- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Rapid Multiplexed Reduced Representation Bisulfite Sequencing Library Prep (rRRBS)
Published: Vol 9, Iss 4, Feb 20, 2019 DOI: 10.21769/BioProtoc.3171 Views: 6797
Reviewed by: Shan JiangAjit ShahAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Oct 2018

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
DNA methylation is a common mechanism of epigenetic regulation involved in transcriptional modulation and genome stability. With the evolution of next-generation sequencing technologies, establishing quantitative genome-wide DNA methylation profiles is becoming routine in many laboratories. However, many of these approaches take several days to accomplish and use subjective PCR methods to amplify sequencing libraries, which can induce amplification bias. Here we propose a rapid Reduced Representation Bisulfite Sequencing (rRRBS) protocol to minimize PCR amplification bias and reduce total time of multiplexed library construction. In this modified approach, the precise quantification of the final library amplification step is accomplished and monitored by qPCR, instead of using standard PCR and gel electrophoresis, to determine the appropriate number of cycles to perform. The main advantages of this rRRBS method are: i) Reduced amount of amplification enzyme used for library prep, ii) Reduced number of PCR cycles resulting in less PCR amplification bias, and iii) Preparation of quality multiplexed rRRBS libraries in only ~2 days.
Keywords: DNA methylationBackground
With the development of high-throughput sequencing technologies, evaluation of genome-wide DNA methylation profiles is more accessible than before. For the human genome, various commercial targeted methyl capture sequencing panels and methylation arrays are available and allow high-throughput, quantitative interrogation of methylation sites at single-nucleotide resolution. However, most of these commercial tools are not offered for other model species. Thus, cost-effective sequencing-based methods for genome-wide methylation analysis, not targeting species-specific genomic sequences, are still highly relevant and valuable especially when profiling large cohorts of samples. One of these techniques is Reduced Representation Bisulfite Sequencing (RRBS). This method enables you to quantitatively investigate the DNA methylation profile of ~10% of the overall CpGs, clustered in fragments composing approximately 1% of the genome, with a preference for CpG rich regions (e.g., CpG islands, promoters) (Meissner et al., 2005 and 2008; Smith et al., 2009; Gu et al., 2011).
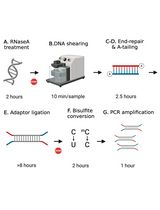
Here, we present a rapid RRBS (rRRBS) protocol (Piché et al., 2018) that significantly decreases hands-on time, allowing to perform the entire protocol in ~2 days compared to the 7 days usually needed (Gu et al., 2011; Boyle et al., 2012). Importantly, by using a qPCR amplification step, we now minimize amplification bias during multiplexed library construction (Figure 1). Also, compared to the original and other RRBS protocols (Gu et al., 2011; Boyle et al., 2012; McGraw et al., 2015) our modified protocol reduces the quantity of reagent and enzyme used as we no longer test for the optimal number of cycles needed before the final library amplification step. This method is suitable for DNA methylation studies from cells or tissues from any common research species (e.g., human, mouse, rat, zebrafish).
Figure 1. Graphical summary of the principal step of the rRRBS procedure
Materials and Reagents
- Pipette tips
- 1.5 ml DNase/RNase-free microtubes
- 0.2 ml DNase/RNase-free PCR strip tubes with individual caps
- QIAamp DNA micro or mini kit (QIAGEN, catalog numbers: 56304 or 51306)
- QuBit dsDNA BR assay kit (Thermo Fisher Scientific, catalog number: Q32853)
- Unmethylated λ phage DNA (Promega, catalog number: D1521)
- MspI 100 U/μl (New England Biolabs, catalog number: R0106M)
- Klenow fragment 5,000 U/ml (New England Biolabs, catalog number: M0212L)
- dNTP set (4 x 100 mM) (Thermo Fisher Scientific, catalog number: 10297018)
- AMPure XP magnetic beads in PEG solution (Beckman Coulter, catalog number: A63881)
- NEBNext® Multiplex Oligos for Illumina® (Methylated Adapter, Index Primers Set 1) (New England Biolabs, catalog number: E7535L)
- T4 DNA ligase 2,000,000 U/ml (New England Biolabs, catalog number: M0202M)
- EpiTect Fast bisulfite kit (QIAGEN, catalog number: 59826)
- SYBR Green, 10,000x (Thermo Fisher Scientific, Invitrogen, catalog number: S7563)
- KAPA HiFi Uracil+ 2x mastermix (Roche, catalog number: KK2802)
- Bioanalyzer High Sensitivity DNA Assay (Agilent, catalog number: 5067-4626)
- DNase/RNase free water
- EB buffer (QIAGEN, catalog number: 19086)
- NEB Buffer 2 (New England Biolabs, catalog number: B7002S)
- Ethanol 100%
Equipment
- P10, P20 pipets
- QuBit fluorometric device (Thermo Fisher Scientific, catalog number: Q33226)
- Vortex
- Mini-centrifuge
- Thermocycler
- Heating block
- DynaMagTM-96 Side Magnet (Thermo Fisher Scientific, catalog number: 12331D)
- DynaMagTM-2 Magnet (Thermo Fisher Scientific, catalog number: 12321D)
- qPCR machine
- qPCR plates2100
- Bioanalyzer instrument (Agilent, catalog number: G2939BA)
- Illumina sequencing apparatus
- -20 °C freezer
Procedure
- DNA extraction and quantification (estimated time: overnight lysis + 2 h)
DNA is extracted from tissues or cultured cells using QIAGEN QIAamp DNA micro or mini kit, depending on the size of the original sample, following manufacturer’s recommendations. The Qubit fluorometric device with dsDNA BR assay kit is then used to quantify DNA samples. To ensure efficiency of bisulfite conversion (Procedure F), 0.5% (2.5 ng) of unmethylated λ phage DNA can be added as a spike-in with 500 ng of sample DNA to provide an internal control. - Enzymatic digestion (estimated time: overnight)
- For each sample, in a 0.2 ml PCR tube from a strip tube with individual caps, dilute 500 ng of DNA in DNase/RNase-free water for a final volume of 26.8 μl.
