- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

In situ, Cell-free Protein Expression on Microarrays and Their Use for the Detection of Immune Responses


(*contributed equally to this work) Published: Vol 9, Iss 3, Feb 5, 2019 DOI: 10.21769/BioProtoc.3152 Views: 6722
Reviewed by: Chiara AmbrogioMauro Sbroggio'Enrico Patrucco
Original research article
The authors used this protocol in:

Jan 2017
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Until recently, whole-proteome microarrays for comprehensive studies of protein interactions were mostly produced by individual cloning and cellular expression of very many open reading frames, followed by protein isolation and purification as well as array production. To overcome this cumbersome process, we have developed a method to generate microarrays representing entire proteomes by a combination of multiple spotting and on-chip, cell-free protein expression. Here, we describe the protocol for the production of bacterial protein microarrays. With slight adaptations, however, the procedure can be applied to the proteome of any organism. Expression constructs of each gene are generated by PCR on bacterial genomic DNA followed by a common secondary amplification that is adding relevant regulative elements to either end of the constructs. The unpurified PCR-products are spotted onto the microarray surface. Full-length proteins are directly expressed in situ in a cell-free manner and stay attached to the surface without further action. As an example of a typical application, we describe here the proteome-wide analysis of the immune response to a bacterial infectious agent by characterizing the binding profiles of the antibodies in patient sera.
Keywords: Whole-proteome microarraysBackground
Protein microarrays are an excellent tool for identifying disease-associated antibody reactivity patterns since they allow the simultaneous detection of antibody binding to a large number of antigens, up to entire bacterial proteomes (Hufnagel et al., 2018) and beyond (Syafrizayanti et al., 2017). Protein microarrays used to be generated with proteins that were isolated from recombinant cellular libraries. This approach is work-intensive, time-consuming and costly, however, since it requires the individual handling of very many cells. Each gene of interest is amplified by a polymerase chain reaction (PCR), followed by cloning into an expression vector. After transformation into a cellular system–frequently Escherichia coli (E. coli)–the cell clones are grown separately from others and characterized. Subsequently, the respective overexpressed protein is isolated and purified, followed by immobilization on the microarray. Our largest array of a non-mammalian gene set consisted of 14,000 proteins (Syafrizayanti et al., 2017), for example. Therefore, producing this array would have required going through the above process 14,000 times.
In order to overcome the complex procedure, means have been developed for the production of protein microarrays by cell-free protein expression directly on the microarray surface. After the initial publication of the method (Protein in situ Arrays, PISA) (He and Taussig, 2001), several adaptations have been reported (Ramachandran et al., 2004; Angenendt et al., 2006; He et al., 2008). All have in common that genes are copied into DNA expression constructs by PCR, which are directly placed onto the microarray surface. Proteins are then synthesized in situ using cell lysates containing all elements and ingredients required for transcription and translation. All steps are done in parallel and in a largely automated manner. No cells are involved throughout. In addition, the approach eliminates the need for protein purification.
Besides many minor variations, our protocol differs from the others mainly by the fact that multiple spotting is applied to produce protein microarrays (Angenendt et al., 2006; Syafrizayanti et al., 2017). First, the DNA expression constructs are placed on planar glass slides. In a second step, each position is revisited to deposit in a second spotting event a cell-free transcription and translation mixture on top of the DNA spots. Proteins are expressed directly on the microarray slide and bind immediately. The double-spotting protocol decreases substantially the volume of cell lysate needed for protein expression since the empty surface in between spots is not covered. Concomitantly, there is less background signal in between the spots eventually. More importantly, however, the reaction space is restricted to the locations of the DNA expression constructs. In consequence, expressed protein cannot float away but binds in situ only. No additional means, such as antibodies, are required to keep the proteins attached to their exact positions. Nevertheless, even extensive washing will not remove them. Previous studies have shown that most proteins are expressed in full-length, and very many fold into a functionally active conformation. Also, the process is versatile and can be adapted to fit to a variety of applications (Syafrizayanti et al., 2017).
Besides studies on microarrays with human or parasite proteins, we have used arrays that represent entire bacterial proteomes to screen clinically characterized patient (and control) sera in order to identify antibody binding patterns that are disease-specific. New marker molecules for disease diagnosis and prognosis were discovered in this way, including markers for cancer entities that are associated with bacterial infections. In a recent study, microarrays were produced that present all proteins of Chlamydia trachomatis (Ct), for example (Hufnagel et al., 2018). Through comparison of the antibody binding patterns on the microarrays, we identified antigens that are reproducibly recognized by the antibodies in Ct-seropositive samples, in addition to molecules that were patient-specific. With samples from cervical cancer patients, we found the common Ct-specific markers and additional antigens, which were shared between the cancer patients only. Large-scale validation experiments using high-throughput suspension bead array serology on hundreds of samples confirmed the significance and relevance of the newly identified markers for both general Ct infection and related cervical cancer. Next to providing meaningful marker molecules, the results strongly support the hypothesis that there is an association of Ct infection with cervical cancer.
