- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Preparing Single-cell DNA Library Using Nextera for Detection of CNV
Published: Vol 9, Iss 4, Feb 20, 2019 DOI: 10.21769/BioProtoc.3175 Views: 6764
Reviewed by: Adler R. DillmanLorrayne SerraAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jul 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Single-cell DNA sequencing is a powerful tool to evaluate the state of heterogeneity of heterogeneous tissues like cancer in a quantitative manner that bulk sequencing can never achieve. DOP-PCR (Degenerate Oligonucleotide-Primed Polymerase Chain Reaction), MDA (Multiple Displacement Amplification), MALBAC (Multiple Annealing and Looping-Based Amplification Cycles), LIANTI (Linear Amplification via Transposon Insertion) and TnBC (Transposon Barcoded) have been the primary choices to prepare single-cell libraries. TnBC library prep method is a simple and versatile methodology, to detect copy number variations or to obtain the absolute copy numbers of genes per cell.
Keywords: Single-cellBackground
Bulk DNA sequencing, although being widely used nowadays, has been proven to be inadequate in analysis of heterogeneous systems, such as cancer tissues, which contain cancer cells of genetic aberrations at various degrees among normal cells with little or no genetic aberrations. Noncancerous cells can contribute a significant portion of the total DNA extracted from tumors, potentially masking important genetic aberrations (Alioto et al., 2015). Even when normal cells are removed, bulk sequencing of cancerous cells still averages out both the heterogeneity of cancerous cells in a tumor tissue and genomic instability over time (Yang et al., 2013; Francis et al., 2014). Single-cell DNA sequencing is believed to be the only method to reveal unequivocally the dynamics of mutations of tumor cell subpopulations in detail over space and time (Navin, 2015). Copy number variations (CNV) is under-detected in bulk sequencing, while they are found to be early events in tumorigenesis (Navin, 2015).
In the past years, several single-cell library preparation methods have been reported, which include DOP-PCR (Baslan and Hicks, 2014), MDA (Fan et al., 2011), MALBAC (Zong et al., 2012), LIANTI (Chen et al., 2017) and TnBC (Xi et al., 2017). Due to the fact that the minute amount of DNA from a single cell is not sufficient for NGS directly for most purposes, the single-cell genome needs to be amplified. Reflecting this requirement, almost all single-cell library preparation methodologies are named after an amplification method. As amplification is involved, the biases and errors associated with amplification inevitably need to be addressed (Xi, 2018). Biased amplification will require deeper sequencing to gain coverages. In an extreme case, under-amplified regions can be falsely identified as deletions. Amplification errors introduced by polymerases may overwhelm authentic mutations, which adds difficulty in mutation-calls. As TnBC methodology employs unique fragment index (UFI), it can handle amplification biases and polymerase-introduced errors better than other methods do (Xi et al., 2017; Xi, 2018).
An engineered Mu transposase was used in our original paper of TnBC library preparation (Xi et al., 2017). Since preparing custom-made transposases is technically demanding and time-consuming, here we report a protocol that utilizes Nextera, a commercially available Tn5 transposase. This protocol is intended to obtain single cell libraries that will be good for CNV detection through shallow sequencing. Due to the proof-reading DNA polymerase that is used in our library amplification, the error rate can be significantly smaller than that from DOP-PCR, MALBAC, or LIANTI. Therefore, the aggregate of sequences from multiple single cells can be used to detect global SNV of the source tissue of the single cells (Knouse et al., 2016).
