- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

An Adjustable Protocol to Analyze Chemical Profiles of Non-sterile Rhizosphere Soil


Published: Vol 9, Iss 10, May 20, 2019 DOI: 10.21769/BioProtoc.3245 Views: 6654
Reviewed by: Joëlle SchläpferYang BaiManish Kumar Patel
Original research article
The authors used this protocol in:

Oct 2017

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
The analysis of chemical diversity in non-sterile rhizosphere soil has been a pressing methodological challenge for years. Rhizosphere-enriched chemicals (i.e., rhizochemicals) include root exudation chemicals, (microbial) breakdown products thereof, and de novo produced metabolites by rhizosphere-inhabiting microbes, all of which can play an important role in plant-soil interactions. The power and resolution of analytical methods and statistical analysis pipelines allow for better acquisition, separation and identification of rhizochemicals, thus providing unprecedented insight into the biochemistry underpinning plant-soil interactions. The current protocol describes a recently developed method to characterize rhizochemical profiles from plants, including crops, and is modular and customizable, allowing for application across a range of different plant-soil combinations. The protocol provides in-depth details about the experimental system for sample collection, data acquisition by liquid chromatography coupled to mass spectrometry, and analytical pipeline, which statistically selects for rhizochemicals by statistical comparison between metabolite profiles from plant-containing soil and plant-free soil. Moreover, the optional addition of chemical standards permits a semi-targeted approach, which improves the annotation of chemical signatures and identification of single rhizochemicals.
Keywords: MetabolomicsBackground
Previous approaches to studying rhizosphere have often focused on sterile and hydroponic growth conditions (van Dam and Bouwmeester, 2016). These approaches limit our understanding of the multi-trophic nature of the rhizosphere, as they fail to provide information about (microbial) breakdown products of root exudation chemicals and de novo produced chemicals by rhizosphere-inhabiting microbes, even though these may drive plant-soil interactions in response to environmental change, such as pathogen attack or abiotic stress. The challenge arises from understanding and simplifying the complex, and often overwhelming, level of chemical diversity that originates from untargeted analyses of non-sterile soil (Figure 1). Nevertheless, untangling this diversity by identifying chemical networks, their origin and function, is critical for obtaining new insight into the ecology of a much under-explored area of plant and soil science. Although not without its caveats, recent advances in mass spectrometry (MS) technology and statistical-analytical techniques permit powerful exploratory methods to uncover the chemistry of natural soils (Pétriacq et al., 2017; Swenson et al., 2015 and 2018). Such techniques are becoming increasingly accessible.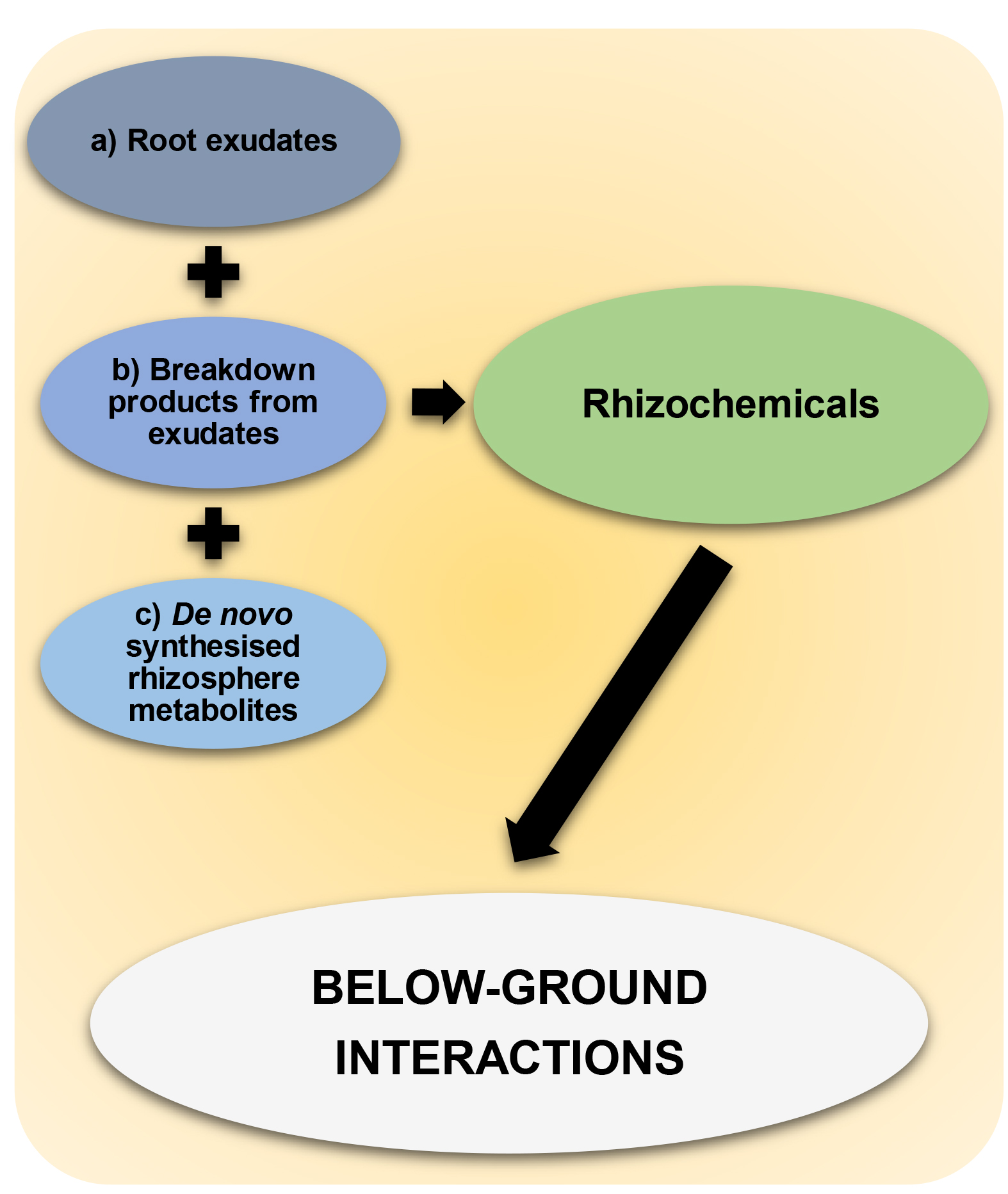