Note: Use 0.2 ml PCR strip tubes with individual caps at every step to avoid contamination of your samples. - Prepare a master mix of MspI enzyme by mixing 3 μl of 10x NEB Buffer 2 and 0.2 μl of 100 U/μl MspI enzyme per reaction (Table 1).
Table 1. Preparation of MspI digestion mix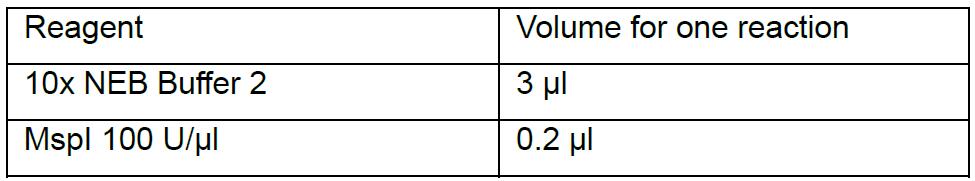
- Add 3.2 μl of the master mix to each sample tube. Mix by pulse vortexing and briefly centrifuge (i.e., spin down) reaction.
- Incubate the digestion reaction in a thermocycler overnight at 37 °C, with lid set at 42 °C.
- For each sample, in a 0.2 ml PCR tube from a strip tube with individual caps, dilute 500 ng of DNA in DNase/RNase-free water for a final volume of 26.8 μl.
- Gap filling and A-tailing (estimated time: 1 h)
Gap filling and A-tailing are achieved using Klenow fragment and a dNTPs mixture (10 mM dATP, 1 mM dCTP, 1 mM dGTP).- Prepare the dNTPs mixture by mixing 176 μl of DNase/RNase-free water, 20 μl of dATP (100 mM), 2 μl dCTP (100 mM) and 2 μl dGTP (100 mM). Aliquots of dNTPs can be stored at -20 °C for future rRRBS experiments.
- For each sample, prepare a master mix by combining 1 μl of the dNTP mixture and 1 μl of Klenow fragment enzyme (Table 2).
Table 2. Preparation of gap filling and A-tailing mix
- Without deactivating MspI digestion, add 2 μl of the master mix in each sample tube. Mix by pulse vortexing and briefly centrifuge.
- Place tubes back in the thermocycler for a cycle of 20 min at 30 °C and 20 min at 37 °C. The lid should not be heated.
- Bead clean-up (estimated time: 1 h 30 min)
After the gap filling and A-tailing step, AMPure XP magnetic beads in PEG solution are used to clean up the reactions from previous reagents. To improve resuspension of the beads later on, warm up EB buffer in a heating block at 55 °C.
Note: Warming up the EB buffer at 55 °C will facilitate the resuspension of the beads, especially if they are over dried.- Add 64 μl of beads (2x ratio of beads [Note 1]) to each sample. Mix by slowly pipetting up and down at least 10 times.
- Incubate mixture for 30 min at room temperature.
- Add 0.2 ml tubes on a DynaMagTM-96 Side magnet for 5 min. At this stage, DNA is bound to the AMPure XP magnetic beads.
- Without disturbing the beads, carefully remove and discard the supernatant.
- Wash the beads by adding 100 μl of 80% freshly prepared ethanol. Incubate for 5 min and remove EtOH without disturbing the beads.
- Repeat Step D5 for a second wash.
- Remove the residual EtOH with a P10 pipet.