Materials and Reagents
- Pipette tips
- 96-well plate
- Read1: TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG (suggested source: custom order from IDT)
- Read2: GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG (suggested source: custom order from IDT)
- TBE buffer: Make 1x from 10x TBE (suggested source: Tris-borate-EDTA Buffer from Fisher Scientific, catalog number: B52)
- Nextera DNA Library Preparation Kit (24 samples) (suggested source: Illumina, catalog number: FC-121-1030)
- Nextera Index Kit (96 Indices) (suggested source: Illumina, catalog number: FC-121-1012)
- Phusion Hot Start II High-Fidelity PCR Master Mix (Thermo Fisher, catalog number: F565L)
- UltraPureTM 0.5 M EDTA, pH 8.0 (suggested source: Thermo Fisher, catalog number: 15575020)
Note: Make 10 fold dilution as working stock (100 mM). - 20x EvaGreen (Biotium, catalog number: 31000)
- Agencourt AMPure XP (Beckmen Courtier, catalog number: A63882)
- Proteinase K, Molecular Biology Grade (New England Biolabs, catalog number: P81075)
- Chymostatin (5 mg) (Sigma-Aldrich, catalog number: C7268-5MG)
Note: Dissolve in 0.83 ml of DMSO to make 6 mg/ml stock solution. - Double-distilled H2O (ddH2O)
- DMSO (Dimethyl sulfoxide)
- Ethanol, freshly make 75% ethanol for Agencourt AMPure XP bead washes
- Tris-HCl, 1 M, pH 8 (suggested source: Thermo Fisher, catalog number: AM9856)
- MgCl2, 1 M (suggested source: Thermo Fisher, catalog number: AM9530G), or equivalent
- DMF (Dimethylformamide)
- CLOROX bleach
- 5x Transposase buffer (see Recipes)
Equipment
- Pipettes
- DNA-free hood
- VeritiTM 96-Well Thermal Cycler (Thermo Fisher, catalog number: 4375786)
- QuantStudio 3 Real-Time PCR System (Thermo Fisher)
- DynaMagTM-96 Side Magnet (Thermo Fisher, catalog number: 12331D)
- Agilent BioAnalyzer
Procedure
- This library prep procedure starts with a 96-well plate with each well containing one cell in 5 μl TBE or ddH2O. (Single cells can be dispensed with a cell sorter according to manuals of manufacturers or by manual means [Knouse et al., 2017]). The overall workflow is shown in the table below:
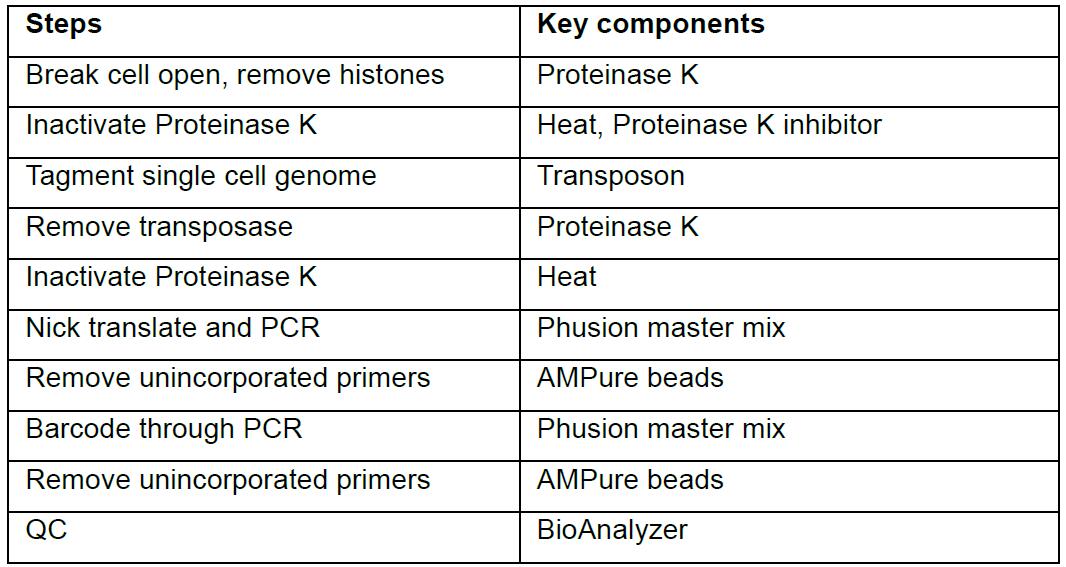
- To each of the well, add 3 μl solution that contains 1 μl of 5x Transposase buffer, 0.5 μl of Proteinase K (NEB), and 1.5 μl of H2O. Spin down to make sure that the protease solution enters cell suspension. Incubate the suspension at 55 °C for 5 h to break the cell open and remove histone proteins, and then 65 °C for 20 min to denature Proteinase K.
- Add 1 μl of 1.5 mg/ml Chymostatin to each well, spin down, incubate at room temperature for 10 min to inactivate remaining Proteinase K activity.
- Add 1.5 μl solution that contains 1 μl of 5x Transposase buffer and 0.5 μl of Tagment DNA Enzyme (TDE) from Illumina’s Nextera DNA Library Preparation Kit. Spin down.
- Keep the tagmentation reaction at 55 °C for 20 min.
