- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Minigene Assay to Evaluate CRISPR/Cas9-based Excision of Intronic Mutations that Cause Aberrant Splicing in Human Cells
Published: Vol 9, Iss 11, Jun 5, 2019 DOI: 10.21769/BioProtoc.3251 Views: 6939
Reviewed by: Raquel Santana da CruzAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Sep 2017

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
The construction of Hybrid minigenes provides a robust and simple strategy to study the effects of disease-causing mutations on mRNA splicing when biological material from patient cells is not available. Hybrid minigenes can be used as splicing reporter plasmids allow RNA expression and heterologous splicing reactions between synthetic splicing signals in the vector and endogenous splicing signals in a cloned genomic DNA fragment that contains one or more introns and exons. Minigene-based assay has been used extensively to test the effect of mutations in the splicing of a target sequence. They can also be used to test the ability of CRISPR/Cas9 and one or more associated gRNAs to target specific sequences in the minigene, and determine the effect of these editing events on splicing. As an example, it is shown that CRISPR/Cas9-based, targeted excision of short intronic sequences containing mutations which create cryptic splice signals, can restore normal splicing in a CFTR Hybrid minigene.
Keywords: Cas9/CRISPRBackground
Our objective was to use CRISPR/Cas9 to correct the effect on splicing of three different intronic mutations in the CFTR gene which cause cystic fibrosis by creating strong alternative splice sites: c.1679+1634a>g[1811+1.6Kba>g], c.3718-2477c>t[3849+10KBc>t] and c.3140-26a>g[3272-26a>g] (Highsmith et al., 1994; Chillón et al., 1995; Beck et al., 1999) and lead to the disruption of normal CFTR mRNA production.
A CRISPR/Cas9-Non-homologous end joining (NHEJ) approach using two gRNAs has been shown to successfully excise small intronic regions of CFTR gene in > 25% of transfected cells (Hollywood et al., 2016). We hypothesized that this approach could be used to remove the strong alternative splice sites described above with high efficiency in cells with these mutations and restore normal splicing of the CFTR mRNA. The efficiency of the NHEJ targeting approach is at least 10-fold higher than previously reported homology direct repair strategies to correct Cystic fibrosis causing mutations in the CFTR gene.
However, the lack of biological samples carrying these mutations of interest prompted us to ask if it is possible to construct hybrid minigene constructs that contain these mutations as a system to test our reagents, and determine if targeted excision could efficiently restore normal splicing. Once PCR was performed to amplify the wt sequences of interest from genomic DNA, these amplicons were cloned in the exon-trapping pSPL3 plasmid and directed mutagenesis was performed to obtain pSPL3 based minigenes containing the mutations of interest. The pSPL3 plasmid contains a small artificial gene composed of an SV40 promotor and exon-intron-exon sequence with splice donor and acceptor sites and a polyadenylation sequence. A multiple cloning site present in the intron allows cloning sequences of interest, and testing of putative heterologous splicing reactions between the plasmid splice sites and the splice sites contained in the cloned sequences (Sanz et al., 2010).
HEK293T cells were transfected with both wt and mutation containing versions of the three minigenes to characterize by RT-PCR the aberrant splicing caused by these three mutations in the context of the minigenes.
HEK293T were also co-transfected with both the mutant minigene and a plasmid expressing both Cas9 and designed gRNAs. After DNA extraction, PCR across the target sequence was performed to determine the percentage excision caused in mutant minigenes by the CRISPR/Cas9 reagents.
RNA extraction from the same samples allows characterizing by RT-PCR the restoration of splicing due to the excision of the target sequences containing the splicing mutations.
In summary, construction of minigenes allows the analysis of how the sequences of interest are edited using designed CRISPR/Cas9 reagents with specific gRNAs, and how this editing changes the splicing process in the minigene, circumventing the need for primary cells or cell lines with the mutation of interest.
Materials and Reagents
- PCR
- Pipette tips
- 0.2 ml PCR Tubes (SARSTEDT, catalog number: 72.737.002)
- Total Human genomic DNA from Lung (Amsbio, catalog number: HG-601)
- Phusion High-Fidelity DNA Polymerase (Thermo Fisher, catalog number: F530S)
- dNTPs Mix (Thermo Fisher, catalog number: R0192)
- Gel electrophoresis
- TAE buffer (40 mM Tris base, 20 mM Acetic Acid, 1 mM EDTA)
- Agarose (Sigma-Aldrich, catalog number: A9539)
- Ethidium bromide (Sigma-Aldrich, catalog number: E7637)
- 1 kb DNA ladder (Thermo Fisher, catalog number: SM0311)
- 100 bp DNA ladder (Thermo Fisher, catalog number: 15628019)
- DNA Gel Loading Dye (Thermo Fisher, catalog number: R0611)
- Cloning
- 1.5 ml Microtubes (SARSTEDT, catalog number: 72.706.700)
- Petri dishes (SARSTEDT, catalog number: 82.1472.001)
- 15 ml polypropylene tubes (SARSTEDT, catalog number: 62.554.002)
- NEB® 5α competent E. coli (NEB, catalog number: C2987I)
- pSPL3 vector (Thermo Fisher, discontinued)
- pSpCas9(BB)-2A-GFP PX458 (Addgene #48138)
- NheI (Thermo Fisher, catalog number: ER0971)
- XhoI (Thermo Fisher, catalog number: ER0691)
- XbaI (Thermo Fisher, catalog number: ER0681)
- BbsI (Thermo Fisher, catalog number: ER1011)
- T4 DNA ligase (NEB, catalog number: M0202S)
- High Pure PCR Product Purification kit (Sigma-Aldrich, catalog number: 11732668001)
- StrataPrep DNA Gel Extraction kit (Agilent, catalog number: 400766)
- QuikChange II Kit (Agilent, catalog number:200523)
- LB Broth Lennox (Sigma-Aldrich, catalog number: L3022)
- LB Broth with Agar Lennox (Sigma-Aldrich, catalog number: L2897)
- Ampicillin Sodium salt (Sigma-Aldrich, catalog number: A0166)
- Mammalian cell culture
- T75 cell culture flasks (SARSTEDT, catalog number: 83.1813.002)
- 12-well plates (SARSTEDT, catalog number: 83.3921.005)
- HEK293T cells, human embryonic kidney cell line (ATCC®, catalog number: CRL-3216)
- CFTE cells, cystic fibrosis tracheal epithelial cell line (Gruenert et al., 2004)
- Dubelcco’s modified Eagle’s Medium (DMEM, Sigma-Aldrich, catalog number: D5796)
- Minimum Essential Medium Eagle (MEM, Sigma-Aldrich, catalog number: M2279)
- Fetal Bovine Serum (Sigma-Aldrich, catalog number: F7524)
- Phosphate-buffered saline PBS (Sigma-Aldrich, catalog number: P5493)
- Penicillin-Streptomycin (Sigma-Aldrich, catalog number: P433)
- Trypsin-EDTA (Sigma-Aldrich, catalog number: T3924)
- Lipofectamine 2000 (Thermo Fisher, catalog number: 11668027)
- Opti-MEM (Thermo Fisher, catalog number: 31985070)
- Plasmidic DNA and Total DNA extraction
- NucleoSpin Plasmid kit (MACHEREY-NAGEL, catalog number: 740588150)
- DNeasy Blood and Tissue Kit (QIAGEN, catalog number: 69506)
- RNA extraction and RT-PCR
- 0.5 ml PCR Tubes (SARSTEDT, catalog number: 70.704.400)
- NucleoSpin RNA (MACHEREY-NAGEL, catalog number: 740955.150)
- High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher, catalog number: 15561175)
- dNTPs Mix (Thermo Fisher, catalog number: R0192)
- Phusion High-Fidelity DNA polymerase (Thermo Fisher, catalog number: F530S)
Equipment
- Pipettes
- PCR Thermocycler Eppendorf Mastercycler
- Gel Doc System (Bio-Rad)
- Water bath
- Bacterial Incubator and shaker
- Mammalian cell incubator (37 °C, 5% CO2)
- Bunsen Burner
- NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, model: NanoDropTM 2000)
- Microcentrifuge
- Electrophoresis tank and power pack
- Autoclave
- Biological safety cabinet
Software
- Primer-blast (https://www.ncbi.nlm.nih.gov/tools/primer-blast/)
- CRISPR-Design (http://crispr.mit.edu/)
- SnapGene Viewer (http://www.snapgene.com/products/snapgene_viewer/)
- Tm Calculator (https://www.thermofisher.com)
- Peak Scanner 2 (https://www.thermofisher.com)
- Eurofins (Oligo synthesis, Sanger sequence and Fragment Length Analysis [FLA] service) (https://www.eurofinsgenomics.eu)
Procedure
- Minigene construction
- To construct wt minigenes, primers were designed (using Primer-Blast web, https://www.ncbi.nlm.nih.gov/tools/primer-blast/) to amplify the regions of interest from a wt DNA sequence. In our case, three intronic regions in the CFTR gene: First, a region of 1,522 bp between intron 12 and intron 13, which includes the nucleotide c.1679+1634. Second, a region of 2,012 bp between intron 18 and intron 20 which includes the nucleotide c.3140-26, and Third a region of 3224 between intron 22 and intron 23 which includes the nucleotide c.3718-2477. Restriction sites were included at the 5′ end of Fw and Rv primers to facilitate cloning into pSPL3 plasmid. We added XhoI sites to the three Fw primers and NheI sites to the first two Rv primers; XbaI was used in the Third one as an NheI site was already present in the genomic sequence. Add six additional nt upstream of the restriction sites to facilitate digestion by restriction enzymes.