- Dry the beads for about 5 min. Do not over dry (i.e., bead ring appears cracked if over dried) as this will significantly decrease elution efficiency (Note 2).
- Remove the tubes from the magnetic rack and resuspend the beads in 26 μl warmed EB buffer by repetitive pipetting, and incubate for 2 min at room temperature (not on the rack).
- Place the tubes back in the magnetic rack and incubate for at least 5 min. Make sure all the beads are bound to the wall of the tube.
- Carefully remove the supernatant without taking any beads and transfer into a new PCR tube.
- Adapter ligation and bead clean-up (estimated time: 2 h)
The next stage is to ligate the adapters for the future qPCR amplification and index fixation steps. Here, the NEBNext® Multiplex Oligos for Illumina® (Methylated Adapter, Index Primers Set 1) kit is used in combination with the T4 DNA ligase.- For each reaction, prepare a master mix of 8.5 μl of DNase/RNase-free water, 5 μl of T4 DNA ligase enzyme 10x buffer, 10 μl of NEB methylated adapters and 1.5 μl of T4 DNA ligase. It is crucial to use methylated adapters to avoid any incompatibility at the amplification step with the index primers. If non-methylated adapters are used, the bisulfite treatment will convert non-methylated cytosine into thymidine, and this will interfere with the recognition of the adapter sequence in future reactions (Table 3).
Table 3. Preparation of adapter ligation mix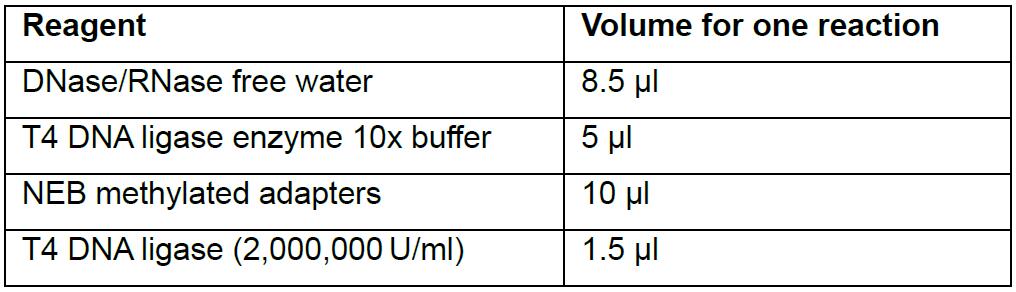
- On ice, add 25 μl of the master mix to each 26 μl DNA sample. Mix by pulse-vortexing and briefly centrifuge.
- Incubate for 20 min at 20 °C in a thermocycler with a non-heated lid.
- Remove the tubes from the thermocycler and add 3 μl of USER enzyme (included in NEBNext® Multiplex Oligos kit) to every reaction. Keep the reaction tubes at room temperature while adding the enzyme. Mix by pulse-vortexing, briefly centrifuge and place the tubes back in the thermocycler for 15 min at 37 °C with a non-heated lid.
Note: A second AMPure XP magnetic bead clean-up step is performed.- Add 55 μl of beads to each sample. Mix by slowly pipetting up and down at least 10 times.
- Incubate for 5 min at room temperature.
- Place the tubes on a DynaMagTM-96 Side magnet for 5 min. At this stage, DNA is captured by the beads.
- Without disturbing the beads, carefully remove and discard the supernatant.
- Wash the beads by adding 100 μl of 80% freshly prepared EtOH. Incubate for 5 min and remove EtOH.
- Repeat Step E9 for a second wash.
- Remove the residual EtOH with a P10 pipet.
- Dry the beads for about 5 min. Do not over dry (Note 2).
- Remove tubes from the magnetic rack, resuspend the beads in 25 μl warmed EB buffer, and incubate for 2 min at room temperature (not on the magnetic rack).
- Place the tubes back in the magnetic rack and incubate for at least 5 min to make sure all the beads stick to the magnet.
- Carefully remove the supernatant without taking any beads and put into a new PCR tube.
Note: The samples can be frozen (-20 °C) at this time to continue later (Note 3).
- For each reaction, prepare a master mix of 8.5 μl of DNase/RNase-free water, 5 μl of T4 DNA ligase enzyme 10x buffer, 10 μl of NEB methylated adapters and 1.5 μl of T4 DNA ligase. It is crucial to use methylated adapters to avoid any incompatibility at the amplification step with the index primers. If non-methylated adapters are used, the bisulfite treatment will convert non-methylated cytosine into thymidine, and this will interfere with the recognition of the adapter sequence in future reactions (Table 3).
- Bisulfite conversion (estimated time: 3 h)
Sodium bisulfite DNA treatment allows for discrimination between methylated and unmethylated cytosines. Compared to previously described RRBS methods, our bisulfite conversion procedure reduces significantly time associated with this step.