- Add 1 μl solution that contains 0.5 μl of 100 mM EDTA and 0.5 μl of Proteinase K. Vortex and spin down. Incubate at 37 °C for 1 h and then 65 °C for 20 min.
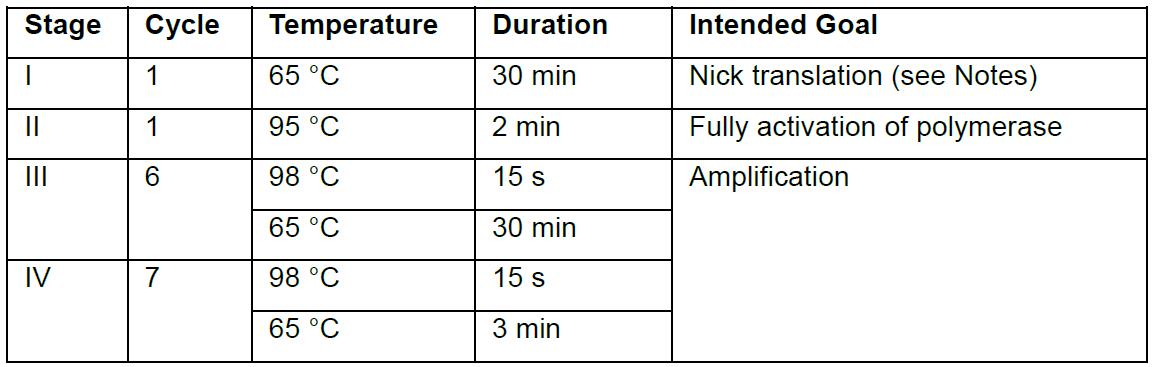
- To each well, add 12.5 μl of 2x Hot-start Phusion Master Mix and 1.25 μl of 100 μM each of Read1 and Read2. Mix well, and run the following PCR protocols: 65 °C for 30 min and 95 °C for 2 min followed by 6 cycles between 15 s at 98 °C and 30 min at 65 °C, plus 7 cycles between 15 s at 98 °C and 3 min at 65 °C. The thermal cycling protocol is summarized in the table below:
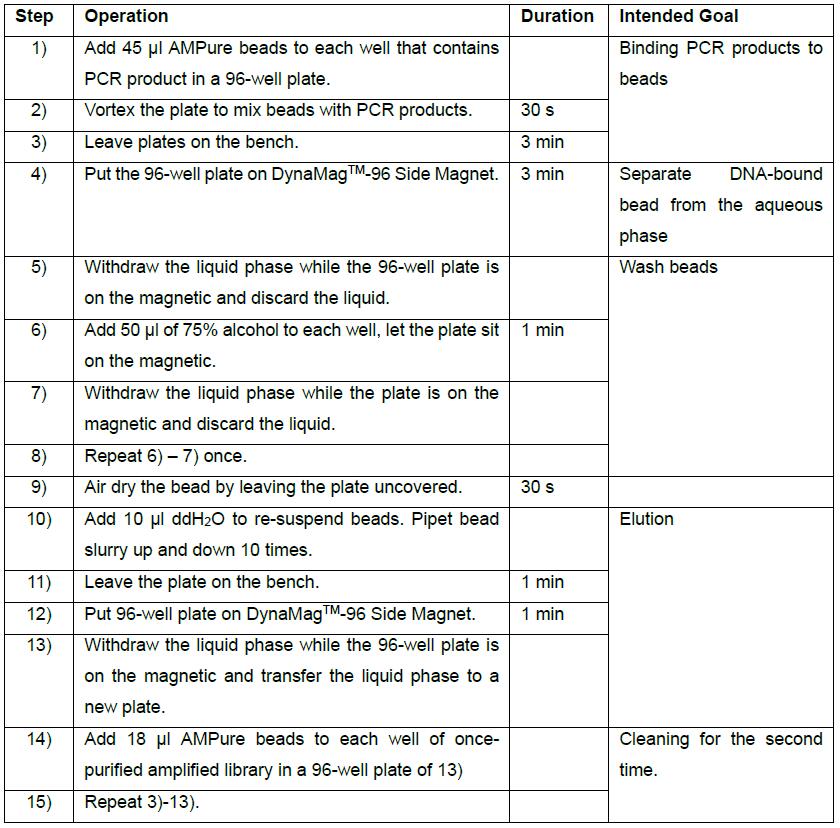
- After PCR, add 45 μl AMPure beads to purify the PCR product according to the manufacturer’s instruction. Elute in 10 μl ddH2O and purify with 18 μl of AMPure beads, and then elute in 10 μl ddH2O. Detailed steps are listed in the table below:
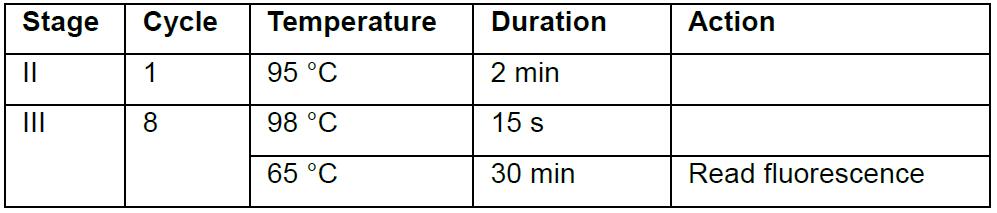
- Use 2.5 μl from last step in 20 μl sample-barcoding PCR which contains 1x Hot-start Phusion Master Mix, 1x EvaGreen, 1 μM each of 5xx and 7xx barcodes for Illumina sequencing, one unique pair for each cell. Amplification is carried out by heating at 95 °C for 2 min, then followed by 8 cycles between 15 s at 98 °C and 3 min at 65 °C, Reactions is optionally monitored at 65 °C. The thermal cycling protocol is summarized in the table below:
- Use 36 μl of AMPure beads to purify barcoded product according to manufacturer’s instruction, and elute in 10 μl ddH2O.
- Take 1 μl for analyses on BioAnalyzer.
- If both the profile and the yield meet the requirements (see Notes), it is ready for sequencing using Illumina’s system.
Notes
- It is critical to avoid contamination and cross-contamination for single-cell operations (Knouse et al., 2017). We use DNA-free hood when pipetting, and eject and submerge pipette tips immediately into about 1% bleach water to minimize the chance of cross-contamination.