Minigenes cloning primers MGin12-in13fw CACACACTCGAGTGTGTTGTCCAGTTTTGGATGA MGin12-in13rv CTCACAGCTAGCACTGGTTTAGCATGAGGCGG MGin18-in20fw CTCACTCTCGAGTGACTAGGAATAGAATGGGGAGAG MGin18-in20rv CACACAGCTAGCACAATGGAAATTCAAAGAAATCACT MGin22-in23fw CACACACTCGAGACAGTACTGGATAGTCCTCTGA MGin22-in23rv CACACATCTAGAGCCTATGAGAAAACTGCACTGG - Amplify Target regions of interest from a wt genomic DNA sample (Genomic DNA-Human adult normal tissue Lung, from a single donor, Amsbio, Abingdon, UK) using Phusion high-fidelity DNA polymerase (Thermo Fisher) and designed primers using the following program in a Mastercycler Thermocycler (Eppendorf).
PCR Amplification of genomic DNA to construct hybrid minigenesDNA template (20 ng/μl) 3 μl 5x Phusion Buffer 4 μl 10 mM dNTPS 0.4 μl 10 μM FW primer 0.5 μl 10 μM RV primer 0.5 μl Phusion polymerase 0.5 μl Nuclease-free water Up to 20 μl
PCR programInitial Denaturation 98 °C 30 s 35 cycles of 98 °C 30 s 57 °C 30 s 72 °C 2 min Final extension 72 °C 10 min Hold 4 °C - Check successful amplification of the desired size by 1.5% agarose gel electrophoresis in TAE buffer, running 100 bp and 1 kb ladders as size standards and purify the PCR product using QIAquick PCR Purification Kit (QIAGEN).
- Digest 1 μg of each Purified product with selected endonucleases and purify by Gel extraction Bands of correct size after electrophoresis in a 1% agarose gel in TAE buffer using StrataPrep DNA Gel Extraction Kit (Agilent).
- Digest 2 μg of Plasmid pSPL3 with XhoI and NheI enzymes and excise and purify pSPL3 plasmid backbone (The bigger band of around 5 kb) from the agarose after electrophoresis (1% agarose in TAE buffer), using the StrataPrep DNA Gel Extraction kit (Agilent).
- Prepare Ligation using 50 ng of each of the three digested amplified PCR products with 150 ng of purified XhoI/Nhei digested pSPL3 backbone using 10 units of T4 ligase (NEB) and incubate for 4 h at 16 °C.
- Use 2 μl of each of the three ligation reactions to transform 1 vial of 50 μl of DH5α competent E. coli (NEB). Seed transformed E. coli in LB-agar Petri dishes containing 100 μg/ml of ampicillin as selection marker for transformed colonies with pSPL3-derived plasmids. Incubate for 16 h at 37 °C
- Pick and grow 5 colonies from each of the cloned amplicons in 5 ml of LB with ampicillin (100 μg/ml) in 15 ml Polypropylene tubes and after 24 h purify plasmidic DNA from cultures using NucleoSpin Plasmid Kit (MACHEREY-NAGEL).
- Sequence plasmid DNA (TubeSeq service, Eurofins) using selected primers SEQpspfw and SEQpsprv and the following PCR setup and program to confirm successful cloning of each wt amplified sequence into pSPL3 plasmid in at least one of the purified plasmids. We will refer to each of these plasmids as wt minigenes, pMG-in12-wt, pMG-in19-wt and pMG-in22-wt, as they contain sequences of the introns 12, 19 and 22 respectively.
Sequencing pSPL3 primersSEQpspfw TCACAGTCTATTATGGGGTACGG SEQpsprv AATTTCTGGGTCCCCTCCTGA
PCR reaction setupExtracted DNA (20 ng/μl) 3 μl 5x Phusion Buffer 4 μl 10 mM dNTPs 0.4 μl 10 μM FW primer 0.5 μl 10 μM RV primer 0.5 μl Phusion polymerase 0.5 μl Nuclease-free water Up to 20 μl
PCR programInitial Denaturation 98 °C 30 s 35 cycles of 98 °C 30 s 57 °C 30 s 72 °C 2 min Final extension 72 °C 5 min Hold 4 °C - Perform site-directed mutagenesis to create mini-gene plasmids containing the desired mutations, site-directed mutagenesis is achieved by using the QuikChange II Kit (Agilent) and selected PCR primers pairs containing the desired mutations.
Directed Mutagenesis MD1811+1.6kbA>Gfw CCTATGTACTTGAGATGTAAGTAAGGTTACTATC MD1811+1.6kbA>Grv GATAGTAACCTTACTTACATCTCAAGTACATAGG MD3272-26A>Gfw GTGTTTATGTTATTTGCAGTGTTTTCTATGG MD3272-26A>Grv CCATAGAAAACACTGCAAATAACATAAACAC MD3849+10kbC>Tfw CTGTTGCAGTATTAAAATGGtGAGTAAGACACCCTGAAAG MD3849+10kbC>Trv CTTTCAGGGTGTCTTACTCaCCATTTTAATACTGCAACAG
Site-Directed mutagenesis reaction setup5x Reaction Buffer 5 μl Pfu Turbo DNA polymerase 1 μl 10 μM MD fw Primer 1.25 μl 10 μM MD rv Primer 1.25 μl Plasmid DNA (25 ng/μl) 1 μl Nuclease-free water up to 50 μl
Directed Mutagenesis ProgramInitial Denaturation 95 °C 30 s 16 cycles of 95 °C 30 s 56 °C 1 min 68 °C 6 min Final Extension 68 °C 2 min Hold 4 °C
Digestion of plasmid template
Add 1 μl of DpnI enzyme directly to each PCR reaction; digest at 37 °C for 1 h. - Use 2 μl of each site-directed mutagenesis reaction, that now contains mini-gene plasmids with the desired mutations, to transform 1 vial of 50 μl of 5-alpha competent E. coli (NEB), and seed the transformed E. coli in LB-agar Petri dishes containing 100 μg/ml of ampicillin as a selection marker for transformed colonies. Incubate for 16 h at 37 °C.