Since sodium bisulfite-treated DNA is more sensitive to freezing and thawing, it is better to perform the bisulfite conversion and the amplification of rRRBS libraries without stopping to avoid any degradation (Note 3). (Table 4)
Table 4. Preparation of sodium bisulfite conversion reaction (from QIAGEN EpiTect Fast bisulfite kit)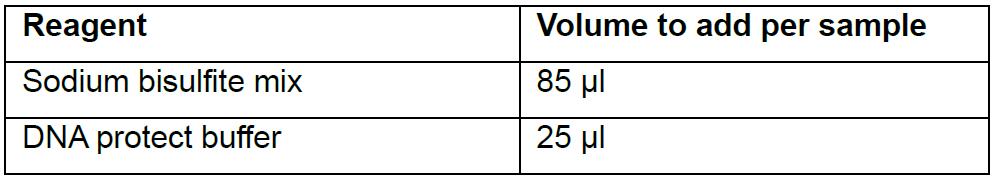
- Add 85 μl of bisulfite mix to each 25 μl DNA sample. Pipet up and down for at least 10 times.
- Add 30 μl of DNA protect buffer. Mix by pulse-vortexing, briefly centrifuge and place tubes in the thermocycler.
- Run 2 cycles: 95 °C for 5 min and 60 °C for 20 min as recommended by the manufacturer’s protocol.
- Amplification of rRRBS libraries
This step, one of the main improvements of our approach, uses a quantitative-PCR (qPCR) method to directly monitor the final amplification of rRRBS libraries to minimize the over cycling effect. In previous protocols, samples were tested with varying PCR cycle numbers to determine the minimal cycle number to be used in the final library preparation. For each sample, the appropriate number of cycles was determined by visualizing the PCR reactions on gel electrophoresis, and then a final library amplification step was performed on the rest of the samples. This protocol is therefore faster and accurately defines the appropriate number of PCR cycles required for library amplification.- Prepare a large-scale PCR mix reaction for each sample following the indication provided in Table 5. NEB Universal primer and Indexing primers are provided with the methylated adapter’s kit. SYBR Green is diluted to 10x in water from 10,000x stock.
Table 5. Preparation of qPCR mix for library amplification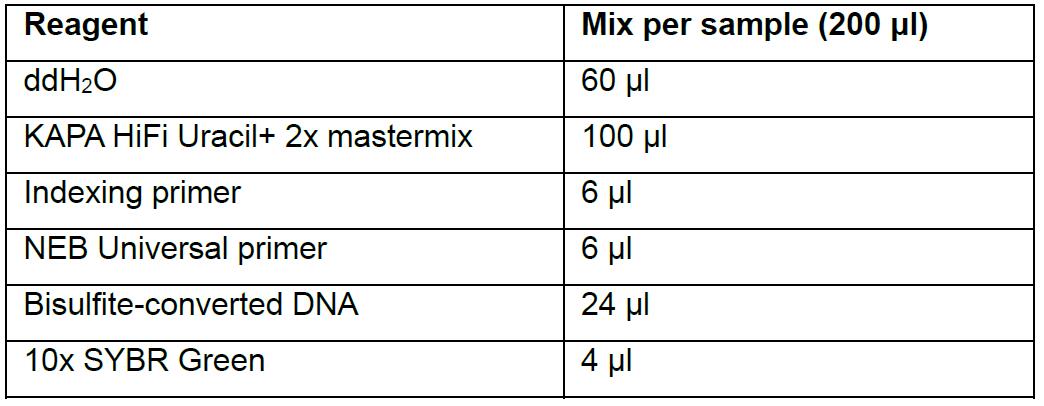
- Divide the 200 μl reaction mix of each sample by distributing 25 μl in individual wells of an 8-strip PCR tube (0.2 ml). Insert PCR strips into the Roche LightCycler96 qPCR instrument and run under conditions indicated in Table 6.
Table 6. qPCR amplification protocol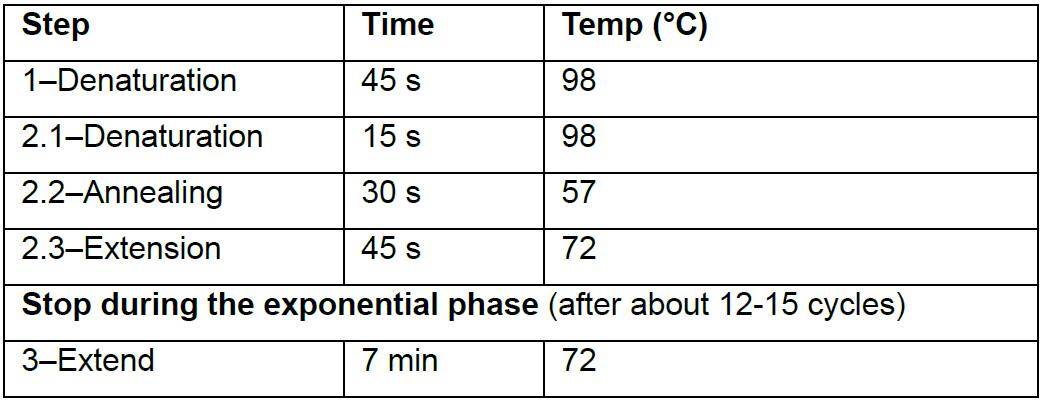
- Stop the qPCR reaction at the end of the linear phase of amplification, after about 12 to 15 cycles, before the plateau phase is reached (Figure 2).
