs = XA × fA + XAB × fAB
where,
XA is raw fluorescence,
fA = 1 - fAB,
fAB = x/(Bt),
x = (-b - sqrt (b2 - 4 × c))/2,
b = -(1/Ka + At + Bt),
c = Bt × At,
Bt = total concentration of protein,
At = total concentration of ligand.
Note the equation above also works if Bt changes (e.g., if a dilution of more than 5% has occurred during the titration and needs to be accounted for).
Recipes
For details about HusA production and haem stock preparation, please see Gao et al., 2018.
- Binding buffer
HusA dissolved in 50 mM Tris, pH 8.0, 150 mM NaCl - Buffer for dissolving ligand
Haem dissolved in 0.1 M NaOH, pH 12
Note: The haem powder is prepared freshly before use by dissolving in 0.1 M NaOH, pH 12, by gentle pipetting and inversion for 5 min at room temperature. Prepared solutions were incubated for 10 min before use. A UV-vis spectra of the dissolved porphyrin can be recorded to confirm the concentration of available porphyrin.
Acknowledgments
This project was supported by a Faculty of Science, University of Sydney Seed funding grant to A.H.K.
Competing interests
The authors declare no competing interests.
References
- Breen, C. J., Raverdeau, M. and Voorheis, H. P. (2016). Development of a quantitative fluorescence-based ligand-binding assay. Sci Rep 6: 25769.
- Fonin, A. V., Sulatskaya, A. I., Kuznetsova, I. M. and Turoverov, K. K. (2014). Fluorescence of dyes in solutions with high absorbance. Inner filter effect correction. PLoS One 9(7): e103878.
- Gao, J. L., Kwan, A. H., Yammine, A., Zhou, X., Trewhella, J., Hugrass, B. M., Collins, D. A. T., Horne, J., Ye, P., Harty, D., Nguyen, K. A., Gell, D. A. and Hunter, N. (2018). Structural properties of a haemophore facilitate targeted elimination of the pathogen Porphyromonas gingivalis. Nat Commun 9(1): 4097.
- Gao, J. L., Nguyen, K. A. and Hunter, N. (2010). Characterization of a hemophore-like protein from Porphyromonas gingivalis. J Biol Chem 285(51): 40028-40038.
- Ghisaidoobe, A. B. and Chung, S. J. (2014). Intrinsic tryptophan fluorescence in the detection and analysis of proteins: a focus on Forster resonance energy transfer techniques. Int J Mol Sci 15(12): 22518-22538.
- Krainer, G. and Keller, S. (2015). Single-experiment displacement assay for quantifying high-affinity binding by isothermal titration calorimetry. Methods 76: 116-123.
- Lakowicz, J. R. (1999). Principles of fluorescence spectroscopy. Kluwer Academic/Plenum Publishers, New York, pp. 1-24.
- Möller, M. and Denicola, A. (2002). Protein tryptophan accessibility studied by fluorescence quenching. Biochemistry and Molecular Biology Education, 30: 175-178.
- Nienhaus, K. and Nienhaus, G. U. (2005). Probing heme protein-ligand interactions by UV/visible absorption spectroscopy. Methods Mol Biol 305: 215-242.
- Teale, F. W. and Weber, G. (1957). Ultraviolet fluorescence of the aromatic amino acids. Biochem J 65(3): 476-482.
- Zelent, B., Kusba, J., Gryczynski, I., Johnson, M. L. and Lakowicz, J. R. (1998). Time-resolved and steady-state fluorescence quenching of N-acetyl-L-tryptophanamide by acrylamide and iodide. Biophys Chem 73(1-2): 53-75.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Yammine, A., Gao, J. and Kwan, A. H. (2019). Tryptophan Fluorescence Quenching Assays for Measuring Protein-ligand Binding Affinities: Principles and a Practical Guide. Bio-protocol 9(11): e3253. DOI: 10.21769/BioProtoc.3253.
Category
Biochemistry > Protein > Interaction > Protein-ligand interaction
Biochemistry > Protein > Fluorescence
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.