- Select 5 colonies coming from each of the Directed mutagenesis reactions, and grow them in 5 ml of LB with ampicillin (100 µg/ml) for 24 h in 15 ml polypropylene tubes, then extract plasmid DNA using NucleoSpin Plasmid Kit (MACHEREY-NAGEL).
- Sequence plasmidic DNA samples (TubeSeq service, Eurofins) using selected Screening primers to confirm desired mutated sequence on each target plasmid in at least one of the colonies.
Screening Primers
Screening Primersin12Ufw AAGAACAGAGTGTGGGGAAGA in12Drv GCATTTTGTTCCTTGTAGTTTCAG in19Ufw ACTCCCAGTGGTAGCCAAGA in19Drv GCACGAAGTGTCCATAGTCCT in22Ufw TCATTCAGTGGGTATAAGCAGCA in22Drv GACATACCCTAAATCTAAGTCAGTG
- To construct wt minigenes, primers were designed (using Primer-Blast web, https://www.ncbi.nlm.nih.gov/tools/primer-blast/) to amplify the regions of interest from a wt DNA sequence. In our case, three intronic regions in the CFTR gene: First, a region of 1,522 bp between intron 12 and intron 13, which includes the nucleotide c.1679+1634. Second, a region of 2,012 bp between intron 18 and intron 20 which includes the nucleotide c.3140-26, and Third a region of 3224 between intron 22 and intron 23 which includes the nucleotide c.3718-2477. Restriction sites were included at the 5′ end of Fw and Rv primers to facilitate cloning into pSPL3 plasmid. We added XhoI sites to the three Fw primers and NheI sites to the first two Rv primers; XbaI was used in the Third one as an NheI site was already present in the genomic sequence. Add six additional nt upstream of the restriction sites to facilitate digestion by restriction enzymes.
- Cloning of gRNA sequences to excise intronic sequences in CFTR
- Use the CRISPR design website tool (http://cripsr.mit.edu) to identify and select gRNAs sequences in the intronic regions of gene of interest upstream and downstream to the position where the variants of interest may occur. We selected three different guide RNA target sequences upstream (U1-U3) and three different guide RNA sequences downstream (D1-D3) to each of the three intronic regions (in12, in19 and in22) we wanted to excise. We have chosen guide RNAs pairs which produce targeted excisions of a size that can be easily detected on a standard agarose gel. However, if a more sensitive detection method is used, such as Fragment Length Analysis (FLA), it is possible to choose guide RNAs that produce excisions of essentially any size. It should be noted that guide RNAs close to the canonical splice sites or the branch point should not be used. The CRISPR design website tool offers a score for each possible guide RNA in a particular region based on putative off-target effects that may assist in the selection of the best guide RNAs to use.
- The CRISPR design website tool will also suggest the sequence of the sense and antisense oligos (Eurofins) to generate a dsDNA fragment encoding the desired gRNA sequence and overhangs to clone using Golden Gate strategy into the BbsI overhangs of the plasmid pSpCas9-(BB)-2A-GFP (Addgene.org PX458). Golden Gate cloning relies on the use of Type IIS restriction enzymes, which cleave outside of their recognition sequence, creating four base flanking overhangs. Oligos are designed to form compatible overhangs when annealed, so they can be cloned in the correct orientation into the plasmid when it is digested by BbsI enzyme. As successful ligation destroys the BbsI recognition site every cycle of BbsI digestion followed by T4 ligation will enrich the successful cloning.
- Anneal sense and antisense oligo (1 μg/μl) by mixing 5 μl of each with 85 μl of nuclease-free water and 5 μl of T4 ligase buffer (NEB) in 1.5 ml tubes, and heating the tubes in a block at 95 °C for 5 min. Turn off the heat block afterward to let the tubes cool down slowly.
Oligos to express gRNAS to excise intron 12 regiongin12U1fw CACCGGAAACTGTGTACATTTTGAT gin12U1rv AAACATCAAAATGTACACAGTTTCC gin12U2fw CACCGAGTATGCAAGAGCTACATAA gin12U2rv AAACTTATGTAGCTCTTGCATACTC gin12U3fw CACCGGACTTTTAAAGTTTTGCCAT gin12U3rv AAACATGGCAAAACTTTAAAAGTCC gin12D1fw CACCGATGTACTTGAGATGTAAGTA gin12D1rv AAACTACTTACATCTCAAGTACATC gin12D2fw CACCGTTACTCATACTTTCCTTATT gin12D2rv AAACAATAAGGAAAGTATGAGTAAC gin12D3fw CACCGTTATCTCATTTCTATTAATA gin12D3rv AAACTATTAATAGAAATGAGATAAC
Oligos to express gRNAS to excise intron 19 regiongin19U1fw CACCGTTTACTTGGCTACCAGAGAT gin19U1rv AAACATCTCTGGTAGCCAAGTAAAC gin19U2fw CACCGAGTTACCCTCTTTTTTTACT gin19U2rv AAACAGTAAAAAAAGAGGGTAACTC gin19D1fw CACCGGTTATTTGCAGTGTTTTCTA gin19D1rv AAACTAGAAAACACTGCAAATAACC gin19D2fw CACCGTTTCTATGGAAATATTTCAC gin19D2rv AAACGTGAAATATTTCCATAGAAAC
Oligos to express gRNAS to excise intron 22 region
gin22U1fw CACCGCATTTTAATACTGCAACAGA gin22U1rv AAACTCTGTTGCAGTATTAAAATGC gin22U2fw CACCGCATCTGTTGCAGTATTAAAA gin22U2rv AAACTTTTAATACTGCAACAGATGC gin22U3fw CACCGCTTGATTTCTGGAGACCACA gin22U3rv AAACTGTGGTCTCCAGAAATCAAGC gin22D1fw CACCGTTGATCCAACATTCTCAGGG gin22D1rv AAACCCCTGAGAATGTTGGATCAAC gin22D2fw CACCGATCCAACATTCTCAGGGAGG gin22D2rv AAACCCTCCCTGAGAATGTTGGATC - Mix 50 ng of each previously annealed gRNA-encoding oligo pair with 150 ng of plasmid pSpCas9-(BB)-2A-GFP, 2 μl of 10x T4 DNA ligase buffer, 1 µl of T4 DNA ligase and 1 μl of BbsI enzyme and water in a total volume of 20 μl. To clone the annealed double strand fragment into the pSpCas9-(BB)-2A-GFP plasmid use the following thermocycler program for golden gate cloning:
Golden gate cloning of Annealed oligos into pSpCas9-(BB)-2A-GFP plasmid10 cycles of 37 °C 5 min 16 °C 10 min 37 °C 20 min 80 °C 20 min Hold 4 °C - Transform 50 μl of competent DH5α E. coli (NEB) using 2 μl of the previous reaction and plate them in LB agar Petri dishes with 100 μg/ml ampicillin. Incubate for 16 h at 37 °C.
- Pick and grow 5 colonies in 5 ml of selective LB medium containing ampicillin in 15 ml Polypropylene tubes and extract plasmid DNA after 24 h from the cultures.