- Saturated transposition (Step 5) is critical for the procedure (Xi et al., 2017). Saturated transpositions minimize sequence bias of transposase.
- Nick translation (Step 7) is necessary to generate “tags” that the libraries can be amplified. The polymerase activity from Hot-start Phusion Master Mix is leaky enough at 65 °C to accomplish the task based on the instruction of the manufacturer.
- We use EvaGreen to monitor the amplification of every single-cell library in barcoding reactions (Step 9). A significant increase in fluorescence starts at Cycle 4.
- We analyze every single cell library on BioAnalyzer (Step 11). We use the profiles to evaluate our procedures. First, the concentration of the library reflects (1) the quantity of starting DNA and (2) amplification efficiency. Starting from 6 pg of DNA, we expect the concentration is at least 10 nM for a library of 10 μl in volume. Using together with the real-time PCR amplification curves, we can optimize cell lysis step and amplification. Second, we expect the profile of the library that is constructed under saturated transposition to look like what is shown in the left panel of Figure 1. This profile is different from what is recommended by Illumina for bulk sequencing. The fragment size is best to range between 170 to 350 base pairs (Xi et al., 2017). Several factors may lead to flat profiles as shown in the right panel of Figure 1: (1) insufficient removal of histone proteins, (2) insufficient amount of transposase, (3) incomplete denaturing of Proteinase K in Steps 2 and 3, leading to degrading of transposase in Step 5, (4) incomplete removal of transposons in Step 6. Larger fragments have relatively lower efficiencies to be sequenced by Illumina’s systems (Xi et al., 2017), leading to undercounting in these regions.