Note: When various libraries are amplified simultaneously, they might not all reach the plateau phase at the same time. Samples with adequate amplification can be quickly removed at the end of an amplification cycle, and transferred to another PCR machine for the final 7 min of extension step. - Perform a final extension step of 7 min at 72 °C.
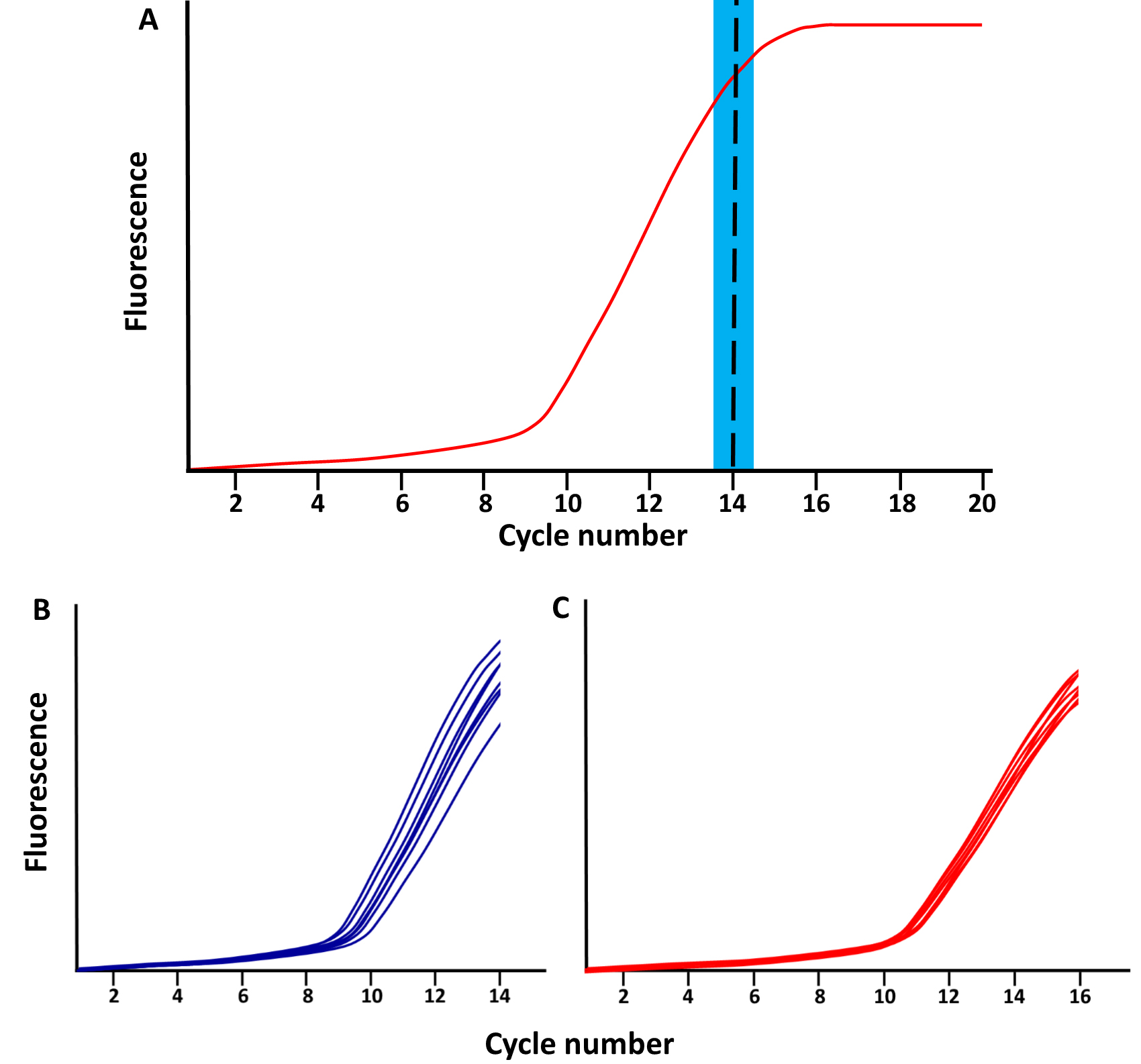
Figure 2. Schematic of qPCR library amplification plots. A. The red line represents the total amplified product accumulation at each qPCR cycle. The blue rectangle represents the period when the qPCR library amplification should be stopped; following the exponential phase and just before the plateau phase. B-C. Examples of library amplifications for 2 sample (each separated in 8 wells) stopped at (B) cycle 14 and (C) cycle 16.
