- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Primary Embryonic Rat Cortical Neuronal Culture and Chronic Rotenone Treatment in Microfluidic Culture Devices
Published: Vol 9, Iss 6, Mar 20, 2019 DOI: 10.21769/BioProtoc.3192 Views: 6663
Reviewed by: Oneil G. BhalalaSébastien GillotinAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Aug 2018
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
In the study of neurodegenerative diseases, it is imperative to study the cellular and molecular changes associated with pathogenesis in the relevant cell type, central nervous system neurons. The unique compartmentalized morphology and bioenergetic needs of primary neurons present complications for their study in culture. Recent microculture techniques utilizing microfluidic culture devices allows for environmental separation and analysis of neuronal cell bodies and neurites in culture. Here, we present our protocol for culture of primary neurons in microfluidic devices and their chronic treatment with the Parkinson’s disease (PD) relevant toxicant rotenone. In addition, we present a method for reuse of devices for culture. This culture methodology presents advantages for evaluating early pathogenic cellular and molecular changes in neurons in a compartment-specific manner.
Keywords: Primary neuron cultureBackground
Studying primary neurons in vitro has long presented challenges due to the unique, diverse morphologies of neurons, and due to neuronal culturing requirements (Millet and Gillette, 2012). Neurons exhibit a unique compartmentalized morphology, with the soma, dendrites, and axons all exhibiting compartment-specific biochemical needs (Van Laar and Berman, 2013). Studying these different microenvironments objectively via large-scale approaches or high-throughput methods can be difficult under traditional plate culture, as cell bodies and neurites growing in proximity often cross, overlap, and functionally connect with one another. The development of microculturing methods incorporating microchannels to restrict cell movement and permit neurite outgrowth has allowed for the culturing of neurons in a manner that environmentally separates axonal and dendritic neurite projections from their cell body, or soma (Millet and Gillette, 2012; Taylor et al., 2003). This innovation permits cellular compartment-specific analyses of neuronal development, biochemistry, and effects of treatments (e.g., treating the axons with a drug or toxicant, but not the soma) (Taylor et al., 2005 and 2015; Park et al., 2006). The commercial availability of such devices now allows for uniform, large-scale studies into comparisons of the axonal environment and the somal environment.
By allowing for prolonged, healthy culture of primary neurons while isolating the microenvironments of the differing cellular compartments, these devices have become essential tools for studies that examine morphological-compartment specific development, senescence, or, in the case of our previous study, chronic drug exposure (Van Laar et al., 2018).
Here, we present our detailed protocol for preparation of embryonic rat primary cortical neurons (Arnold et al., 2011; adapted from Ghosh and Greenberg, 1995), culturing the neurons in Xona Microfluidics® brand microfluidic culture devices, and chronically treating the neurons with sublethal levels of the PD-relevant toxicant rotenone, as described in our recent publication in Van Laar et al. (2018). Our microfluidic device plating protocol, adapted from work originally presented by Park et al. (2006), provides details to (1) better ensure isolation of the soma from the axonal compartment side during the initial plating and (2) to promote neuronal health during prolonged culturing and treatment requiring media changes. We also present our protocol for the cleaning and reuse of microfluidic culture devices. This helps prevent the waste of devices in the event that initial platings of neuronal cultures fail to thrive or are otherwise unusable for experimental analyses.
Materials and Reagents
- Materials
- 6 cm sterile culture Petri dishes (Fisher, catalog number: AS-4052)
- 10 cm sterile culture Petri dishes (Corning, catalog number: 430167)
- 15 cm sterile culture Petri dishes (Corning, catalog number: 430599)
- 10 ml sterile syringes (BD, catalog number: 309604)
- PES 0.22 µm-pore sterile syringe filters (Millipore, catalog number: SLGP033RS)
- Sterile 0.22 µm Bottle-Top Filters (Fisher, catalog number: 09-761-112)
- Media Storage Bottles, 500 ml, 1,000 ml (Fisher, catalog numbers: 06-414-1C, 06-414-1D)
- Hydrion pH paper (Fisher, catalog number: 14-853-150N)
- 15 ml Falcon Conical Tubes (Fisher, catalog number: 14-959-70C)
- 50 ml Falcon Conical Tubes (Fisher, catalog number: 14-959-49A)
- 40 µm Sterile Cell Strainer (Fisher, catalog number: 22363547)
- Animal
E18 Pregnant female Sprague-Dawley rat - Primary neuron dissection
- Sodium sulfate (Na2SO4) (Sigma, catalog number: S5640-500G)
- Potassium sulfate (K2SO4) (Sigma, catalog number: P8541-1KG)
- Magnesium chloride (MgCl2), 1 M solution (Sigma, catalog number: M1028-100ML)
- Calcium chloride, anhydrous (CaCl2) (Sigma, catalog number: C5670-500G)
- HEPES, 1 M solution (Gibco, catalog number: 15630-080)
- Glucose (Gibco, catalog number: 15023-021)
- Phenol Red (Sigma, catalog number: P0290-100ML)
- Sodium Hydroxide (NaOH), 10 N (Ricca Chemical, catalog number: 7470-32)
- Cell-culture grade sterile water (Hyclone, catalog number: SH30529.03)
- Hanks’ Balanced Salt Solution (HBSS), 10x (Sigma, catalog number: H1641-500ML)
- Sodium bicarbonate (NaHCO3) (Sigma, catalog number: S5761-500G)
- L-Cysteine hydrochloride monohydrate (Cysteine-HCl) (Sigma, catalog number: C7880-100G)
- Papain (Worthington Biochemical Corp, catalog number: LS003126)
- Cell-culture grade sterile water (Hyclone, catalog number: SH30529.03)