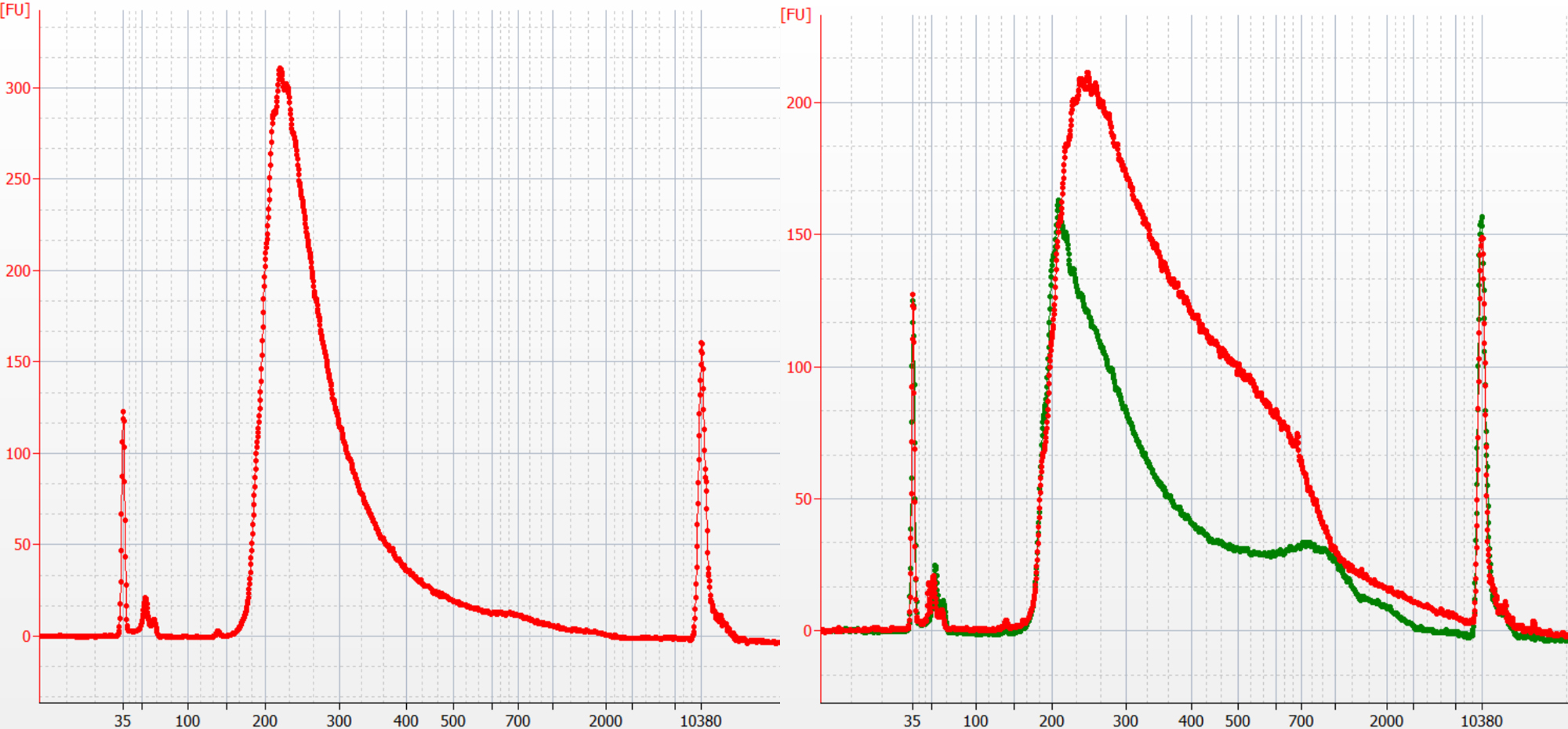
Figure 1. Using BioAnalyzer to examine if transposition is saturated (left panel) or unsaturated (right panel) in addition to the quantitation - After BioAnalyzer analysis, the concentration of the library can be adjusted for next-gen sequencing. A sequencing depth of 0.5%x to 5%x of genome is good for CNV analysis. The resolution (i.e., bin size) depends on the number of unique fragments (counts) per bin. To obtain statistically sound copy number call, we use at least 50 counts per bin. For example, if the intended resolution is 1,000 kilobases, for a library with the average insert size of 100 bases, the minimal coverage = (50 counts x 100 bases)/1,000,000 = 0.5%. If the resolution is set to be 100 kilobases, the minimal coverage = (50 counts x 100 bases)/100,000 = 5%.
- We have used the protocol on single cells of K562 cell line, BJ, human lymphocyte GM01202 cell line, human intestine cells, human skin cells, mouse skin cells, and mouse brain cells.
Recipes
- 5x Transposase buffer (Picelli et al., 2014)
50 mM Tris-HCl, 25 mM MgCl2, 50% (v/v) DMF (pH 8.0) at 25°C
Competing interests
Digenomix is commercializing the technology.