- Sequence plasmids (Tubeseq, Eurofins) using pcas9SEQfw and pcas9SEQrv primers to check for successful cloning of annealed oligos in at least one of the selected colonies.
pcas9 sequence primerspcas9SEQfw GAGGGCCTATTTCCCATGATTCC pcas9SEQrv GTCTGCAGAATTGGCGCAC
- Cotransfection of CFTE cells with pairs of constructed gRNAs/Cas9 expressing plasmids
- Prior to transfection, CFTE cells were kept in culture and passaged twice a week with Trypsin-EDTA in a T75 cell culture flask with MEM + 10% FBS + 1% Penicillin/Streptomycin at 37 °C, 5% CO2.
- Seed 100,000 CFTE cells in 12-well plates in free antibiotic MEM + 10% FBS medium 24 h prior to transfection. For other cell types seed enough cells to have around 90% confluency after 24 h.
- Transfect cells with paired Cas9/gRNA expressing plasmids constructed in Procedure B using one plasmid that express a guideRNA upstream and other downstream of the mutation to get an excision of the region comprised between the two gRNA target sequences. It is worthwhile to test different combinations of gRNA pairs, as the excision efficiency of the different pairs may be different. We used 4 μl of Lipofectamine 2000 and 0.8 μg of each plasmid. A different transfection reagent or different transfection conditions may be necessary for other cell types.
- Extract total DNA from transfected cells 48 h after transfection using DNeasy Blood and Tissue kit. Check Total Nucleic acid concentration using Nanodrop 2000.
Note: RNase may be added as described in the DNeasy kit protocol to remove RNA from the sample for a more accurate measuring of DNA concentration. - Perform PCR of the three different targeted intronic regions using the following set-up reaction and PCR program to detect excisions produced in genomic DNA from CFTE cells. Use the Screening primers described in Procedure A and the following PCR conditions.
PCR reaction setupExtracted DNA (~20 ng/μl) 3 μl 5x Phusion Buffer 4 μl 10 mM dNTPS 0.4 μl 10 μM FW primer 0.5 μl 10 μM RV primer 0.5 μl Phusion polymerase 0.5 μl Nuclease-free water Up to 20 μl
PCR ProgramInitial Denaturation 98 °C 30 s 35 cycles of 98 °C 30 s 57 °C 30 s 72 °C 1 min Final extension 72 °C 5 min Hold 4 °C - Perform 1.5% agarose gel electrophoresis in TAE buffer to see products of smaller size corresponding to the different produced excisions (Figure 1). Select the most efficient plasmid pairs in producing excision on each of the three target intronic regions using densitometry analysis software (Bio-Rad) from the Bio-Rad Gel Doc System.
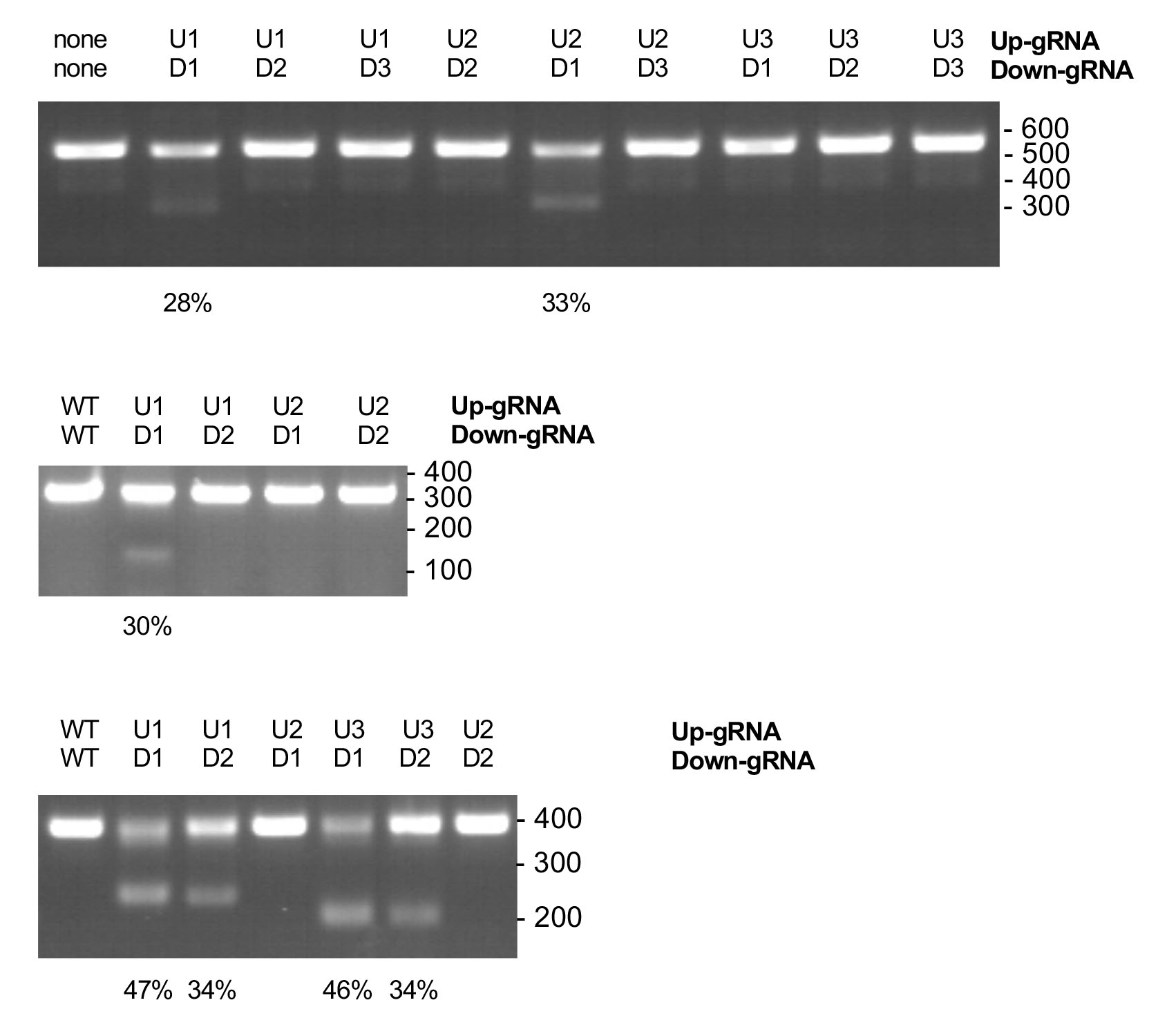
Figure 1. Excisions produced by different pairs of Cas9/gRNA expressing plasmids in intron 12 (Top), intron 19 (mid) or intron 22 (bottom) of genomic CFTR. gin12U2 and gin12D1 were identified as most efficient pair to excise selected sequence in intron 12, meanwhile gin19U1 and gin19D1 pair, and gin22U1 and gin22D1 pair were identified as the most efficient to excise target sequences in intron 19 and 22 respectively.
- (Optional) Construction of pTandem plasmids
Once the most efficient pair of gRNAs that excise the particular intronic region of interest have been identified, it is possible to clone the expression cassettes from plasmids expressing gRNAs targeting the sequence upstream (gin12U2, gin19U1 and gin22U1) and sub-clone them into NheI/XbaI sites of plasmids expressing gRNAs targeting sequences downstream in the same intronic region (gin12D1, 19D1 and gin22D1) respectively, to obtain three plasmids (pTandem-in12, pTandem-in19 and pTandem-in22) that express the two gRNAs from each pair. This refinement is optional as the excision efficiency of using the two plasmids expressing one gRNA as a pair is similar in all the three regions of interest.- PCR amplify the gRNA expression cassettes from the plasmids that express the upstream gRNA (gin12U2, gin19U1 and gin22U1) using primers that incorporate NheI at 5′ and XbaI at 3′ of each PCR product respectively.