- Bovine Serum Albumin (BSA), Fraction V (Fisher, catalog number: BP1600-100)
- Soybean Trypsin Inhibitor (Sigma, catalog number: T6522-1G)
- Phosphate Buffered Saline (PBS), 10x Solution (Fisher, catalog number: SH3037803)
- 1x PBS Solution (diluted from 10x PBS using cell-culture grade sterile water)
- Paraformaldehyde (PFA) (Fisher, catalog number: AC416780030)
- HBSS Dissection Media (store at 4 °C for up to 3 months) (see Recipes)
- Dissociation Media (store at 4 °C for up to 3 months) (see Recipes)
- L-Cysteine 50x Stock (see Recipes)
- Papain Dissociation Solution and Inhibitor Solutions for Isolation of Primary Rat Cortical Neurons (make fresh on the day of use) (see Recipes)
- Enzyme Solution
- Heavy Inhibitor Solution
- Light Inhibitor Solution
- 4% PFA Solution (see Recipes)
- Plating and culture media
- Neurobasal Media (Gibco, catalog number: 21103-049)
- Fetal Bovine Serum, Heat-Inactivated (Hyclone, catalog number: SH30070.03HI)
- B27 Media Supplement, 50x (Gibco, catalog number: 17504-044)
- Glutamax (Gibco, catalog number: 35050-061)
- Penicillin/Streptomycin Antibiotic Solution (Gibco, catalog number: 15140-122)
- Cortical Neuron Media for Seeding (store each at 4 °C for up to 10 days) (see Recipes)
- Cortical Neuron Media for Feeding (store each at 4 °C for up to 10 days) (see Recipes)
- Microfluidic device preparation, culture, and reuse
- Xona Microfluidics® microchannel culturing microfluidic devices
- 450 μm microgroove barrier Standard Neuron Device (Xona Microfluidics, catalog number: SND450)
- 900 μm microgroove barrier Standard Neuron Device (Xona Microfluidics, catalog number: SND900)
- Corning cover glass coverslip (Corning, catalog number: 2975-244)
- Poly-D-Lysine hydrobromide, MW 70,000-150,000 (Sigma, catalog number: P0899-50MG)
- Boric Acid (Fisher, catalog number: S79802)
- Borax, Anhydrous (MP Biomedicals, catalog number: 190309)
- HCl, 12 N (Fisher, catalog number: A144-500)
- Cell-culture grade sterile water (Hyclone, catalog number: SH30529.03)
- RBS detergent solution (Thermo, catalog number: 27950)
- 100% Ethanol (Decon Labs, catalog number: 2705HC)
- 0.1 M Borate Buffer Solution (see Recipes)
- Poly-D-Lysine 2.5 mg/ml Stock Solution (see Recipes)
- Poly-D-Lysine 100 µg/ml Coating Solution (see Recipes)
- Xona Microfluidics® microchannel culturing microfluidic devices
- Rotenone treatment
- DMSO (Sigma, catalog number: D2438)
- Rotenone (Sigma, catalog number: R8875-1G)
- Rotenone Stock Solution (see Recipes)
- Rotenone 2x and 1x solutions (see Recipes)
- DMSO Vehicle Control 2x and 1x solutions (see Recipes)
Equipment
- CO2 tank
- CO2 single-stage flowmeter regulator (Western Medica, model: M1-320-12FM)
- CO2 gas anesthetizing Box (Braintree Scientific, model: AB-2)
- Motic Dissection microscope (Motic, model: SMZ-168)
- Roboz surgical instrument dissection tools
- Dumont #5 Forceps, Straight tip (Roboz, catalog number: RS-5060)
- Dumont #5 Forceps, 45 Degree tip (Roboz, catalog number: RS-5005)
- Straight, Sharp-Blunt Operating Scissors (Roboz, catalog number: RS-6812)
- Extra Fine Micro Dissecting Scissors (Roboz, catalog number: RS-5882)
- Micro Dissecting Spring Scissors (Roboz, catalog number: RS-5603)
- Sorvall Legend T table-top centrifuge (Thermo, model: 75004367)
- Nuaire Class II Type A2 Laminar-flow sterile culture hood with vacuum waste system (Nuaire, model: NU-425-400)
- Leica Fiberlight light source (Leica, model: EEG2823)
- Olympus light microscope (Olympus, model: CKX31)
- HeraCell CO2- and Temperature-Controlled culture incubator (Thermo, model: 51022394)
- Isotemp 110 Heated Water Bath (Fisher Scientific, model: 15-460-10)
- Wheaton Glass Slide Staining Jar (Fisher Scientific, model: 22-309-247)
- Disposable transfer pipettes (Fisher Scientific, catalog number: 13-711-20)
- Rocking Shaker Plate (Labnet, model: S-2035)
- Heated Stir Plate (Corning, model: PC-420)
- Accumet basic pH meter (Fisher Scientific)
- Vortex Mixer (Labnet, model: VX100)
- Scale (Denver Instruments, model: APX-100)
- Millipore Milli-Q Ultrapure Water System
- Assorted mechanical pipettes and corresponding sterile tips
- Hemocytometer and Cell Counter
- Thermometer
- Autoclave
- Chemical Fume Hood
- 4 °C refrigerator
- -20 °C freezer
- -80 °C freezer
Procedure
- Prepare in advance of dissection and plating
- Prepare Borate Buffer solution, the Dissociation Media and the HBSS Dissection Media according to the Recipes below.
- Prepare the necessary amounts of Seeding Media and Feeding Media according to the Recipes below. The amount prepared will depend on the size of the planned experiment.
- Each full neuronal preparation needs approximately 25 ml of Seeding Media, and each device needs approximately 5 ml of Feeding Media for plating and maintenance.
- Prepared media can be kept for up to 10 days, and could be used for subsequent neuronal preparations. It is therefore reasonable to make enough media for subsequent uses.
- Prepare 2-days before dissection and plating
- Thaw Poly-D-Lysine
- Pull the necessary number of aliquots of 2.5 mg/ml Poly-D-Lysine Stock Solution (400 µl) from -80 °C.
- Place on ice, and thaw overnight at 4 °C.
- Acid-wash and sterilize coverslips
- In a sterile culture hood, place the required number of coverslips (1 per device to be plated, plus 2 extras) into a glass slide staining jar.
- Fill with 50 ml of 1 N HCl (prepared in sterile cell culture water).
- Place the cover on the staining jar, and place the jar on a shaker plate outside the hood. Agitate the coverslips gently on a shaker plate for 1 h.
- Return the jar to the sterile culture hood and remove the HCl from the jar via pipette or aspiration.
- Add sterile cell culture water to the jar, cover, and place the jar on a shaker plate outside the hood. Repeat this water rinse 3 x 10 min.