- Prepare a large-scale PCR mix reaction for each sample following the indication provided in Table 5. NEB Universal primer and Indexing primers are provided with the methylated adapter’s kit. SYBR Green is diluted to 10x in water from 10,000x stock.
- Clean-up and size selection of rRRBS libraries (estimated time: 1 h 30 min)
After amplification of rRRBS libraries by qPCR, the last clean-up step is performed using AMPure XP magnetic beads to remove reagent and primer-dimers.- For each sample, pool all 8 PCR reactions into a 1.5 ml low-binding DNase/RNase-free tube.
- Calculate the precise volume of each reaction and add the same volume of beads to the PCR reaction to have a 1x ratio (Note 1) (i.e., add 200 μl of beads if 200 μl of reaction remains after the PCR). Mix by pipetting up and down. The DNA/bead 1x ratio will remove most fragments below 200 bp and all smaller fragments below 100 bp.
- Incubate for 15 min at room temperature and gently tap the tube every 5 min to mix beads and DNA.
- Place tubes in a magnetic rack and let stand 10 min.
- Carefully remove and discard the supernatant without disturbing the beads.
- By keeping the tubes on the magnetic rack, wash the beads by adding 1 ml of 80% freshly prepared EtOH. Incubate for 5 min and remove EtOH.
- Repeat Step H6 for a second wash.
- Remove the residual EtOH with a P20 pipet.
- Dry the beads for about 10 min. Do not over dry (Note 2).
- Remove the tube from the magnetic rack and resuspend the beads in 40 μl of pre-warmed EB buffer. Mix by pipetting, and incubate for 5 min at room temperature (not on the rack).
- Put the tubes back in the magnetic rack and incubate for at least 5 min to ensure all the beads stick to the magnet.
- Carefully remove the supernatant without taking any beads and transfer the solution into a new 1.5 ml tube.
- Quality control (estimated time: 1 h 30 min)
Assess the quality and concentration of the sample by performing a High Sensitivity DNA Assay on the 2100 Bioanalyzer instrument. Concentration varies depending on the original amount of input DNA and the efficiency of the different steps. The peak should be between 200 bp and 300 bp, and concentration at approximatively 5 ng/μl or higher. If a peak of primer-dimer is visible on the Bioanalyzer, at a length of about 75 bp or 150 bp (see Figure 3), perform a second DNA/beads cleanup and repeat the Bioanalyzer quality assessment before sequencing.
Note: If you observe a peak at around 75 bp or 150 bp, perform a second DNA/beads clean-up with a 1x ratio of beads to get rid of all the primer-dimer as those smaller fragments can affect the quality of the sequencing.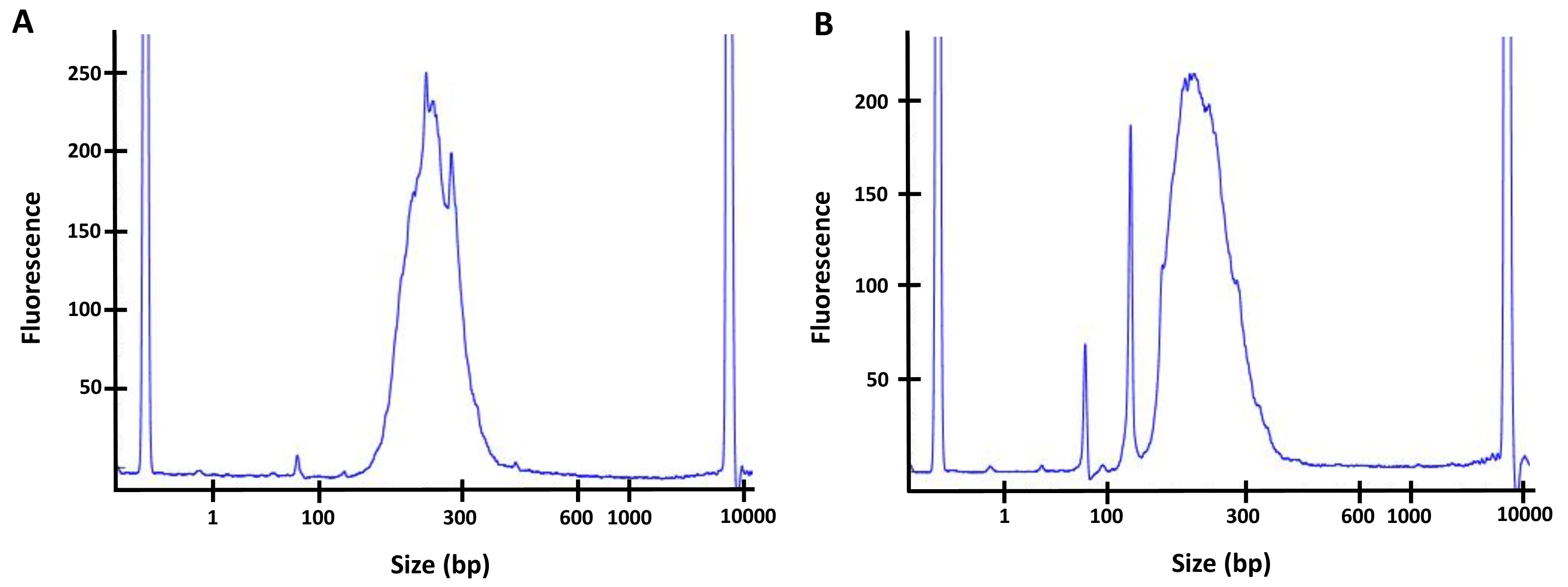
Figure 3. Quality controls of the final libraries by Bioanalyzer. A. Example of a good library with the major peak between 200 bp and 300 bp, no significant peak is observed at a lower size. B. Example of a library containing primer dimers (significant peak at ~75 bp and/or 150 bp). For this sample, a second bead clean-up (with 1x ratio of beads) would have to be performed to remove the primer dimers before sequencing the library. - Sequencing