Figure 1. Rhizochemicals that shape below-ground interactions. Rhizochemicals are compounds that are enriched in the rhizosphere and include a) root exudation chemicals, b) microbial breakdown products of root exudation chemicals, c) de novo synthesized metabolites by rhizosphere-inhabiting microbes. Rhizochemicals can have important signaling activities thereby shaping below-ground interactions between plants and soil microbes.
The protocol presented here describes an adjustable experimental system for sample collection and sample preparation, followed by data acquisition using ultra-performance liquid chromatography coupled to quadrupole time-of-flight mass spectrometry (UPLC-Q-TOF). Subsequent statistical analysis allows separating chemical signatures by retention time and mass to charge (m/z) ratios between plant-free soil and plant-containing soil, which ultimately identifies profiles of rhizosphere-enriched chemical (i.e., rhizochemicals). These profiles can be further annotated using chemical databases, such as METLIN (Smith et al., 2005). Moreover, during mass spectrometry examination, running a library of relevant standards can increase the accuracy of this approach and allow focus of the analysis on specific metabolite classes.
The protocol outlined here, and originally used in Pétriacq et al. (2017), was initially developed for the analysis of rhizosphere chemistry in the model plant species Arabidopsis thaliana (Arabidopsis; Figures 2 and 3) and has been adapted for maize. For other plant species, a similar experimental design can be adapted and implemented, but method optimization is advised. Particularly, we recommend further optimization when insufficient separation is obtained between rhizochemical signatures from the plant-containing soil samples and the plant-free soil samples (see Figure 4). These adjustments involve i) testing different soils, ii) using different extraction solutions (e.g., with varying MeOH concentrations), iii) increasing root biomass in collection tubes, iv) adjusting soil moisture, v) increasing the time of sample extraction from the collection tubes, and vi) varying MS settings. Further attention should be paid to the fact that extraction solutions with high MeOH concentrations can cause damage to microbial and/or root cells, in which case specific assays can be used to assess the extent of cell damage, as detailed in Petriacq et al. (2017). If cell damage occurs, we recommend using a lower MeOH concentration in the extraction solution and/or shorter extraction times. If insufficient sample yield is obtained with shorter extraction times, a pressurized collection system can be used, as is illustrated in Figure 2D.
Materials and Reagents
Note: All chemicals and solvents used for metabolomics were of mass spectrometry grade (Sigma-Aldrich, Germany). Other solvents were of analytical grade.