- Remove final water wash via pipette or aspiration, and add 50 ml of 70% ethanol solution to sterilize coverslips. Cover the jar and agitate gently on a shaker plate for 30 min.
- Remove ethanol wash via pipette or aspiration, and add 50 ml of sterile culture water to the coverslips. Cover the jar and agitate gently on a shaker plate for 10 min.
- In the sterile culture hood, remove coverslips from the staining jar, and rest in an open 10 cm Petri dish to air dry. Prop the edges of the coverslips up on the sides of the petri dish–do not let the coverslips lay flat.
- Once dry, sterilize the coverslips by exposing them to the ultraviolet (UV) light of the sterile culture hood for 20 min.
- After UV exposure, place the coverslips inside the Petri dish and cover with the lid. Store overnight in a closed container or in the sterile culture hood without the UV light on.
- Thaw Poly-D-Lysine
- Prepare 1-day before dissection and plating
- Coat coverslips with Poly-D-Lysine
- Carry this procedure out in a sterile culture hood.
- Arrange coverslips, so they are lying flat, not overlapping, on the bottom of a 10 cm Petri dish.
- Prepare 6 ml of 100 µg/ml Poly-D-Lysine Coating Solution per 10 cm dish by adding the 2.5 mg/ml Poly-D-Lysine Stock Solution to sterile 0.1 M Borate Buffer in the ratio of 400 µl, 2.5 mg/ml Poly-D-Lysine stock to 9.6 ml, 0.1 M Borate Buffer.
- Mix and sterile filter the 100 µg/ml Poly-D-Lysine Coating Solution.
- Cover the coverslips with the 100 µg/ml Poly-D-Lysine Coating Solution, place the lid on the Petri dish, and allow the solution to coat coverslips overnight at 4 °C.
- Sterilize microfluidic devices
- Working in a sterile culture hood, remove devices from packaging.
- Place devices groove-side up (i.e., upside-down) in a sterile 10 cm Petri dish.
- Submerse the devices in 100% ethanol for 30 min.
- Remove ethanol, and submerse devices in 70% ethanol for 30 min.
- Remove devices from ethanol, and submerse in a dish of sterile cell culture water.
- Set devices at an angle on the edge of a sterile Petri dish and allow to fully dry in the sterile culture hood.
- Place in the Petri dish and cover. Store overnight in a closed container or in the sterile culture hood in the dark, without either UV light or the fluorescent light on.
- Do not expose the devices to UV light at any time.
- Coat coverslips with Poly-D-Lysine
- Day of dissection
- Prepare papain solution and inhibitor solutions according to the Recipes below.
- Place Feeding Media, Seeding Media, the Papain Digestion Solution, and the papain inhibitor solutions in a 37 °C water bath.
- Place HBSS Dissection Solution on ice. Aliquot approximately 150 ml HBSS to be used at the bench, and 25 ml HBSS to remain sterile for use in the hood.
- Rinse and dry coverslips
- Working in a sterile culture hood, use sterile forceps to remove coverslips from the Poly-D-Lysine solution and place angled on the edge of a sterile Petri dish to dry.
- Once dry, dip each coverslip 3 x in sterile culture water to rinse, allowing water to drip off between dips.
- Dip the coverslip in sterile 1x PBS, let solution drip off, and return to the Petri dish, propped up against the edge of the dish and allowed to dry fully. Do not expose to UV light.
- Attach microfluidic devices to coverslips and prime with Feeding Media–see Figure 1 for device placement and orientation
- Carry this procedure out in a sterile culture hood.
- Using sterile forceps, place a single Poly-D-Lysine-coated coverslip onto the bottom of a sterile 6 cm Petri dish, making sure to have the Poly-D-Lysine-coated side up.
- Using a second set of sterile forceps, lift a microfluidic device and place it, microgroove-side-down, onto the center of the coverslip. (Figure 1A)
- Use the blunt end of the forceps to gently press the device onto the coverslip, making sure to gently press out any air bubbles to ensure a tight seal.
- Place the lid on the Petri dish to maintain sterility, and examine the device assembly under a light microscope to ensure a tight seal around the microgrooves and feeding reservoirs.
- Return to the sterile culture hood and prime the device with warmed Feeding Media as follows (Figures 1B and 1C):
- Add 100 µl of warm Feeding Media to the top-left reservoir. Watch to make sure the media enters and fills the channel connecting the two left reservoirs.
- Wait 2 min to ensure the media has filled the axonal channel and the microgrooves.
- Next, add 100 µl of warm media to the top-right reservoir.
- Wait 2 min to ensure the media has filled the somal channel.
- Add 100 µl of warm media each to the bottom-left and bottom-right reservoirs.
- Place the lid on the Petri dish holding the device, and place in a cell culture incubator (37 °C, 5% CO2) for at least 1 h before plating neurons.