The quick and efficient proteome-wide immunoassay can easily be adapted to other microorganisms in all areas of infection research. By reverse transcribing RNA to DNA-templates, the protocol is applicable to any transcript isolate. We also have utilized established ORF libraries, such as the Human ORFeome library (The ORFeome Collaboration, 2016), by which the process of microarray production is simplified further since only one vector-specific primer pair is required for the initial PCR. Here, we provide a detailed protocol for the production of whole-proteome bacterial microarrays and their application to screening patient sera for the immune response in infected people.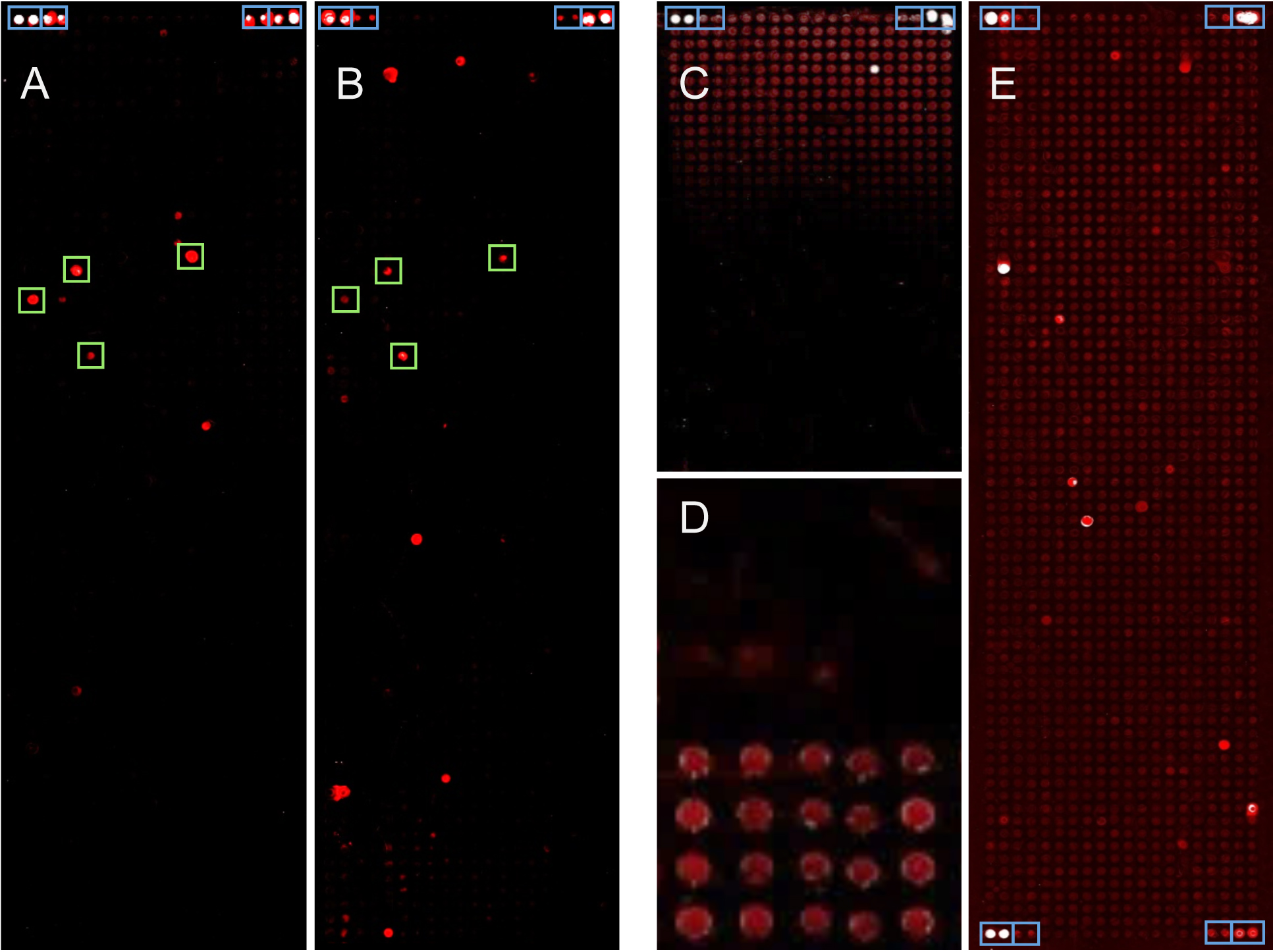
Figure 1. Images of protein microarrays made from bacterial genes. About 1,500 proteins were produced on each array. Blue boxes indicate controls. A-B. Two examples of analyses are presented that were performed using the protocol described below. Signals that were common to a group of patients are labeled by green boxes. C-E. Few examples of technical problems are presented. C. An artifact is shown, which was due to a technical fault of the spotting device, resulting in a signal gradient across the microarray. D. In a close-up, background signals on 40 microarray positions are shown. The only difference between the top and bottom half was the quality of the DNA-polymerase used for PCR. After spotting unpurified PCR-products, incubation with patient serum and subsequent labeling produced significant differences in background signal. E. A complete protein microarray is shown after incubation with patient serum and detection of antibody binding. Because of inadequate blocking, a relatively high level of background signal was produced.