Oligos for p-Tandem plasmids constructionPc9tand fwNheI CACACAGCTAGCGAGGGCCTATTTCCCATGATT Pc9tand rvXbaI CACACATCTAGATCTAGCTCTAAAACAAAAAAGC
PCR reaction setup to amplify gRNA expression cassettesPlasmid (~20 ng/μl) 3 μl 5x Phusion Buffer 4 μl 10 mM dNTPS 0.4 μl 10 μM Pc9tand fwNheI 0.5 μl 10 μM Pc9tand rvXbaI 0.5 μl Phusion polymerase 0.5 μl Nuclease free water Up to 20 μl
PCR programInitial Denaturation 98 °C 30 s 35 cycles of 98 °C 30 s 57 °C 30 s 72 °C 30 s Final extension 72 °C 2 min Hold 4 °C - Purify PCR product with High Pure PCR product purification kit (Sigma-Aldrich).
- Digest 1 μg of each of the three PCR products and 1 μg of the plasmids expressing the corresponding downstream gRNA for that region (gin12D1, gin19D1 and gin22D1) with NheI and XbaI enzymes.
Enzyme digestionDNA (1 μg/μl) 1 μl XbaI (10 U/μl) 1 μl NheI (10 U/μl) 1 μl Tango buffer 1 μl Nuclease free water Up to 10 μl - Purify digested PCR and plasmids using High Pure PCR product purification kit (Sigma-Aldrich)
- Set up Ligation reactions, each purified digested PCR product coming from each of the upstream gRNA expression plasmids (gin12U2, gin19U1 and gin22U1) with the corresponding digested downstream expression plasmid (gin12D1, gin19D1 and gin22D1) respectively. Keep reactions at 16 °C for 4 h.
Ligation reaction setupPurified Digested PCR product 1 μl (50 ng/μl) Purified Digested plasmid 1 μl (150 ng/μl) T4 ligase buffer 1 μl T4 ligase 1 μl Nuclease free water Up to 10 μl - Transform a vial of 50 μl of competent E. coli (NEB) with 3 μl of each of the ligation reactions, and seed in LB agar dishes (100 μg/ml of Ampicillin). Incubate for 16 h at 37 °C.
- Pick five colonies for each transformation and grow in 5 ml of LB medium in 15 ml Polypropylene tubes (100 μg/ml of Ampicillin) for 24 h. Extract plasmid DNA after that.
- Sequence extracted plasmid DNA using specific primers to confirm the correct sequence of the cloned construct in at least one colony.
Oligos for p-Tandem plasmids sequencingpcas9tandemSEQfw TTTGTGATGCTCGTCAGGGG pcas9tandemSEQrv TGGAAAGTCCCTATTGGCGT
- PCR amplify the gRNA expression cassettes from the plasmids that express the upstream gRNA (gin12U2, gin19U1 and gin22U1) using primers that incorporate NheI at 5′ and XbaI at 3′ of each PCR product respectively.
- Cotransfection of HEK293T cells with wt, mutant minigenes and mutant minigenes and selected pairs of Cas9/gRNAs expressing plasmids
- Prior to transfection, HEK293T cells were kept in culture and passaged twice a week with Trypsin-EDTA in a T75 cell culture flask with DMEM + 10% FBS + 1% Penicillin/Streptomycin at 37 °C and 5% CO2.
- Seed 1 x 105 HEK293T cells in 12-well plates in DMEM + 10% FBS medium without antibiotics 24 h prior to transfection.
- For each intronic variant, transfect HEK293T cells with (i) just wild type minigene construct, (ii) just mutant minigene construct, or (iii) the mutant minigene construct and corresponding pTandem plasmid. Use 4 μl of Lipofectamine 2000 and 1.6 μg of total plasmid DNA, with an equimolar distribution when more than one plasmid is used. Table 1 shows a summary of the different experimental conditions.
Table 1. Summary of different experimental conditions in minigenes assay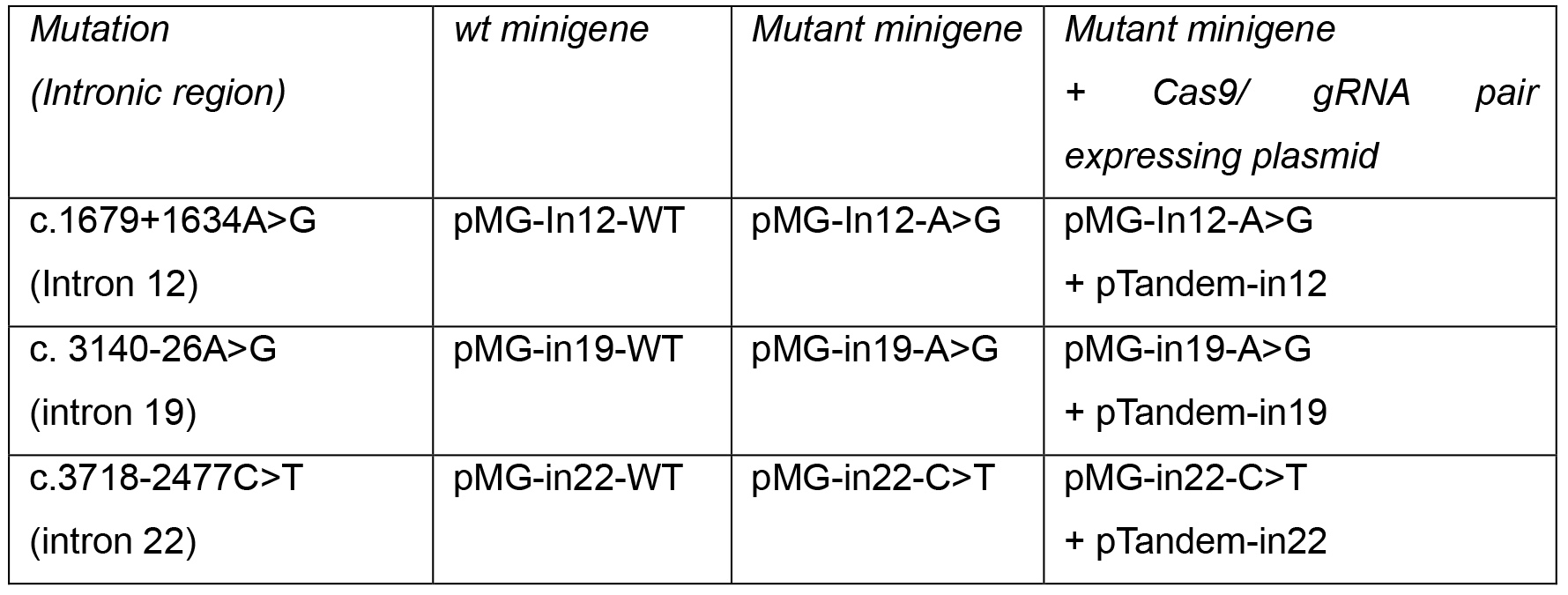
- Forty-eight hours after transfection, Extract total DNA and total RNA from each transfected HEK293T sample using DNeasy Blood and Tissue Kit (QIAGEN) and NucleoSpin RNA (MACHEREY-NAGEL) respectively.
- Extract total DNA from cells transfected with both mutant minigene and corresponding pTandem plasmid.