- Tips
- Millipore miracloth, pore size of 22-25 µm (VWR, catalog number: 475855-1)
- Petri dish (lids) NunclonTM Delta, 8.8 cm2 (Thermo Fisher Scientific, catalog number: 150318)
- 15 ml FalconTM conical centrifuge tubes (Thermo Fisher Scientific, catalog number: 10468502)
- 50 ml FalconTM conical centrifuge tubes (Thermo Fisher Scientific, catalog number: 10788561)
- 50 ml syringe, without needle (Thermo Fisher Scientific, catalog number: 11901563)
- 5 ml conical preparation tube (Starlab, catalog number: E1450-1100)
- 2 ml microcentrifuge tube (Starlab, catalog number: I1420-2600)
- 30 ml SterilinTM universal container (Thermo Fisher Scientific, catalog number: 11309143)
- Aluminum foil
- Filter paper
- 0.2 µm disposable Merck cartridge (Thermo Fisher Scientific, catalog number: 10740365)
- 1.5 ml Glass screw neck vials, with screw caps and micro inserts (VWR, catalog numbers: 548-0018, -0020, -0106)
- Levington M3 dry compost (Spunhill, catalog number: YEV05K)
- Non staining silica sand (Sweepfast, catalog number: CH52)
- Arabidopsis seeds
- Maize seeds (Zea mays)
- Acetonitrile (Hypergrade, Sigma-Aldrich, catalog number: 1000291000)
- Methanol (Hypergrade, Sigma-Aldrich, catalog number: 1060351000)
- Formic acid (FA, Sigma-Aldrich, catalog number: F0507-500ML)
- Liquid Nitrogen (BOC)
- Bleach Mexcel (5-10%) (SLS, catalog number: CLE0300)
- Hydrochloric acid (37%) (Sigma-Aldrich, catalog number: H1758)
- Sodium formate (0.01 M NaOH/1% FA (1/1, v/v) ten-fold diluted with acetonitrile/water (80/20, v/v) (Sigma-Aldrich, catalog number: 71539-500G)
- Leucine enkephalin (2 ppm in acetonitrile/water (50/50, v/v) with 0.1% of formic acid) (Sigma-Aldrich, catalog number: L9133-25MG)
- Ice-cold extraction solution MeOH 50% + FA 0.05% (see Recipes)
Equipment
- Pipettes (P-5000, P-1000) (Starlab)
- Growth tubes (make these bespoke, see below)
- Pressure lids (make these bespoke, see below)
- Speedvac vacuum concentrator (Thermo Fisher Scientific, catalog number: SPD120)
- Freeze drier (Modulyo benchtop freeze dryer) (Edwards, catalog number: D-230)
- BEH C18 column for UPLC (2.1 x 50 mm, 1.7 μm, Waters) with a guard column (VanGuard, 2.1 x 5 mm, 1.7 µm, Waters)
- SYNAPT G2si Q-TOF (Waters)
- Centrifuge (that fits 5 ml tubes)
- Microcentrifuge
- Ultrasonic cleaning bath
- Soldering iron
- Drill (7 mm multipurpose drill bit)
- Growth cabinets (Fitotron, SANYO)
- Tweezers
- 4 °C refrigerator
- -80 °C freezer
Software
- MassLynx, version 4.1 with Data bridge, https://www.waters.com
- R with XCMS package, version 3.2, https://www.r-project.org
- Multiple experiment viewer–MeV, version 4.9.0, http://mev.tm4.org/#/welcome
- MetaboAnalyst version 3.0, http://www.metaboanalyst.ca
- METLIN, https://metlin.scripps.edu
Procedure
- Equipment preparation
- Growth tubes for plant development and subsequent chemical sampling (Figure 2A).
To create growth tubes, in a fume-hood use a pre-heated soldering iron to melt 7 mm holes in the base of 30 ml plastic tubes. This step will need to be repeated for as many tubes are required. Growth tubes can be thoroughly washed with pure distilled water and reused several times. Fit tubes with 40 mm2 miracloth at the bottom to hold soil. Fill tube with desired soil matrix (~40 ml), for instance 9:1 (v/v) mixture of sand and dry compost (see Materials and Reagents for more information).
Note: For larger plant species, such as 4-week-old maize seedlings, growing in clay agricultural soil, 50 ml Falcons were suitable with similar sized holes (Figure 2C). - Pressurized lid (for larger Falcons) to aide in pushing the liquid through the growth tube (if necessary; Figure 2D).
Create these lids by drilling a hole, smaller than the diameter of the syringe, and forcing the syringe through, securing with strong and watertight epoxy. Applying silicon to the edges and thread of the lid will ensure a tight seal to allow sufficient pressure. This set-up may be required for soil that retains more water, in order to shorten the time of flushing the soil with extraction solution.
Note: Simultaneous assays can be performed using the same experimental set-up. For instance, non-destructive rhizosphere or bulk soil can be sampled prior to extraction solution application to allow elemental analysis, or DNA/RNA extractions for determination of microbial community compositions and soil functional analysis.
- Growth tubes for plant development and subsequent chemical sampling (Figure 2A).
- Experimental set-up of growth system (Arabidopsis; Figures 2A and 2B)
- Stratify Arabidopsis seeds for 2 days in the dark in autoclaved water at 4 °C.
Note: Seeds can be surface sterilized if appropriate for the experimental design. Such techniques are effective and straightforward (Lindsey et al., 2017). - Wrap soil-filled growth tubes in tin foil to limit algal growth and stand growth tubes in individual Petri dishes to prevent cross-contamination between bio-replicates.