Paired-end sequencing (> 100 bp read length) of libraries is performed on Illumina apparatus. The number of multiplex libraries per sequencing lane depends on the sequencing technology used and the species investigated (i.e., the total number of CpG of species) (Doherty and Couldrey, 2014). One should aim to obtain at least 20-30 million reads per sample for downstream bioinformatics analysis.
Data analysis
Various bioinformatics tools are widely available to analyze genome-wide DNA methylation sequencing data, and are usually customized depending on how data will be interpreted and visualized (e.g., single CpG methylation, tilling-window approach). Details of basic analysis are detailed in previous papers (Magnus et al., 2014; McGraw et al., 2015; Piché et al., 2018). Briefly, Trim Galore (Krueger, 2015) is used for sequence trimming and quality control, then reads are aligned to the reference genome using BSMAP (Xi and Li, 2009), which is specific for the alignment of bisulfite-treated sequenced DNA. If λ DNA was spike-in, you can calculate the bisulfite conversion rate by mapping this genome and determining the percentage of unmethylated cytosine (sequenced as thymine) at the position of a cytosine in the λ phage reference genome (Lister et al., 2011). Methylation calls are obtained using BSMAP and differentially methylated regions across two conditions are obtained using the R package methylKit (Akalin et al., 2012). Differentially methylated sequences are annotated using HOMER v4.10 (Heinz et al., 2010). Final output processed data can be used in various other downstream analyses, such as functional annotation tools to highlight enrichment of specific biological [e.g., DAVID (Dennis et al., 2003), Metascape (Tripathi et al., 2015)], or converted into specific formats, such as a bedGraph, in order to view methylation levels in various genome browsers [e.g., USCS genome browser, IGV (Thorvaldsdottir et al., 2013)].
Notes
- The ratio of beads is crucial to ensure a good clean-up. To make sure to add the right volume of beads, you can measure the exact volume of sample using a pipet at every clean-up step.
- Bead ring appears cracked if you over dry it. The beads will be harder to resuspend and this will decrease nucleic acid elution efficiency.
- Stopping point: It is recommended to stop after the adapter ligation step (Procedure E) or to go through all the protocol in the same day.
Acknowledgments
Research was funded by an NSERC discovery grant to SM. LML received scholarship from Fondation du CHU Sainte-Justine and Réseau Québécois en Reproduction. SM is supported by Chercheur-boursier Junior 1 from the Fonds de recherche du Québec – Santé (FRQS). We thank Elizabeth Maurice-Elder for editing.
Competing interests
The authors declare no conflict of interest.
References
- Akalin, A., Kormaksson, M., Li, S., Garrett-Bakelman, F. E., Figueroa, M. E., Melnick, A. and Mason, C. E. (2012). methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol 13(10): R87.
- Boyle, P., Clement, K., Gu, H., Smith, Z. D., Ziller, M., Fostel, J. L., Holmes, L., Meldrim, J., Kelley, F., Gnirke, A. and Meissner, A. (2012). Gel-free multiplexed reduced representation bisulfite sequencing for large-scale DNA methylation profiling. Genome Biol 13(10): R92.