- Set up the following PCR amplification to detect excisions produced in each mutant minigene by the corresponding pTandem plasmid. In order to have PCR products coming from excised and non-excised minigenes that can be distinguished in an agarose-based electrophoresis, use one sequencing primer of the vector (Step A9) and one screening primer of the corresponding cloned sequence (Step A13) as indicated:
Primers used to detect excisions in mutant minigenes
In pMG-in12 minigene: pspSEQrv and in12Drv
In pMG-in19 minigene: in19Ufw and pspSEQrv
In pMG-in23 minigene: pspSEQfw and in22Drv
PCR reaction setupExtracted DNA (~20 ng/μl) 3 μl 5x Phusion Buffer 4 μl 10 mM dNTPS 0.4 μl 10 μM fw primer 0.5 μl 10 μM rv primer 0.5 μl Phusion polymerase 0.5 μl Nuclease free water Up to 20 μl
PCR programInitial Denaturation 98 °C 30 s 35 cycles of 98 °C 30 s 57 °C 30 s 72 °C 30 s Final extension 72 °C 2 min Hold 4 °C - Excisions produced by pTandem plasmids in mutant minigenes can be detected on a 1.5% agarose gel in TAE buffer, as a smaller amplification product (Figures 2F, 3F and 4F). Due to the existent homology between excised and non-excised PCR products, extra bands corresponding to heteroduplexes products may appear in the electrophoresis.
- Produce cDNA using High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher) from extracted RNA samples. Check produced cDNA concentration using Nanodrop 2000.
- PCR amplify cDNA samples using specific primers for the exonic sequences in pSPL3.
RT-PCR pSPL3 primersRTpspfw TCTGAGTCACCTGGACAACC RTpsprv ATCTCAGTGGTATTTGTGAGC 5′ FAM- RTpspfw FAM-TCTGAGTCACCTGGACAACC
PCR reaction setupcDNA (~20 ng/μl) 3 μl 5x Phusion Buffer 4 μl 10 mM dNTPS 0.4 μl 10 μM RTpspfw 0.5 μl 10 μM RTpsprv 0.5 μl Phusion polymerase 0.5 μl Nuclease free water Up to 20 μl
PCR programInitial Denaturation 98 °C 30 s 35 cycles of 98 °C 30 s 57 °C 30 s 72 °C 30 s Final extension 72 °C 2 min Hold 4 °C - Run 1.5% agarose gel electrophoresis of RT-PCR products, and gel extract and purify the different-sized fragments using StrataPrep kit (Agilent).
- Sequence the extracted bands using RTpspfw and RTpsprv primers to determine the effect on splicing of the introduced mutations in the minigenes when compared to wt minigene, and to know the effect of Cas9/gRNA-based excision in restoring aberrant splicing on each mutant minigene.
- Repeat RT-PCR as above but using a 5′ FAM RTpspfw labeled primer (Eurofins) and non-labeled standard RTpsprv to obtain 5′ FAM labeled PCR reactions.
- Purify 5′ FAM labeled PCR reactions using high pure PCR product purification (Sigma-Aldrich) and perform fragment length analysis (Eurofins) to quantify the percentage of each RT-PCR product as the area of the peak. In fragment length analysis fluorescent-labeled PCR products are separated using capillary electrophoresis and sized by comparison to a size standard.
Data analysis
Experiments were performed in triplicate.
Targeted excisions produced by the gRNA pairs in CFTE cells were quantified using GelDoc Software analysis (Bio-Rad) after PCR and Agarose gel Electrophoresis. The selected gRNA pairs were able to produce excisions in genomic CFTR gene with an efficiency of 33%, 30% and 47%, measured as the percentage of PCR products of reduced size (Figure 1).
Figures 2F, 3F and 4F show the percentage of amplicon of reduced size coming from the excised plasmid; this percentage was quantified using GelDoc Software analysis (Bio-Rad).
The FLA electropherograms in Figures 2-4 shows the size and percentage of the transcripts from (i) the wt minigene (2B, 3B, 4B), (ii) from the mutated minigene (2D, 3D, 4D), and (iii) the mutated minigene treated with the corresponding excision maker pTandem-In plasmid (2G, 3G, 4G). Those percentages were supplied by the FLA service and correspond to the area of the said peaks compared to the total area of the peaks in the electropherogram. All the three mutations of interest produce aberrant splicing with no production (2D, 3D) or partial production (4D) of normal transcript; this aberrant splicing is almost completely restored with up to 90% (2G, 3G, 4G) of normal transcript produced when the pTandem-in plasmids excise the intronic regions containing the target mutation.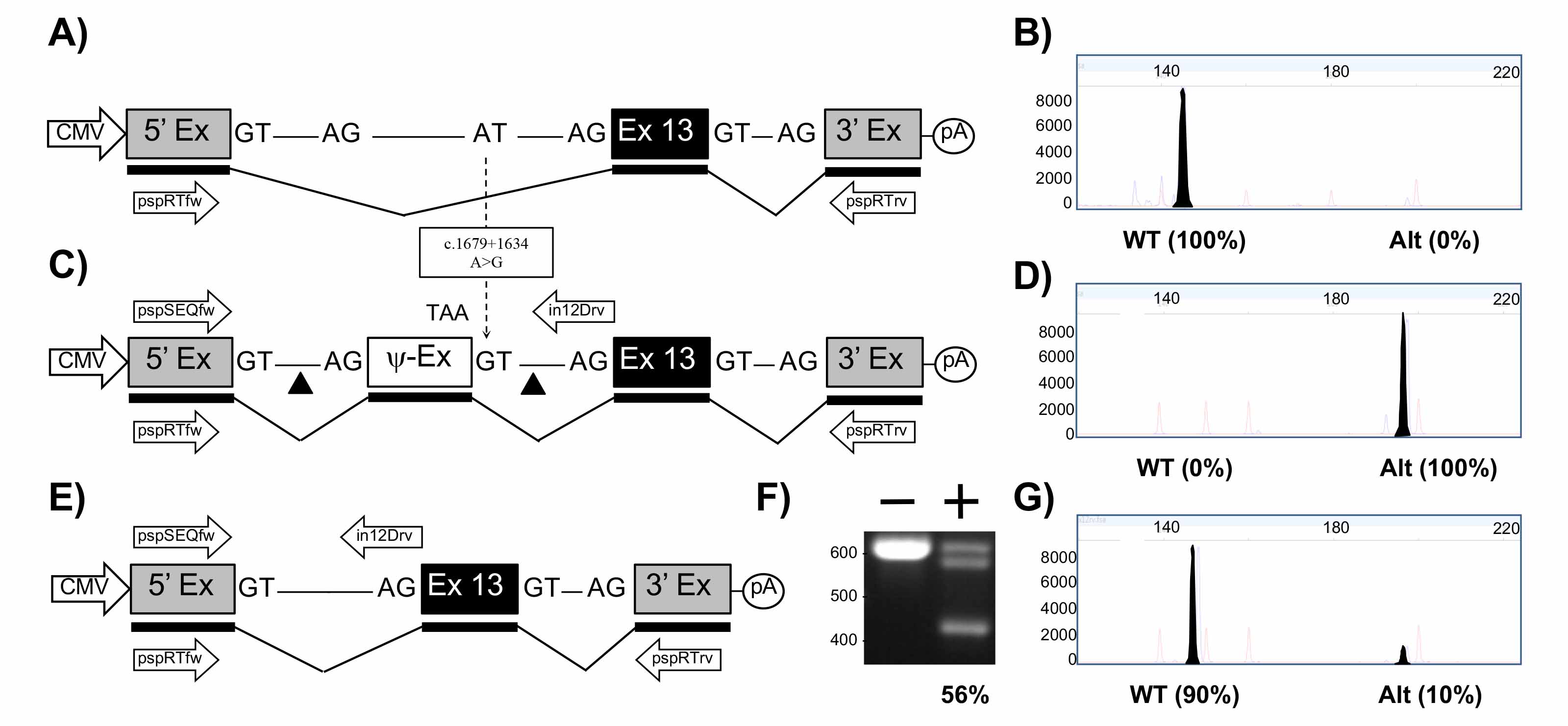