- Seal tubes to the Petri dish with masking tape to provide stability. Stand tubes in a tray.
- Apply 15 ml of water to individual Petri dishes to saturate the soil.

Figure 2. Experimental set-up for sampling rhizochemicals. A-C. Photos show collection tubes containing plant-free and plant-containing soils (1. Arabidopsis and 2. Maize). Tubes were covered with foil to prevent algal growth and were placed onto small Petri dishes to prevent cross-contamination. D. Example of the pressurized lid that connects to an epoxy-secured syringe. This allows the application of pressure during sample collection, in order to increase (this word was pluralized) the speed and yield of sample collection while minimizing exposure of living cells to potentially damaging organic solvents. When using larger plants such as maize, the shoot is gently removed with scissors just before applying the pressure.- Pipette four seeds onto the soil surface of individual tubes. If desirable, prepare unseeded soil-only samples to act as controls.
- Cover trays with a lid and place into a growth cabinet with the appropriate growth conditions.
- Petri dishes should be supplied with 5-10 ml of water bi-weekly.
- After seedling emergence, carefully thin seedlings using tweezers to leave one seedling per pot.
- Stratify Arabidopsis seeds for 2 days in the dark in autoclaved water at 4 °C.
- Experimental set-up of growth system (Maize; Figure 2C)
- Imbibe seeds overnight in autoclaved, sterile water before placing on Petri dishes containing sterile, damp filter paper in the dark at 23 °C for two days.
- Plant germinated seeds in soil-filled growth tubes, 1.5 cm from the soil surface.
- Wrap growth tubes in foil and cover the surface with black plastic beads to limit algal growth.
- Place the tubes in a growth chamber with the desired environmental conditions.
- Metabolite extraction from control and Arabidopsis/maize soil (Figure 3)
- Collect plant soil samples from tubes containing one 5-week-old Arabidopsis plant, or one 17-day-old maize plant (timings can depend on experimental design).
Note: The timings used in this protocol assume similar growth conditions to those mentioned above. - For Arabidopsis, with the seedling intact, apply ice-cold extraction solution (5 ml), to the top of the tubes and avoid any disturbance of the soil surface.
Note: With larger plant, you may want to sever, or simultaneously sample the seedling. If doing so, be careful to avoid contaminating the extraction solution with damaged plant material. Furthermore, when using larger plants such as maize, the shoot can be gently removed with scissors just before applying the pressure. - After 1 min, 4-4.5 ml can be collected from the drainage hole in 5 ml centrifuge tubes.
- For use with maize, gently slit the seedling with a blade around 0.5 cm of the soil surface, and subsequently apply 15 ml of the extraction solution to the soil and induce pressure to the top of the pot, using a modified lid containing a syringe.