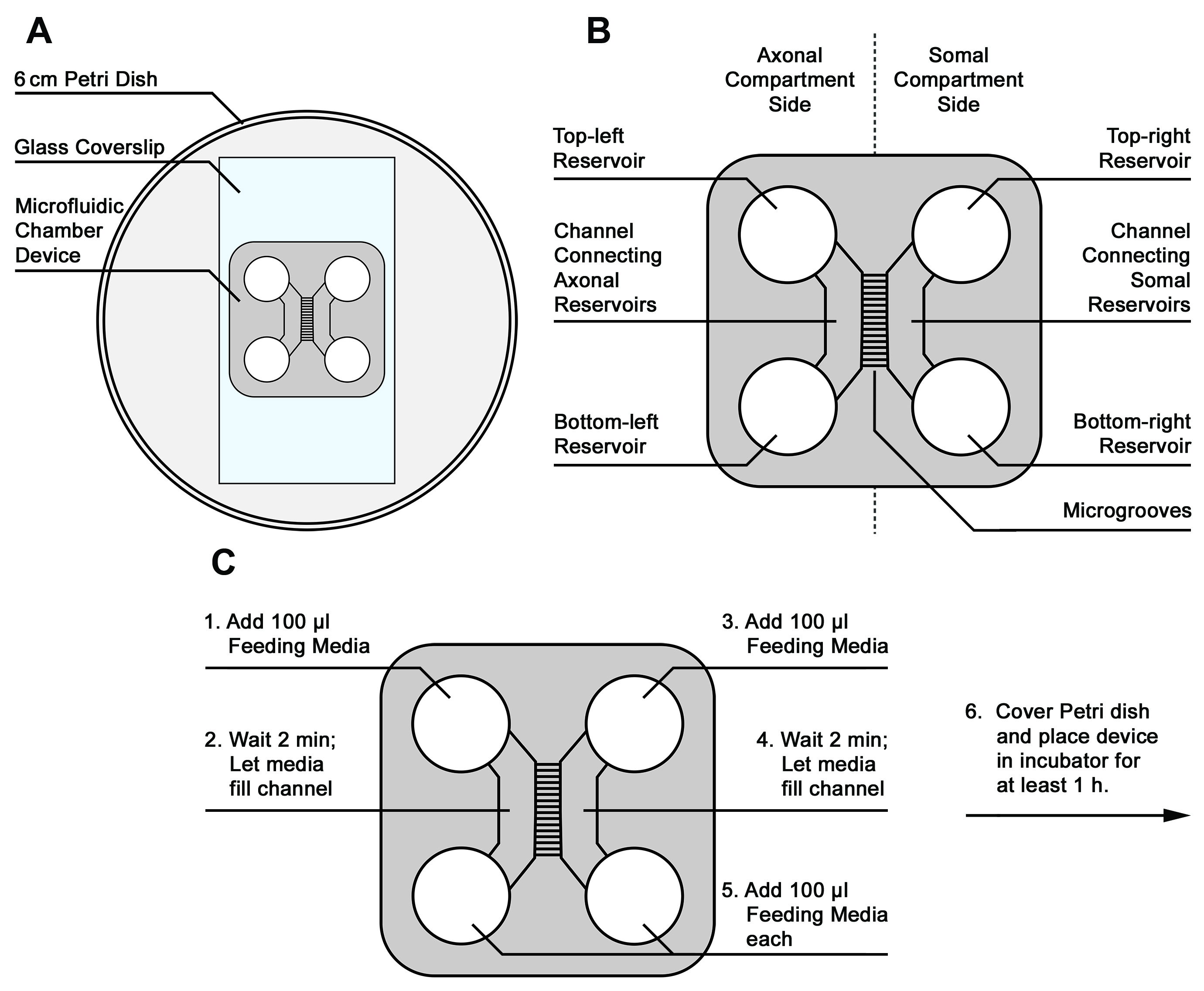
Figure 1. Schematic of microfluidic device mounting, orientation, and priming with culture media. A. Placement of microfluidic device on coverslip. B. Device features and orientation. C. Order of media addition to prime device. - Primary cortical neuron dissection and cell preparation
At the bench- An E18 pregnant female rat is humanely sacrificed and embryonic rat primary cortices dissected (meninges removed) per established lab methods and according to an approved animal protocol adhering to the requirements of the University of Pittsburgh Department of Laboratory Animal Research (DLAR) and Institutional Animal Care and Use Committee (IACUC). These requirements will vary between institutions. Our methods were adapted from previous work as described by Ghosh and Greenberg (1995), and as previously described by our lab (Arnold et al., 2011). Cell yield varies and is approximately 0.5-1.0 x 106 cells per embryo.
- Gently transfer all cortices to a 15 ml conical tube on ice containing 9 ml HBSS dissection media.
- After the dissections are complete, move to a sterile culture hood to continue the procedure.
- Gently remove HBSS from cortices using a transfer pipette and discard HBSS.
- Gently wash cortices 3 x using 5 ml cold HBSS for each wash. After each wash, gently remove HBSS from cortices using a transfer pipette and discard.
- After the last HBSS wash, promptly add 5 ml of pre-warmed Enzyme Solution to the cortices and incubate at 37 °C for 20 min.
- Remove Enzyme Solution using a transfer pipette and immediately add the pre-warmed Light Inhibitor Solution (10 ml) to cortices for 1 min.
- Remove the Light Inhibitor Solution using a transfer pipette and immediately add the Heavy Inhibitor Solution (5 ml) to cortices for 2 min at 37 °C.
- Remove the Heavy Inhibitor Solution using a transfer pipette and gently wash the cortices 3 x using 5 ml of 37 °C Seeding Media for each wash. After each wash, gently remove media from cortices using a transfer pipette and discard.
- Add fresh 5 ml Seeding Media to the cortices and use a P1000 (1 ml) mechanical pipette and triturate by pipetting up-and-down approximately 20 times to break up the tissue and cell clumps into a homogeneous cell suspension. You may need to triturate for longer (5-10 more times) if the cells are still in clumps, but do not triturate excessively. The cell suspension should look cloudy and there should be no obvious large particles or cell clumps remaining. Alternatively, the cells can be run through a 40 µm sterile cell strainer sieve after the 20-times trituration.
- Count cells
- Combine a 10 µl aliquot of suspended cells with 90 µl of Seeding Media.
- Use hemocytometer to count cells and calculate cells/ml in suspension.
- Plating microfluidic devices
Primary cortical neurons are plated at 50,000 cells per device for standard 2-compartment microfluidic neuron devices with 450 µm- or 900 µm-width microgroove barriers (Xona microfluidics). To help ensure that the plating doesn’t result in cells being forced through the microgrooves to the axonal, non-cell side of the device, proceed as follows (Figure 2):- Adjust cell suspension to 2.5 x 106 cells/ml in Seeding Media. Prepare enough for 20 µl of cell solution per device to be plated. Set aside.
- Remove the Petri dishes holding mounted microfluidic devices from the incubator, and place them in the sterile culture hood.
- Remove all of the media used to prime the device from all reservoirs.
- Add 100 µl of Feeding Media to the top-left reservoir (axonal side) of each device.
- Add 100 µl of Feeding Media to the connected bottom-left reservoir (axonal side). Wait 1 min for media to equilibrate on the axonal side of the device. (see Notes)
- On the somal side of the device, add 10 µl of cell suspension to the top-right reservoir (somal side).