Figure 2. Mini-gene analysis of c.1679+1634>G mutation. A. Schematic of pMG-in12-WT plasmid comprising a contiguous ~1.5 kb region of CFTR containing part of intron 12, exon 13 and part of intron 13 cloned between the 5′ and 3′ exons of the pSPL3 vector. Exons are shown as boxes, introns as lines, and predicted transcript as thicker line shown underneath. Arrows below transcript represent pspRTfw and pspRTrv primers for RT-PCR. B. Electropherogram analysis of RT-PCR products (filled peaks) generated from HEK293T cells transfected with pMG-in12-WT and size markers (unfilled peaks). C. Schematic of pMG-in12-A>G mutation which differs from pMG-in12-WT by the A>G change shown with dotted arrow. The A>G change creates a pseudo exon (Ψ-EX) which contains an in-frame stop codon (TAA). The triangles flanking the Ψ-EX indicate target sites of Cas9/gRNAs encoded by pTandem-in12 vector. Arrows above DNA represent pspSEQfw and in12Drv primers for PCR. D. Electropherogram analysis of RT-PCR products generated from HEK293T cells transfected with pMG-in12-A>G. E. Schematic of pMG-in12-A>G mutation following targeted excision and repair. F. Agarose gel electrophoresis analysis of targeted deletions in pMG-in12-A>G measured by PCR. “-” lane is PCR products generated from HEK293T cells transfected with pMG-in12-A>G only, “+” lane is PCR products generated from HEK293T cells transfected with pMG-in12-A>G and pTandem-in12. The top band is generated from untargeted DNA, the bottom band is generated from DNA containing the targeted deletion, and the middle band is most likely heteroduplexes formed during the late stages of PCR. G. Electropherogram analysis of RT-PCR products generated from HEK293T cells co-transfected with pMG-in12-A>G and pTandem-in12 vector.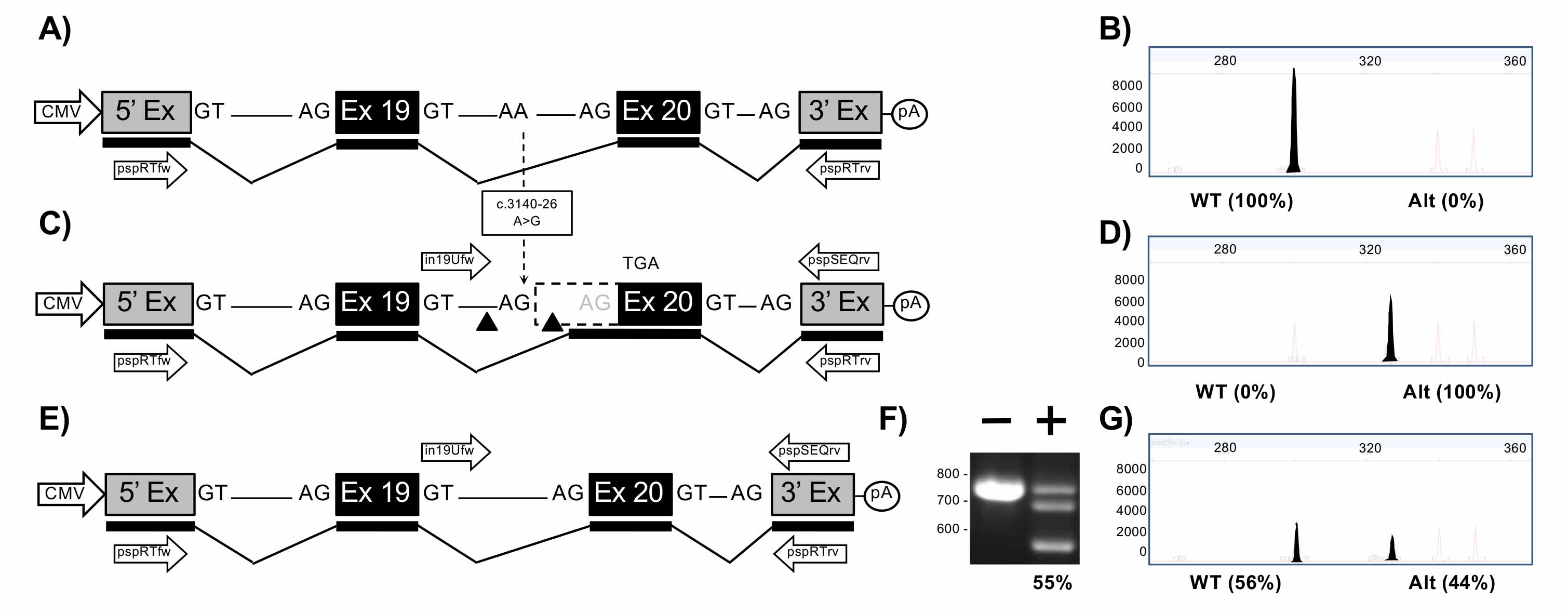
Figure 3. Mini-gene analysis of c.3140-26A>G mutation. A. Schematic of pMG-in19-WT plasmid comprising a contiguous ~2.0 kb region of CFTR containing part of intron 18, exon 19, all of intron 19, exon 20 and part of intron 20 cloned between the 5′ and 3′ exons of the pSPL3 vector. B. Electropherogram analysis of RT-PCR products generated from HEK293T cells transfected with pMG-in19-WT. C. Schematic of pMG-in19-A>G mutation which differs from pMG-in19-WT by the A>G change shown with dotted arrow. The A>G change extends exon 20 by 25 bp which shifts the reading frame and eventually results in-frame stop codon (TGA). The triangles indicate target sites of Cas9/gRNAs encoded by pTandem-in19 vector. Arrows above DNA represent in19Ufw and pspSEQrv primers for PCR. D. Electropherogram analysis of RT-PCR products generated from HEK293T cells transfected with pMG-in19-A>G. E. Schematic of pMG-in19-A>G mutation following targeted excision and repair. F. Agarose gel electrophoresis analysis of targeted deletions in pMG-in19-A>G measured by PCR. “-” lane is PCR products generated from HEK293T cells transfected with pMGin19-A>G only, “+” lane is PCR products generated from HEK293T cells transfected with pMG-in19-A>G and pTandem-in12. G. Electropherogram analysis of RT-PCR products generated from HEK293T cells co-transfected with pMG-in19-A>G and pTandem-in19 vector.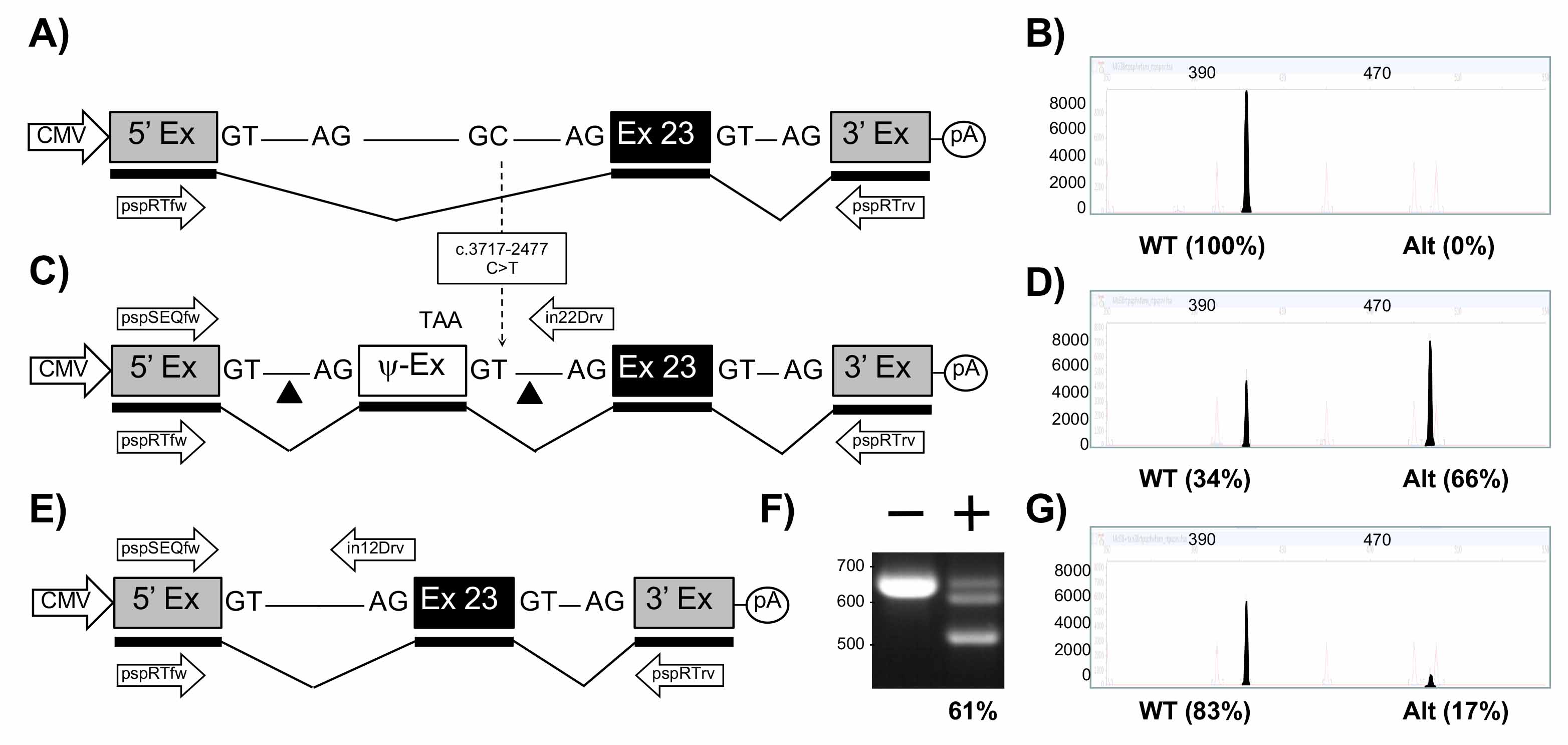