- After 1 min, 5-10 ml are collected in centrifuge tubes.
- Centrifuge to pellet soil residues (5 min, 3,500 x g), aliquot 4 ml of supernatant into a new centrifuge tube.
- Flash-freeze in liquid nitrogen and freeze-dry until complete dryness.
- Samples can now be stored at -80 °C until chemical analysis.
- If the soil is not saturated, less flow-through can be collected. The longer the buffer is in contact with the plant the more chance there is of tissue damage and bias through cellular metabolites. Limit time for which samples are exposed to MeOH is less than 1 min, to prevent the risk of root damage. Irrelevant for sampling with sterile distilled water.
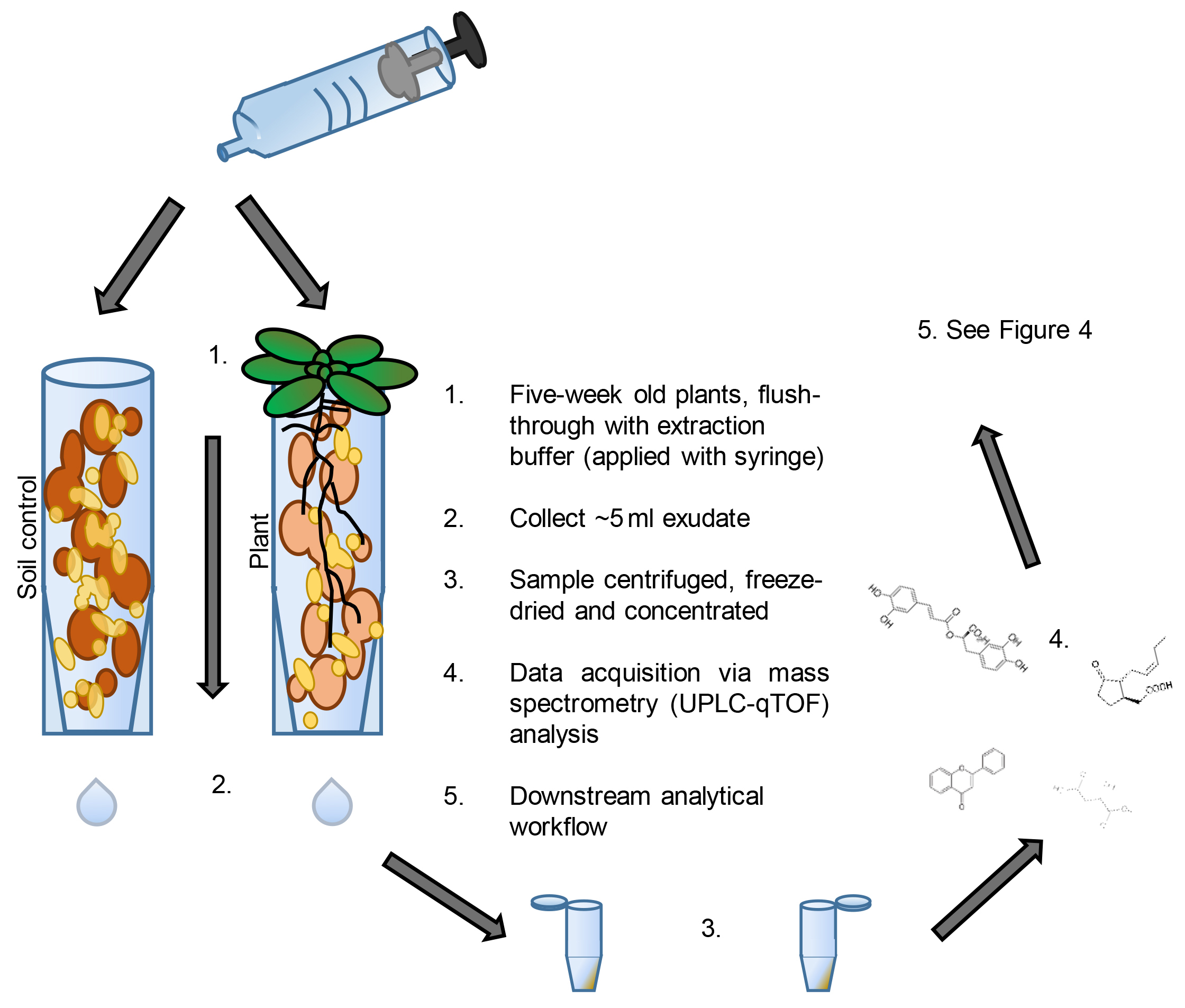
Figure 3. Sampling workflow. See figure for notes. - Collect plant soil samples from tubes containing one 5-week-old Arabidopsis plant, or one 17-day-old maize plant (timings can depend on experimental design).
- Preparing samples for UPLC-qTOF analysis
- Resuspend dried aliquots into 100 µl of methanol:water:formic acid (50:49.9:0.1, v/v).
- Sonicate at 4 °C for 20 min, vortex and centrifuge (15 min, 14,000 x g, 4 °C) to remove potential particles that could block the UPLC column. Alternatively, filter the samples through 0.2 µm disposable cartridge obtained from analytical suppliers.
- Transfer final supernatants (80 µl) into glass vials containing a glass insert before injection through the UPLC system.
- During this process, generate a quality control (QC) sample by mixing 5 µl of each sample into one single vial. This will act as a control for analytical reliability of the LCMS system, and for optional normalization of the data in case of MS intensity drift when running a high number of samples (Broadhurst et al., 2018).
- UPLC-qTOF Set-up
- LC and MS characteristics are fully detailed in Pétriacq et al. (2017). Using a SYNAPT G2si HDMS Q-TOF mass spectrometer (Waters), coupled to a UPLC BEH C18 column (2.1 x 50 mm, 1.7 μm, Waters) with a guard column (VanGuard, 2.1 x 5 mm, 1.7 µm, Waters) for separation of compounds at a flow rate of 400 μl min−1, prepare the mobile phases as A; water with 0.05% formic acid, and B; acetonitrile with 0.05% formic acid with the following gradient: 0-3 min 5-35% B, 3-6 min 35-100% B, holding at 100% B for 2 min, 8-10 min, 100-5% B.
- Set the column temperature to 45 °C and use an injection volume of 10 μl.
- Use a mass range of 50-1,200 Da and a scan time of 0.2 s (ESI- and ESI+) with the instrument operating in sensitivity mode for the MS full scan (i.e., without collision energy).
- Ramp collision energy in the transfer cell from 5 to 45 eV (MSE), using appropriate voltage conditions for ESI- mode (e.g., Capillary, -3 kV; Sampling cone, -25 V; Extraction cone, 4.5 V) and ESI+ mode (e.g., Capillary, +3 kV; Sampling cone, +25 V; Extraction cone, 10 V).
- Source temperature for each mode should be 120 °C, desolvation temperature at 350 °C, desolvation gas flow at 800 L h-1 and cone gas flow at 60 L h-1.