Materials and Reagents
- Corning Costar Stripette serological pipettes (Corning, Sigma-Aldrich, catalog number: CLS4488-50EA)
- Epoxysilane-coated slides (Schott, catalog number: 1066643)
- Microarray hybridization cassettes (Arrayit Corporation, blue: AHCTZ, green: AHCP, gold: AHCC)
- ProPlate Slide Modules (Grace Bio-Labs, catalog number: GBL246861-2EA)
- Standard 96-well PCR plate (Steinbrenner Laborsystem, catalog number: SL-PP96-4L)
- Microplates, 384-well (Greiner Bio-One, catalog number: 781281)
- PCR seal (4titude, catalog number: 4ti-0500)
- quadriPerm chambers (SARSTEDT, catalog number: 94.6077.307)
- Escherichia coli BL21 wild-type bacteria (GE Healthcare)
- 6x DNA loading dye (Thermo Scientific, catalog number: R0611)
- Alexa Fluor 647-conjugated goat anti-human IgA, IgG, IgM (Jackson Immuno Research, catalog number: 109-605-064)
- Agarose NEEO Ultra-Quality (Carl Roth, catalog number: 2267.4)
- Betaine monohydrate (Sigma-Aldrich, catalog number: 14300)
- Bovine serum albumin (Carl Roth, catalog number: 8076.3)
- Calcium chloride (Carl Roth, catalog number: CN93.2)
- Deoxynucleotide (dNTP) solution set (New England Biolabs, catalog number: N0446S)
- Disodium phosphate (Neolab Migge GmbH, catalog number: 4770.1000)
- DL-Dithiothreitol (DTT) (Thermo Scientific, catalog number: R0861)
- Ethylenediaminetetraacetic acid (Carl Roth, catalog number: 8043.2)
- GeneRuler 1 kb DNA Ladder (Thermo Scientific, catalog number: SM0311)
- Glacial acetic acid (Sigma-Aldrich, catalog number: 695084)
- Glycine (Gerbu, catalog number: 56406)
- Nickel(II) sulfate hexahydrate (Sigma-Aldrich, catalog number: 227676)
- Nitrilotriacetic acid (NTA) (Sigma-Aldrich, catalog number: 72559)
- Nuclease-free water (Life Technologies, catalog number: AM9922)
- Monoclonal anti-V5-Cy3 antibody produced in mouse (Sigma-Aldrich, catalog number: V4014-100UG)
- Monopotassium phosphate (Carl Roth, catalog number: 3904.1)
- Penta-His Alexa Fluor 647 Conjugate (QIAGEN, catalog number: 35370)
- peqGreen (VWR, peqLab brand, catalog number: 732-3196)
- Potassium chloride (Carl Roth, catalog number: 6781.1)
- Q5 High-Fidelity DNA polymerase, 100 Units, and buffers (New England Biolabs, catalog number: M0491S)
- S30 T7 High-Yield Protein Expression Kit (Promega, catalog number: L1110)
- SuperBlock (PBS) blocking buffer (Thermo Scientific, catalog number: 37515)
- Taq DNA Polymerase (1,000 U) and buffers (QIAGEN, catalog number: 201205)
- Tris base (Sigma-Aldrich, catalog number: SLBP5070V)
- Sodium carbonate (Na2CO3) (Carl Roth, catalog number: A135.1)
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S9888-1KG-M)
- Sodium hydrogen carbonate (NaHCO3) (Carl Roth, catalog number: 6885.1)
- Sodium hydroxide (NaOH) (Sigma-Aldrich, catalog number: 1310-73-2)
- Tryptone (Sigma-Aldrich, catalog number: T7293-250G)
- Tween-20 (Sigma-Aldrich, catalog number: P1379)
- Yeast extract (Sigma-Aldrich, catalog number: Y1625)
- Complete Protease inhibitor (Roche, catalog number: 04693132001)
- KH2PO4 (Merck, catalog number: 1048711000)
- MgCl2 (Carl Roth, catalog number: KK36.1)
- Acetic acid (Sigma-Aldrich, catalog number: A6283)
- Glycerol (Sigma-Aldrich, catalog number: G5516)
- BSA-blocking buffer (see Recipes)
- NTA-solution (see Recipes)
- Nickel-solution (see Recipes)
- LB-medium (see Recipes)
- Opti-Solution (see Recipes)
- 10x PBS (see Recipes)
- PBST (see Recipes)
- TAE (50x) (see Recipes)
Equipment