References
- Alioto, T. S., Buchhalter, I., Derdak, S., Hutter, B., Eldridge, M. D., Hovig, E., Heisler, L. E., Beck, T. A., Simpson, J. T., Tonon, L., Sertier, A. S., Patch, A. M., Jager, N., Ginsbach, P., Drews, R., Paramasivam, N., Kabbe, R., Chotewutmontri, S., Diessl, N., Previti, C., Schmidt, S., Brors, B., Feuerbach, L., Heinold, M., Grobner, S., Korshunov, A., Tarpey, P. S., Butler, A. P., Hinton, J., Jones, D., Menzies, A., Raine, K., Shepherd, R., Stebbings, L., Teague, J. W., Ribeca, P., Giner, F. C., Beltran, S., Raineri, E., Dabad, M., Heath, S. C., Gut, M., Denroche, R. E., Harding, N. J., Yamaguchi, T. N., Fujimoto, A., Nakagawa, H., Quesada, V., Valdes-Mas, R., Nakken, S., Vodak, D., Bower, L., Lynch, A. G., Anderson, C. L., Waddell, N., Pearson, J. V., Grimmond, S. M., Peto, M., Spellman, P., He, M., Kandoth, C., Lee, S., Zhang, J., Letourneau, L., Ma, S., Seth, S., Torrents, D., Xi, L., Wheeler, D. A., Lopez-Otin, C., Campo, E., Campbell, P. J., Boutros, P. C., Puente, X. S., Gerhard, D. S., Pfister, S. M., McPherson, J. D., Hudson, T. J., Schlesner, M., Lichter, P., Eils, R., Jones, D. T. and Gut, I. G. (2015). A comprehensive assessment of somatic mutation detection in cancer using whole-genome sequencing. Nat Commun 6: 10001.
- Baslan, T. and Hicks, J. (2014). Single cell sequencing approaches for complex biological systems. Curr Opin Genet Dev 26: 59-65.
- Chen, C., Xing, D., Tan, L., Li, H., Zhou, G., Huang, L. and Xie, X. S. (2017). Single-cell whole-genome analyses by Linear Amplification via Transposon Insertion (LIANTI). Science 356(6334): 189-194.
- Fan, H. C., Wang, J., Potanina, A. and Quake, S. R. (2011). Whole-genome molecular haplotyping of single cells. Nat Biotechnol 29(1): 51-57.
- Francis, J. M., Zhang, C. Z., Maire, C. L., Jung, J., Manzo, V. E., Adalsteinsson, V. A., Homer, H., Haidar, S., Blumenstiel, B., Pedamallu, C. S., Ligon, A. H., Love, J. C., Meyerson, M. and Ligon, K. L. (2014). EGFR variant heterogeneity in glioblastoma resolved through single-nucleus sequencing. Cancer Discov 4(8): 956-971.
- Knouse, K. A., Wu, J. and Amon, A. (2016). Assessment of megabase-scale somatic copy number variation using single-cell sequencing. Genome Res 26(3): 376-384.
- Knouse, K. A., Wu, J. and Hendricks, A. (2017). Detection of copy number alterations using single cell sequencing. J Vis Exp(120). Doi: 10.3791/55143.
- Navin, N. E. (2015). Delineating cancer evolution with single-cell sequencing. Sci Transl Med 7(296): 296fs229.
- Picelli, S., Bjorklund, A. K., Reinius, B., Sagasser, S., Winberg, G. and Sandberg, R. (2014). Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res 24(12): 2033-2040.
- Xi, L., Belyaev, A., Spurgeon, S., Wang, X., Gong, H., Aboukhalil, R. and Fekete, R. (2017). New library construction method for single-cell genomes. PLoS One 12(7): e0181163.
- Xi, L. (2018). Single-Cell DNA Sequencing: From Analog to Digital. Cancer Research Frontiers. 3(1): 161-169.
- Yang, L., Luquette, L. J., Gehlenborg, N., Xi, R., Haseley, P. S., Hsieh, C. H., Zhang, C., Ren, X., Protopopov, A., Chin, L., Kucherlapati, R., Lee, C. and Park, P. J. (2013). Diverse mechanisms of somatic structural variations in human cancer genomes. Cell 153(4): 919-929.
- Zong, C., Lu, S., Chapman, A. R. and Xie, X. S. (2012). Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science 338(6114): 1622-1626.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Xi, L., Leong, P. and Mihajlovic, A. (2019). Preparing Single-cell DNA Library Using Nextera for Detection of CNV. Bio-protocol 9(4): e3175. DOI: 10.21769/BioProtoc.3175.
Category
Molecular Biology > DNA > DNA sequencing
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.