- Calibration of the qTOF mass spectrometer
- Prior to analysis, calibrate the Q-TOF detector with a solution of sodium formate.
- During each run, accurate mass measurements can be ensured by infusing leucine enkephalin peptide as an internal reference (i.e., lock mass) with 10 s scan frequency, cone voltage of 40 V and a capillary voltage of 3 kV.
- Inject 6 QCs samples at the beginning of the analytical run, then every 10 injections, and finish with QC sample at the end of the analytical run.
- Inject blank samples (50% methanol, v/v) between each treatment and between ESI- and ESI+ ionization modes for stabilization of the electrospray ionization source.
- Processing of MS data for statistical analysis (see Figure 4 for a schematic of the subsequent analytical steps) and deconstruction of rhizochemicals
- After the run has finished, convert raw files, here obtained from MassLynx, into CDF or mwXML format, using the Databridge function in MassLynx, or ProteoWizard (MsConvert).
- For subsequent alignment and integration of metabolic peaks, use R with the XCMS package installed (Smith et al., 2006) and the standard script described in Supplemental Data S1.
- Peaks can be retained for analysis when present in all bio-replicates (n = 3, in an example where there are 3 bio-replicates), at a threshold intensity of 10 (I = 10) and at maximum resolution range of 20 ppm. XCMS parameters should be adjusted according to the mass spectrometer used in the study (here this is q-TOF), and nuances of the particular dataset being investigated.
- Normalize peak values from each run against total ion current (TIC). For each sample, normalized peak values will generate separate datasets for ESI+ and ESI- ionization modes.
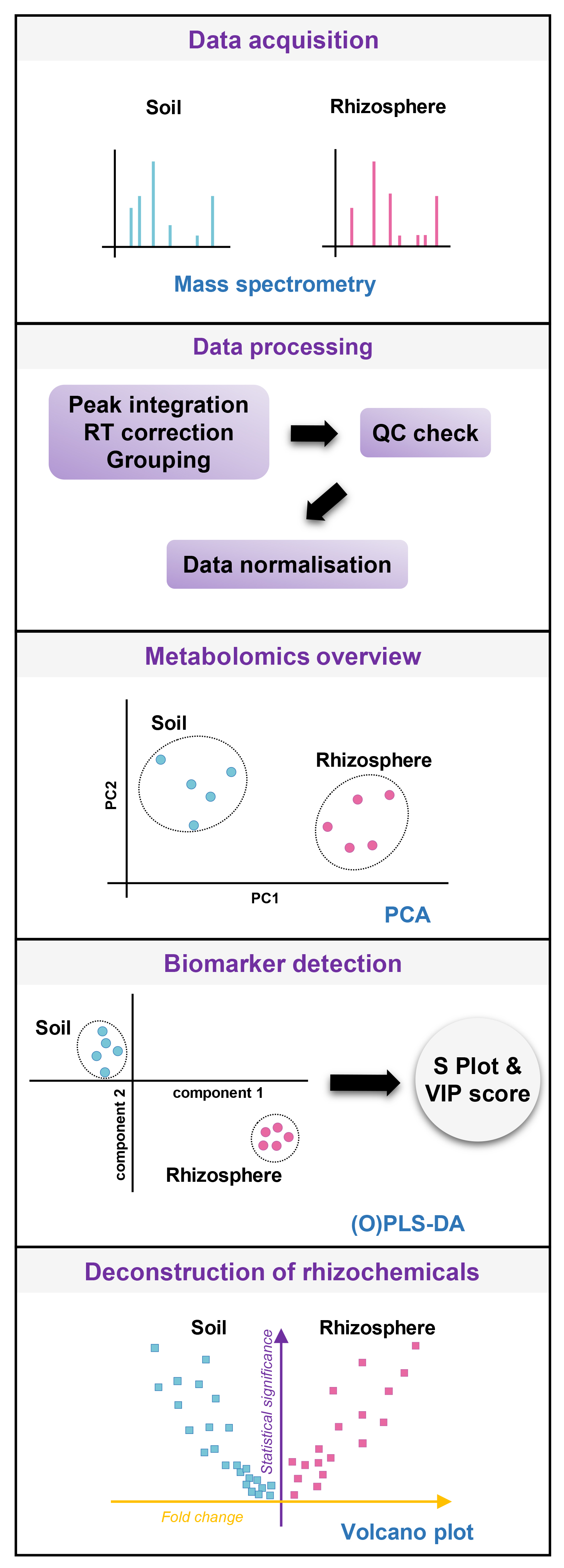
Figure 4. Analytical workflow for the identification of rhizosphere metabolites. Mass spectrometry data are processed using XCMS (Smith et al., 2006) for peak identification (RT-m/z features) and alignment, grouping, and retention time (RT) correction, then checked for quality control and normalized (Median-centered, cube-root transformation and Pareto scaling). Metabolomics overview is given by principal component analysis (PCA), which is crucial to verify sufficient metabolomics separation between extracts from plant-containing and plant-free tubes. If the global differences are not convincing by PCA, the rhizosphere chemistry in the plant-containing soil might be too much diluted by bulk soil chemistry, and further adjustments of collection systems are necessary (e.g., bigger plants, other soil, different extracting solutions, and longer sample extraction). In the specific case of binary comparison (plant-free versus plant-containing tubes), biomarker detection can be performed with S Plot obtained from (Orthogonal) Partial Least Square-Discriminant Analysis (O)PLS-DA (Worley and Powers, 2013). This supervised technique provides Variable Important for the Projection (VIP) scores that reflect the discriminant statistical power of each variable for the PLS discriminant model. Ultimately, univariate statistical comparison via volcano plots (fold change vs. statistical significance) allows for straightforward identification of metabolic markers that are specific to the rhizosphere (i.e., rhizochemicals). - Statistical analysis of MS data
For this experimental set-up, the strength in identifying ‘rhizosphere’ chemical signatures comes from a combination of the experimental design, i.e., free soil vs. planted soil, and the subsequent binary statistical comparison, obtained through a volcano plot showing statistical significance against biological significance. Further statistical tests (e.g., ANOVA, t-tests with consideration for multiple comparisons, such as false discovery rate–FDR or Bonferroni corrections, where appropriate) can be used for the quantification and annotation of markers that are affected by the treatment. Such pipelines depend on the conditions used in the initial structure of the experiment and question being asked.