Figure 4. Mini-gene analysis of c.3718-2477C>T mutation. A. Schematic of pMG-in22-WT plasmid comprising a contiguous ~3.2 kb region of CFTR containing part of intron 22, exon 23, and part of intron 23 cloned between the 5′ and 3′ exons of the pSPL3 vector. B. Electropherogram analysis of RT-PCR products (filled peaks) generated from HEK293T cells transfected with pMG-in22-WT. C. Schematic of pMG-in22-C>T mutation which differs from pMG-in22-WT by the C>T change shown with dotted arrow. The C>T change creates a pseudo exon (Ψ-EX) which contains an in-frame stop codon (TAA). The triangles flanking the Ψ-EX indicate target sites of Cas9/gRNAs encoded by pTandem-in22 vector. Arrows above DNA represent pspSEQfw and in22Drv primers for PCR. D. Electropherogram analysis of RT-PCR products generated from HEK293T cells transfected with pMGin22-C>T. E. Schematic of pMG-in22-C>T mutation following targeted excision and repair. F. Agarose gel electrophoresis analysis of targeted deletions in pMG-in22-C>T measured by PCR. “-” lane is PCR products generated from HEK293T cells transfected with pMG-in22-C>T only, “+” lane is PCR products generated from HEK293T cells transfected with pMG-in22-C>T and pTandem-in22. G. Electropherogram analysis of RT-PCR products generated from HEK293T cells co-transfected with pMG-in22-C>T and pTandem-in22 vector.
Acknowledgments
This protocol was used for the work previously published in PLOS ONE (Sanz et al., 2017). We want to thank Dieter Gruenert for the CFTE cell line and to Feng Zhang (Broad Institute, Boston, MA) for the pSpCas9(BB)-2A-GFP plasmid. Cystic Fibrosis Foundation (Bethesda, MD, USA) and Cystic Fibrosis Trust (London, UK) funded this work.
Competing interests
The authors declare no conflict of interest.
References
- Beck, S., Penque, D., Garcia, S., Gomes, A., Farinha, C., Mata, L., Gulbenkian, S., Gil-Ferreira, K., Duarte, A., Pacheco, P., Barreto, C., Lopes, B., Cavaco, J., Lavinha, J. and Amaral, M. D. (1999). Cystic fibrosis patients with the 3272-26A-->G mutation have mild disease, leaky alternative mRNA splicing, and CFTR protein at the cell membrane. Hum Mutat 14(2): 133-144.
- Chillón, M., Dork, T., Casals, T., Gimenez, J., Fonknechten, N., Will, K., Ramos, D., Nunes, V. and Estivill, X. (1995). A novel donor splice site in intron 11 of the CFTR gene, created by mutation 1811+1.6kbA-->G, produces a new exon: high frequency in Spanish cystic fibrosis chromosomes and association with severe phenotype. Am J Hum Genet 56(3): 623-629.
- Gruenert, D. C., Willems, M., Cassiman, J. J. and Frizzell, R. A. (2004). Established cell lines used in cystic fibrosis research. J Cyst Fibros. 3 Suppl 2:191-6.
- Highsmith, W. E., Burch, L. H., Zhou, Z., Olsen, J. C., Boat, T. E., Spock, A., Gorvoy, J. D., Quittel, L., Friedman, K. J., Silverman, L. M. and et al. (1994). A novel mutation in the cystic fibrosis gene in patients with pulmonary disease but normal sweat chloride concentrations. N Engl J Med 331(15): 974-980.
- Hollywood, J. A., Lee, C. M., Scallan, M. F. and Harrison, P. T. (2016). Analysis of gene repair tracts from Cas9/gRNA double-stranded breaks in the human CFTR gene. Sci Rep 6: 32230.
- Sanz, D. J., Acedo, A., Infante, M., Duran, M., Perez-Cabornero, L., Esteban-Cardenosa, E., Lastra, E., Pagani, F., Miner, C. and Velasco, E. A. (2010). A high proportion of DNA variants of BRCA1 and BRCA2 is associated with aberrant splicing in breast/ovarian cancer patients. Clin Cancer Res 16(6): 1957-1967.
- Sanz, D. J., Hollywood, J. A., Scallan, M. F. and Harrison, P. T. (2017). Cas9/gRNA targeted excision of cystic fibrosis-causing deep-intronic splicing mutations restores normal splicing of CFTR mRNA. PLoS One 12(9): e0184009.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Sanz, D. J. and Harrison, P. T. (2019). Minigene Assay to Evaluate CRISPR/Cas9-based Excision of Intronic Mutations that Cause Aberrant Splicing in Human Cells. Bio-protocol 9(11): e3251. DOI: 10.21769/BioProtoc.3251.
Category
Molecular Biology > DNA > Chromosome engineering
Molecular Biology > RNA > RNA splicing
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.