Note: Place the 10 µl cell suspension at the junction of where the reservoir joins the channel (see Figure 2). - Cover the Petri dish holding the device, and examine under a light microscope to ensure that cells are entering the channel between the top-right and bottom-right reservoirs. (see Notes)
- After 1 min, Add another 10 µl of cell suspension to the connected bottom-right reservoir (somal side).
- Place the lid on the Petri dish holding the device and return it to the incubator. Allow 10 min for the cells to attach.
- After 10 min, remove the device from the incubator and return to the sterile culture hood.
- Gently add an extra 100 µl of Feeding Media to the top-left reservoir (axonal side).
- Gently add an extra 100 µl of Feeding Media to the connected bottom-left reservoir (axonal side).
- Immediately after Step E12, gently add 200 µl of Feeding Media to the cell-side top-right reservoir (somal side).
- Immediately following, gently add 200 µl of Feeding Media to the connected bottom-right reservoir (somal side).
Notes:- Be slow and gentle when adding media; adding media forcefully or too fast will wash the cells out of the channel.
- Now each reservoir on the left has 200 µl of Feeding Media, and each reservoir on the right has 210 µl of Feeding Media.
- Place the lid on the 6 cm Petri dish holding the device.
- Place the covered 6 cm Petri dishes holding devices into a 10 cm Petri dish. Up to three 6 cm dishes will fit into one 10 cm dish. Place the lid on the 10 cm Petri dish and return devices to the incubator. This dish-within-a-dish system is used to help reduce evaporation and maintain media levels in the devices during the culture period.
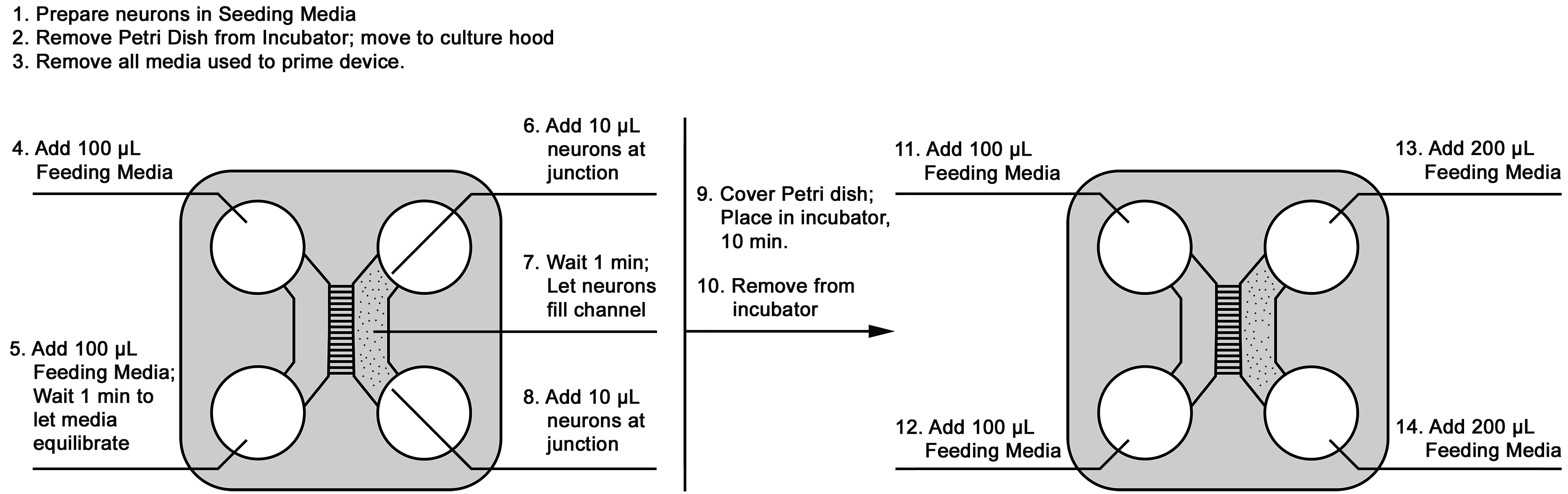
Figure 2. Schematic of seeding neurons into the microfluidic device
- Feeding maintenance and rotenone treatment
- Maintain cells
- After plating, add 10 µl of Feeding Media to each of the four reservoirs on each device every 3-4 days to maintain media levels. Add warmed media to devices in the sterile culture hood.
- No media changes occur until the initial treatment.
- Chronic rotenone and DMSO vehicle treatment
On day in vitro 7 (DIV7), add the initial treatment with rotenone or DMSO vehicle control, via a ½ media change in both the axonal side and somal side reservoirs as follows:- Prepare a 2x concentration of the desired final rotenone concentration in warm Feeding Media (see Recipes for solution preparation).
- In parallel, prepare warm Feeding Media with a corresponding 2x concentration of DMSO as a vehicle control (see Recipes for solution preparation).
- Remove the Petri dish holding microfluidic devices from the incubator.
- In the sterile culture hood, remove 100 µl of media from each of the four reservoirs on each device.
- For each device, add 100 µl of the 2x rotenone or 2x DMSO solution to each of the four reservoirs.
- The final concentration will be 1x rotenone or 1x DMSO throughout the device.
- Treatments continue for 1-2 weeks with ½ media changes every 3-4 d using 1x concentration media.
- Prepare a 1x concentration of the desired final rotenone concentration and a 1x concentration of DMSO vehicle control each in warm Feeding Media (see Recipes for solution preparation).
- Repeat the ½ media change for each reservoir as described above.
- Maintain cells
- Fixing neurons (Figure 3)
- At the end of the experiment, remove media from all reservoirs.
- Immediately add 100 µl of warm 1x PBS to the top-left and top-right reservoirs.
- Allow PBS to flow to bottom reservoirs for 1 min.
- Immediately remove all PBS.
- Add 100 µl of warm 4% PFA into each top reservoir.
- Allow cells to fix for 20 min at room temperature.
- Remove all PFA, and dispose of properly.
- Add 200 µl of PBS to each top reservoir.
- Rinse for 5 min.
- Remove PBS rinse.
- Repeat PBS rinse 2 x.
- Fill each reservoir with PBS.
- Cover device, and store at 4 °C until use for further analyses.