Prior to analysis, an appropriate normalization is required. Median-normalization, cube-root transformation and Pareto scaling of the data seem to perform best on metabolomics datasets (van den Berg et al., 2006): the original structure of the dataset is conserved while the influence of peaks with high intensity is softened compared to peaks with low intensity.- Global differences in metabolic signals between treatment/point combinations can be visualized for anions (ESI-) and cations (ESI+) separately, or jointly where both datasets are concatenated, by principal component analysis (PCA), using MetaboAnalyst online (v. 3.0; http://www.metaboanalyst.ca; (Xia et al., 2015) on median-normalized, cube-root-transformed and Pareto-scaled data.
- For quantification of the number of ions showing quantitative differences between binary treatments (specifically the comparison between plant-free soil and plant-containing soil), volcano plots can be created on median normalized, Pareto-scaled and cube-root transformed data, with a cut-off value of > 2 fold-change (Log2 > 1) and a statistically significant threshold of P < 0.05 (Welch’s t-test; MetaboAnalyst). These markers can then be selected for identification. When a condition of ‘rhizosphere’ chemicals is screened against the corresponding soil, then volcano plots can deconstruct the markers that are specific to semiochemicals of the rhizosphere. When soil chemistry is the target, conventional heatmap or cluster analysis could be performed and the relevant markers further retained for subsequent analysis (such as ANOVA with subsequent Hierarchal clustering through Pearson’s correlation (Williams et al., 2018). It is highly recommended that you adhere to the guidelines laid out by the Metabolomics Standards Initiative (Fiehn et al., 2008; Goodacre et al., 2007; Sumner et al., 2007).
- Annotation of putative metabolic markers
Putative annotation of the selected ion markers can be assigned, based on accurately detected m/z values at a mass accuracy < 30 ppm), using METLIN chemical database (Smith et al., 2005), or other chemical/metabolite database available online, or in house. PubChem can then be used to check the predicted pathway or class annotation (https://pubchem.ncbi.nlm.nih.gov/).
Notes
When applying this method to larger species with greater soil volumes, it is important to note that the water content of the soil will dilute the extraction solution, which needs to be taken into account in the design of the extraction solution and the amount of flush-through collected from the sample.
In addition, the extraction solution suggested here (50% MeOH) limits tissue damage to the sample, but the actual design of this solution can be adapted depending on the chemical profile of interest. It is worth noting that both lower and higher concentrations of methanol can be appropriate. Thus, it is possible that other extraction buffers may be preferable (such as water, 70% MeOH, 10% MeOH etc.). It is highly recommended that method optimization is performed regardless, but especially in the instance of applying a different extraction solution as to those discussed.
Recipes
- Extraction solution
50% methanol (hypergrade):49.95% ultra-purified water: 0.05% Formic acid
Notes:- Use clean equipment to produce this solution to the desired volume. Store in the fridge–shelf life is less than a week.
- Extraction solution can contain different levels of MeOH, depending on the type of chemistry that is desired for collection.