- Dennis, G., Jr., Sherman, B. T., Hosack, D. A., Yang, J., Gao, W., Lane, H. C. and Lempicki, R. A. (2003). DAVID: database for annotation, visualization, and integrated discovery. Genome Biol 4(5): P3.
- Doherty, R. and Couldrey, C. (2014). Exploring genome wide bisulfite sequencing for DNA methylation analysis in livestock: a technical assessment. Front Genet 5: 126.
- Gu, H., Smith, Z. D., Bock, C., Boyle, P., Gnirke, A. and Meissner, A. (2011). Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat Protoc 6(4): 468-481.
- Heinz, S., Benner, C., Spann, N., Bertolino, E., Lin, Y. C., Laslo, P., Cheng, J. X., Murre, C., Singh, H. and Glass, C. K. (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38(4): 576-589.
- Krueger, F. (2015). Trim Galore!: A wrapper tool around Cutadapt and FastQC to consistently apply quality and adapter trimming to FastQ files: 0.4.
- Lister, R., Pelizzola, M., Kida, Y. S., Hawkins, R. D., Nery, J. R., Hon, G., Antosiewicz-Bourget, J., O'Malley, R., Castanon, R., Klugman, S., Downes, M., Yu, R., Stewart, R., Ren, B., Thomson, J. A., Evans, R. M. and Ecker, J. R. (2011). Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature 471(7336): 68-73.
- Magnus, N., Garnier, D., Meehan, B., McGraw, S., Lee, T. H., Caron, M., Bourque, G., Milsom, C., Jabado, N., Trasler, J., Pawlinski, R., Mackman, N. and Rak, J. (2014). Tissue factor expression provokes escape from tumor dormancy and leads to genomic alterations. Proc Natl Acad Sci U S A 111(9): 3544-3549.
- McGraw, S., Zhang, J. X., Farag, M., Chan, D., Caron, M., Konermann, C., Oakes, C. C., Mohan, K. N., Plass, C., Pastinen, T., Bourque, G., Chaillet, J. R. and Trasler, J. M. (2015). Transient DNMT1 suppression reveals hidden heritable marks in the genome. Nucleic Acids Res 43(3): 1485-1497.
- Meissner, A., Gnirke, A., Bell, G. W., Ramsahoye, B., Lander, E. S. and Jaenisch, R. (2005). Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res 33(18): 5868-5877.
- Meissner, A., Mikkelsen, T. S., Gu, H., Wernig, M., Hanna, J., Sivachenko, A., Zhang, X., Bernstein, B. E., Nusbaum, C., Jaffe, D. B., Gnirke, A., Jaenisch, R. and Lander, E. S. (2008). Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454(7205): 766-770.
- Piché, J., Gosset, N., Legault, L.-M., Pacis, A., Oneglia, A., Caron, M., Chetaille, P., Barreiro, L., Liu, D., Qi, X., Nattel, S., Leclerc, S., Breton-Larrivée, M., Andelfinger, G., Bakkers, J., Loeys, B., Pucéat, M., McGraw, S. and Andelfinger, G. (2018). Molecular signature of CAID syndrome: noncanonical roles of SGO1 in regulation of TGF-β signaling and epigenomics. CMGH Doi:10.1016/j.jcmgh.2018.10.011
- Smith, Z. D., Gu, H., Bock, C., Gnirke, A. and Meissner, A. (2009). High-throughput bisulfite sequencing in mammalian genomes. Methods 48(3): 226-232.
- Thorvaldsdottir, H., Robinson, J. T. and Mesirov, J. P. (2013). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14(2): 178-192.
- Tripathi, S., Pohl, M. O., Zhou, Y., Rodriguez-Frandsen, A., Wang, G., Stein, D. A., Moulton, H. M., DeJesus, P., Che, J., Mulder, L. C., Yanguez, E., Andenmatten, D., Pache, L., Manicassamy, B., Albrecht, R. A., Gonzalez, M. G., Nguyen, Q., Brass, A., Elledge, S., White, M., Shapira, S., Hacohen, N., Karlas, A., Meyer, T. F., Shales, M., Gatorano, A., Johnson, J. R., Jang, G., Johnson, T., Verschueren, E., Sanders, D., Krogan, N., Shaw, M., Konig, R., Stertz, S., Garcia-Sastre, A. and Chanda, S. K. (2015). Meta- and orthogonal integration of influenza "OMICs" data defines a role for UBR4 in virus budding. Cell Host Microbe 18(6): 723-735.
- Xi, Y. and Li, W. (2009). BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics 10: 232.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Legault, L., Chan, D. and McGraw, S. (2019). Rapid Multiplexed Reduced Representation Bisulfite Sequencing Library Prep (rRRBS). Bio-protocol 9(4): e3171. DOI: 10.21769/BioProtoc.3171.
Category
Molecular Biology > DNA > DNA sequencing
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.