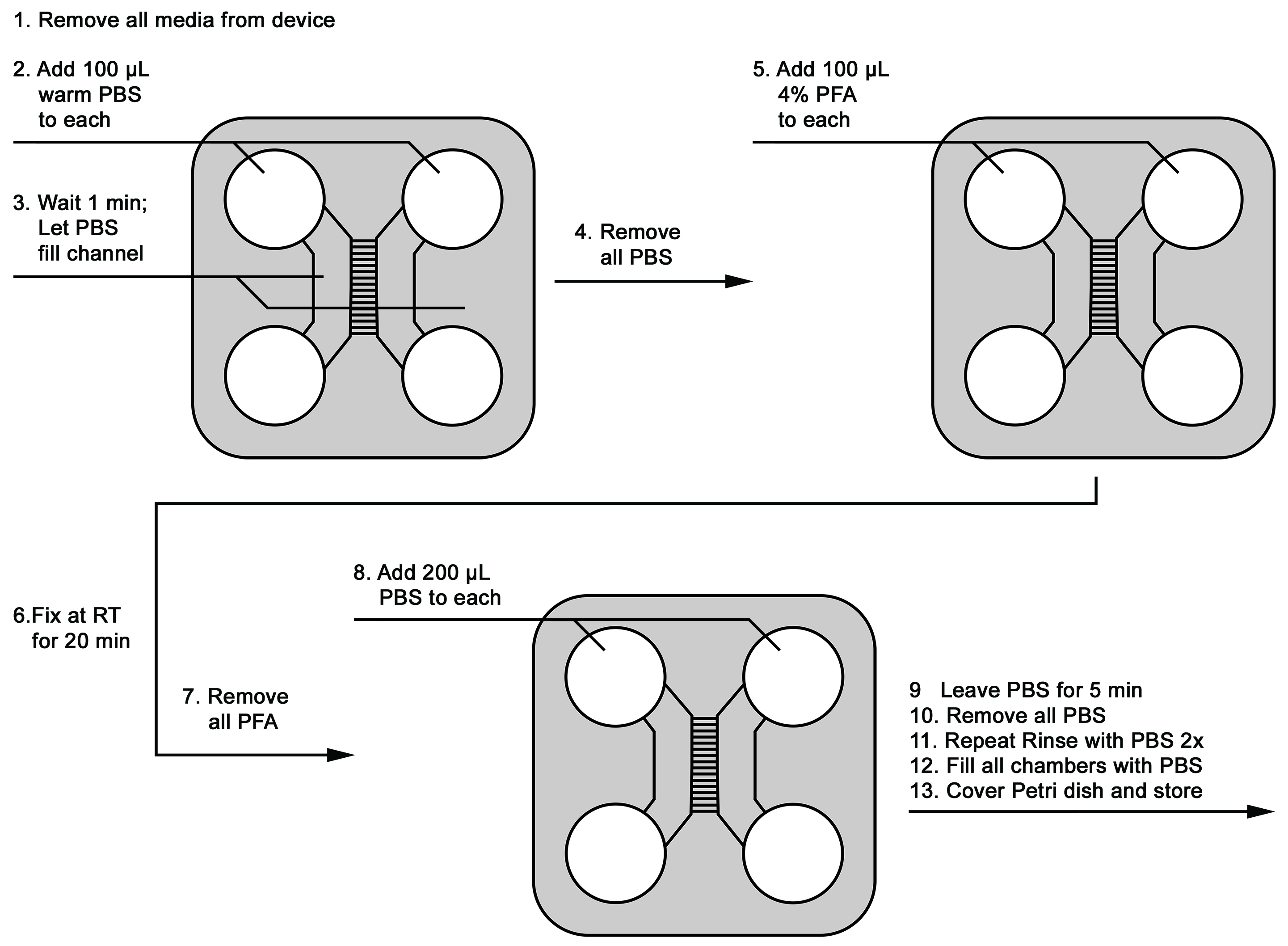
Figure 3. Schematic of fixing neurons in the microfluidic device - Reusing microfluidic devices
In the event that a culture dies before experimental treatments begin, the device was not appropriately mounted and cells entered the non-cell-body side of the device, or the culture is otherwise unusable for experiments, we have found that the device can be reused successfully. Do not reuse devices that have been exposed to experimental treatments, such as drugs or toxicants (see Notes). Starting from a mounted microfluidic device with an active culture, follow the following steps for device reuse:- Remove the media from the device reservoirs using a pipette.
- Place a finger on the corner of the coverslip, then use forceps to gently grip the edge of the device at a reservoir and gently pull up at an angle until the device is free from the coverslip.
- Rinse the device gently with distilled water, then place groove-side-up in a Petri dish.
- Submerge the device in 1x RBS detergent solution. Cover the dish, and gently agitate on a shaker overnight at room temperature.
- Remove the RBS detergent and wash the device using culture grade water 3 x for 1 h each, with gently agitation on a shaker at room temperature.
- After the final wash, with the device still submerged in water, examine the device under a light microscope to verify that all cellular debris has been removed from the reservoirs and grooves.
- Remove the final wash, and carry out the remaining procedure in a sterile culture hood.
- Transfer the device, groove side up, to a sterile Petri dish, then submerge the device in sterile culture grade water. Cover the dish, and gently agitate on a shaker overnight at room temperature.
- In the sterile culture hood, remove the device from the sterile wash and either air dry in a sterile culture dish to store for future use, or carry out the sterilization procedure as listed above under Step C2 for immediate use.
Data analysis
Prior to use for experiments, data acquisition, and data analyses, carefully examine the devices and culture quality. Device cultures in which the cells appear in obvious distress prior to treatments should not be used for experiments or further analyses. Despite the design of the devices being such that cells should not be able to cross the microchannels, some cells occasionally do cross the microchannels. Glia in particular are able to readily cross. An occasional neuronal soma is not a concern. If after a week of culture you notice there are large populations of neuron somata in the axonal side, discontinue use of this device culture. If after your experiment you notice many somata on the axonal side, do not use this device culture for data analyses. Large numbers of somata on the axonal side may indicate there were issues with plating (see Notes) or that the device was not properly attached to the coverslip.