Acknowledgments
We would like to thank Anne Cotton and Roland Schwarzenbacher for collecting images to illustrate the collection system. JT acknowledges support by a consolidator grant from the European Research Council (ERC; no. 309944 “Prime-A-Plant”), a Research Leadership Award from the Leverhulme Trust (no. RL-2012-042) and a BBSRC-IPA grant (BB/P006698/1). PP is grateful to the MetaboHUB (ANR-11-INBS-0010) project, University of Bordeaux and P3 Research Institute for financial support.
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Broadhurst, D., Goodacre, R., Reinke, S. N., Kuligowski, J., Wilson, I. D., Lewis, M. R. and Dunn, W. B. (2018). Guidelines and considerations for the use of system suitability and quality control samples in mass spectrometry assays applied in untargeted clinical metabolomic studies. Metabolomics 14(6): 72.
- Fiehn, O., Wohlgemuth, G., Scholz, M., Kind, T., Lee, D. Y., Lu, Y., Moon, S. and Nikolau, B. (2008). Quality control for plant metabolomics: reporting MSI-compliant studies. Plant J 53(4): 691-704.
- Goodacre, R., Broadhurst, D., Smilde, A. K., Kristal, B. S., Baker, J. D., Beger, R., Bessant, C., Connor, S., Capuani, G., Craig, A., Ebbels, T., Kell, D. B., Manetti, C., Newton, J., Paternostro, G., Somorjai, R., Sjöström, M., Trygg, J. and Wulfert, F. (2007). Proposed minimum reporting standards for data analysis in metabolomics. Metabolomics 3(3): 231-241.
- Lindsey, B. E., 3rd, Rivero, L., Calhoun, C. S., Grotewold, E. and Brkljacic, J. (2017). Standardized method for high-throughput sterilization of Arabidopsis seeds. J Vis Exp(128).
- Petriacq, P., Williams, A., Cotton, A., McFarlane, A. E., Rolfe, S. A. and Ton, J. (2017). Metabolite profiling of non-sterile rhizosphere soil. Plant J 92(1): 147-162.
- Smith, C. A., O'Maille, G., Want, E. J., Qin, C., Trauger, S. A., Brandon, T. R., Custodio, D. E., Abagyan, R. and Siuzdak, G. (2005). METLIN: a metabolite mass spectral database. Ther Drug Monit 27(6): 747-751.
- Smith, C. A., Want, E. J., O'Maille, G., Abagyan, R. and Siuzdak, G. (2006). XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal Chem 78(3): 779-787.
- Sumner, L. W., Amberg, A., Barrett, D., Beale, M. H., Beger, R., Daykin, C. A., Fan, T. W., Fiehn, O., Goodacre, R., Griffin, J. L., Hankemeier, T., Hardy, N., Harnly, J., Higashi, R., Kopka, J., Lane, A. N., Lindon, J. C., Marriott, P., Nicholls, A. W., Reily, M. D., Thaden, J. J. and Viant, M. R. (2007). Proposed minimum reporting standards for chemical analysis chemical analysis working group (CAWG) metabolomics standards initiative (MSI). Metabolomics 3(3): 211-221.
- Swenson, T. L., Jenkins, S., Bowen, B. P., Northen, T. R. (2015). Untargeted soil metabolomics methods for analysis of extractable organic matter. Soil Biol Biochem 80: 189-198
- Swenson, T. L., Karaoz, U., Swenson, J. M., Bowen, B. P. and Northen, T. R. (2018). Linking soil biology and chemistry in biological soil crust using isolate exometabolomics. Nat Commun 9(1): 19.
- van Dam, N. M. and Bouwmeester, H. J. (2016). Metabolomics in the rhizosphere: tapping into belowground chemical communication. Trends Plant Sci 21(3): 256-265.
- van den Berg, R. A., Hoefsloot, H. C., Westerhuis, J. A., Smilde, A. K. and van der Werf, M. J. (2006). Centering, scaling, and transformations: improving the biological information content of metabolomics data. BMC Genomics 7: 142.
- Williams, A., Petriacq, P., Schwarzenbacher, R. E., Beerling, D. J. and Ton, J. (2018). Mechanisms of glacial-to-future atmospheric CO2 effects on plant immunity. New Phytol 218(2): 752-761.
- Worley, B. and Powers, R. (2013). Multivariate analysis in metabolomics. Curr Metabolomics 1(1): 92-107.
- Xia, J., Sinelnikov, I. V., Han, B. and Wishart, D. S. (2015). MetaboAnalyst 3.0--making metabolomics more meaningful. Nucleic Acids Res 43(W1): W251-257.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Williams, A., Ton, J. and Pétriacq, P. (2019). An Adjustable Protocol to Analyze Chemical Profiles of Non-sterile Rhizosphere Soil. Bio-protocol 9(10): e3245. DOI: 10.21769/BioProtoc.3245.
Category
Plant Science > Plant biochemistry > Other compound
Plant Science > Plant physiology > Metabolism
Biochemistry > Other compound > Soil chemicals
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.