Notes
Plating neurons in microfluidic devices (Procedure E)
- The plating of the cells is a crucial step to maintaining the isolation between soma and axons. Be sure that the reservoirs and channel on the axonal compartment side of the device are filled with Feeding Media 1-2 min before adding cells to the reservoirs on the somal compartment side of the device. If the axonal compartment is empty when cells are added, the fluid dynamics will force the detached cells through the microchannels and into the axonal channel space. If this occurs, the device and its neurons cannot be used for further experimentation as the axons will no longer be an isolated population. (Steps E2-E3)
- In Steps E4-E6, when adding the cells to the somal side of the device, it is advised to always check that cells are entering the somal compartment side channel via microscope. Fluid dynamics should pull the cells into the channel. If you do not observe this, return to the sterile culture hood and try gently removing the cells with a pipette. Make sure all media is removed from both top-right and bottom-right reservoirs, and again try placing the cells in the top-right reservoir where the reservoir joins the channel.
- Only reuse devices that have not been exposed to any exogenous treatment not typically found in culture media, such as drugs, toxicants (e.g., rotenone), or tissue fixative such as PFA. If the device has already been used during a treatment experiment or if the cells and device were already exposed to fixative, the device cannot be reused. Thus, any device in which an experiment was completed cannot be reused for another experiment.
- This reuse method only applies to microfluidic devices that were pressed onto an extracellular-matrix coated coverslip. If the devices were bound to the coverslip using plasma bonding techniques (see the manufacturer’s instructions), they cannot be reused.
- In our studies, we found that cultures from new devices and reused devices did not exhibit any discernable differences. Further, we observed no differences in the results obtained from experiments completed in new devices and experiments completed in reused devices.
Recipes
- 0.1 M Borate Buffer Solution
- 1.24 g boric acid and 1.002 g anhydrous borax in 400 ml cell culture grade sterile H2O
- Cover and stir on a stir plate overnight at RT
- Adjust pH to 8.4
- In sterile culture hood, filter sterilize into autoclaved media bottle
- Store at 4 °C for up to 3 months
- Poly-D-Lysine 2.5 mg/ml Stock Solution
- Thaw frozen lyophilized powder overnight at 4 °C on ice
- Prepare the stock in a sterile culture hood
- Add 20 ml of 0.1 M sterile Borate Buffer solution to 50 mg Poly-D-Lysine
- In sterile culture hood, filter sterilize the stock solution using a 10 ml syringes and 0.22 µm PES syringe filter
- Aliquot 400 µl stocks into sterile microfuge tubes
- Store at -80 °C
- Poly-D-Lysine 100 µg/ml Coating Solution
- Thaw an aliquot of Poly-D-Lysine Stock Solution (400 µl, 2.5 mg/ml) overnight at 4 °C on ice
- In sterile culture hood, add to 9.5 ml sterile 0.1 M Borate Buffer
- Final concentration: 100 µg/ml Poly-D-Lysine
- Cortical Neuron Media for Seeding
Note: Prepare all the solutions in a sterile culture hood.Reagent % vol/vol For 500 ml Neurobasal Media 91.5% 457.5 ml B27 supplement 2% 10 ml Glutamax 100x 1% 5 ml Pen/Strep 0.5% 2.5 ml FBS 5% 25 ml - Combine all the reagents in a sterile culture hood, filter sterilize into an autoclaved media bottle
- Store at 4 °C for up to 10 days
- Cortical Neuron Media for Feeding
Note: Prepare all the solutions in a sterile culture hood.Reagent % vol/vol For 500 ml Neurobasal Media 96.5% 457.5 ml B27 supplement 2% 10 ml Glutamax 100x 1% 5 ml Pen/Strep 0.5% 2.5 ml - Combine reagents in a sterile culture hood, filter sterilize into an autoclaved media bottle
- Store at 4 °C for up to 10 days
- HBSS Dissection Media
Bring to 1 L with cell culture grade sterile H2OReagent Amt. Final Conc. 10x HBSS 100 ml 1x NaHCO3 350 mg 4.2 mM HEPES, 1 M 10 ml 10 mM Glucose 4.3 g 35 mM
Notes:
- Do not adjust pH.
- Prepare all the solutions in a sterile culture hood.
- Combine reagents in a sterile culture hood, filter sterilize into an autoclaved media bottle.
- Store at 4 °C for up to 3 months
- Dissociation Media
Bring to 250 ml with cell culture grade sterile H2OReagent Amt. Na2SO4, 1 M 20.44 ml K2SO4, 0.5 M 15 ml MgCl2, 1 M 1.45 ml CaCl2, 100 mM 0.625 ml HEPES, 1 M 250 µl Glucose, 1 M 5 ml Phenol Red, 0.5% 0.5 ml NaOH, 0.1 N 0.5 ml
Notes:
- Do not adjust pH.
- Prepare all the solutions in a sterile culture hood.
- Combine reagents in a sterile culture hood, filter sterilize into an autoclaved media bottle
- Store at 4 °C for up to 3 months
- L-Cysteine 50x Stock
- Weigh out 160 mg, then prepare in sterile culture hood
- Dissolve in 10 ml of sterile Dissociation media
- Aliquot in sterile microcentrifuge tubes at 225 µl and store at -80 °C
- Papain Dissociation Solution and Inhibitor Solutions for Isolation of Primary Rat Cortical Neurons
Notes:
- Make these solutions fresh on the day of use.
- Begin by aliquoting dissociation media into sterile 15 ml conical tubes in the sterile culture hood for each solution below.
- Prepare solutions at the bench with pH meter.
- After solutions are prepared, return to the sterile culture hood.
- For each solution, use a 10 ml sterile syringe and a syringe filter to sterile-filter the solution into a new, sterile 15 ml conical tube.
- Following preparation and final pH adjustment, filter-sterilize all solutions into new, sterile 15 ml conical vials under the sterile culture hood using 10 ml syringes and 0.22 µm PES syringe filters.
- Enzyme Solution
10 ml Dissociation Media
0.2 ml of 50x Cysteine-HCl
200 U of Papain Solution
Pulse on a vortex mixer
Adjust pH to 7.4 with 0.1 N NaOH - Heavy Inhibitor Solution
6 ml Dissociation Media
Add 60 mg BSA (4 °C) to 6 ml of DM
60 mg of Soybean Trypsin Inhibitor (4 °C)
Pulse on a vortex mixer, then place on a shaker to mix until fully in solution
Adjust pH to 7.4 with 0.1 N NaOH - Light Inhibitor Solution
9 ml Dissociation Media
Add 1 ml of Heavy Inhibitor Solution
Pulse on a vortex mixer
Adjust pH to 7.4 with 0.1 N NaOH
- Rotenone Stock Solution
- Use fresh rotenone and DMSO to prepare stocks
- Prepare 5 mM rotenone in DMSO
- Aliquot into 100 µl aliquots and store at -20 °C
- Prepare enough stocks for all the experiments
- Rotenone 2x and 1x solutions
- Make fresh on the day of use, immediately before use
- Prepare in sterile culture hood
- Thaw an aliquot of 5 mM stock rotenone at RT protected from light
- Dilute directly into neuron Feeding Media to 2x of the final desired treatment concentration for the first treatment, and to 1x for subsequent treatments
- DMSO Vehicle Control 2x and 1x solutions
- Make fresh on the day of use, immediately before use
- Prepare in sterile culture hood
- Dilute DMSO directly into neuron Feeding Media to 2x of the final desired treatment concentration for the first treatment, and to 1x for subsequent treatments
- Vehicle control media should be prepared to have the same final concentration of DMSO as in rotenone-treatment media
- 4% PFA Solution
To prepare 2 L of 4% PFA
200 ml of 10x PBS
80 g Paraformaldehyde
Milli-Q Ultrapure Water to 2 L- Prepare in a chemical fume hood
- Add 1 L of ultrapure water to a 2 L flask with large stir bar
- Add 112.5 µl of 10 N NaOH
Notes:
i.Paraformaldehyde solubilizes in base better than in acid.
ii.Leave out PBS at this stage to eliminate buffering. - Suspend a thermometer in solution and place flask on a heated stir plate and stir
- Bring the solution to just below 60 °C and continue to stir until PFA is solubilized
Note: Monitor closely; do not let the paraformaldehyde solution go above 60 °C. - When PFA is solubilized solution pH to 7.4 with pH paper
Note: Do not use pH meter. Adjust pH as necessary with HCl and NaOH. - Add 200 ml of 10x PBS
- Bring the solution to 2 L with ultrapure water
- Recheck pH with pH paper to confirm that the final pH is 7.4
- Filter sterilize and prepare 40 ml aliquots in 50 ml conical tubes
- Store at -20 °C until use
Acknowledgments
This work was supported by a grant from the NIH (R01NS077954, S.B.B.) and a University of Pittsburgh Physicians Academic Foundation Research Grant (S.B.B).
The microfluidic device plating and culture methods were adapted from previous work as described by Park et al. (2006).
The chronic, low-dose rotenone exposure treatment methods were modified from our previous work as described in Arnold et al. (2011).
Competing interests
The authors have no conflicts of interest to disclose.
Ethics
All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Pittsburgh and are in accordance with guidelines put forth by the National Institutes of Health in the Guide for the Care and Use of Laboratory Animals. All euthanasia methods are consistent with the most updated American Veterinary Medical Association (AVMA) Guidelines for the Euthanasia of Animals.
References
- Arnold, B., Cassady, S. J., VanLaar, V. S. and Berman, S. B. (2011). Integrating multiple aspects of mitochondrial dynamics in neurons: age-related differences and dynamic changes in a chronic rotenone model. Neurobiol Dis 41(1): 189-200.
- Ghosh, A. and Greenberg, M. E. (1995). Calcium signaling in neurons: molecular mechanisms and cellular consequences. Science 268(5208): 239-247.
- Millet, L. J. and Gillette, M. U. (2012). Over a century of neuron culture: from the hanging drop to microfluidic devices. Yale J Biol Med 85(4): 501-521.
- Park, J. W., Vahidi, B., Taylor, A. M., Rhee, S. W. and Jeon, N. L. (2006). Microfluidic culture platform for neuroscience research. Nat Protoc 1(4): 2128-2136.
- Taylor, A. M., Blurton-Jones, M., Rhee, S. W., Cribbs, D. H., Cotman, C. W. and Jeon, N. L. (2005). A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat Methods 2(8): 599-605.
- Taylor, A. M., Menon, S. and Gupton, S. L. (2015). Passive microfluidic chamber for long-term imaging of axon guidance in response to soluble gradients. Lab Chip 15(13): 2781-2789.
- Taylor, A. M., Rhee, S. W., Tu, C. H., Cribbs, D. H., Cotman, C. W. and Jeon, N. L. (2003). Microfluidic multicompartment device for neuroscience research. Langmuir 19(5): 1551-1556.
- Van Laar, V. S. and Berman, S. B. (2013). The interplay of neuronal mitochondrial dynamics and bioenergetics: implications for Parkinson's disease. Neurobiol Dis 51: 43-55.
- Van Laar, V. S., Arnold, B., Howlett, E. H., Calderon, M. J., St Croix, C. M., Greenamyre, J. T., Sanders, L. H. and Berman, S. B. (2018). Evidence for compartmentalized axonal mitochondrial biogenesis: mitochondrial DNA replication increases in distal axons as an early response to Parkinson's disease-relevant stress. J Neurosci 38(34): 7505-7515.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Van Laar, V. S., Arnold, B. and Berman, S. B. (2019). Primary Embryonic Rat Cortical Neuronal Culture and Chronic Rotenone Treatment in Microfluidic Culture Devices. Bio-protocol 9(6): e3192. DOI: 10.21769/BioProtoc.3192.
- Van Laar, V. S., Arnold, B., Howlett, E. H., Calderon, M. J., St Croix, C. M., Greenamyre, J. T., Sanders, L. H. and Berman, S. B. (2018). Evidence for compartmentalized axonal mitochondrial biogenesis: mitochondrial DNA replication increases in distal axons as an early response to Parkinson's disease-relevant stress. J Neurosci 38(34): 7505-7515.
Category
Neuroscience > Cellular mechanisms > Tissue isolation and culture
Cell Biology > Cell isolation and culture > Microfluidic culture
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.