- Pipettes (Gilson, PIPETMAN Classic, catalog numbers: F144566, F144057M, F144055M)
- Slide staining and storage systems (Sigma-Aldrich, catalog number: BAF441800000)
- Biofuge, Pico (Hereaus Instruments, DJB Labcare 75003235)
- Magnetic stirrer (IKA, IKAMAG REO)
- Centrifuge 5810R (Eppendorf, model: 5810R, catalog number: 5811000010)
- Gel documentation system (Azure C200 Biosystems, catalog number: 170-8195)
- Homogenizator EmulsiFlex-C5 (AVESTIN, catalog number: 0506394)
- Laminar hood (Hereaus Instruments, model: LaminAir HB2436)
- Microcomputer electrophoresis power supply (Consort, model: E835)
- Mixmate Microplate Mixer (Eppendorf, model: 5353 000.014)
- NanoDrop spectrophotometer ND-1000 (Thermo Scientific, model: 7934)
- Nanoplotter 2.0 (GeSIM, model: 060-308)
- Orbital Shaker Unimay 1010 (Heidolph, model: 543-12310-00)
- Power Scanner (Tecan, model: 30038387)
- LifeEco Thermal Cycler (BioER, model: 685115)
- Ventilated oven (neoLab, catalog number: 7-9155)
- 4 °C refrigerator (neoLab, catalog number: 10064)
- -20 °C freezer (Bosch, model GIV21AF30)
Software
- Fuzznuc (EMBOSS, http://emboss.sourceforge.net/apps/cvs/emboss/apps/fuzznuc.html)
- Software NPC16 V2.15 (GeSiM, Radeberg, Germany)
- PowerScanner V1.2 (Tecan Trading AG, Switzerland)
- GenePix Pro 6.0 (Molecular Devices, Sunnyvale, USA)
- R Studio (R version 3.3.2, https://www.rstudio.com/)
- MELTTEMP (http://www.biology.wustl.edu/gcg/melttemp.html)
- Excel (Microsoft Cooperation, Redmond, USA)
Procedure
- Primers and primer design
- For the initial PCR, design gene-specific primers in a way that each primer pair is binding at the beginning and end of each ORF and is yielding an in-frame PCR-product. Primer length should be between 16 and 24 nucleotides (nt).
Note: For primer design, a Perl script can be generated to design primers using the ORF table information text file and fasta sequence(s) of the reference genome. - Calculate the melting temperature of possible primers of different length with MELTTEMP (http://www.biology.wustl.edu/gcg/melttemp.html).
Note: Other software packages or websites could be used instead. However, this software was found to be useful for calculating melting temperatures of many primers required for whole-proteome projects. - Choose the best fitting primers within a range of the melting temperatures of either 45 °C-55 °C or 55 °C-65 °C.
Note: Primers used in a single microtiter plate should have a similar melting temperature. If no suitable primers are found that represent the very end of an ORF, consider primer sequences that start one or two triplets downstream of the ATG start codon of the gene of interest. - Check and calculate the uniqueness and the product length of the primers with fuzznuc (EMBOSS, http://emboss.sourceforge.net/apps/cvs/emboss/apps/fuzznuc.html).
Note: Again, other software packages could be used instead. As before, this package was found to be rather handy in handling a large number of primers. - Add the following 15 nt adaptor sequences to all 5’-ends: forward primers: 5’-atgcaccaaacccaa-3’; reverse primers: 5’-cgcactggcatcatc-3’.
- Primers should be synthesized in 96-well plates for easy handling; we used oligonucleotides produced by the commercial provider biomers.net (Ulm, Germany).
- The common primers for the second PCR contain all sequences necessary for transcription and translation [T7 promoter, untranslated region (UTR), ribosome binding site (RBS), start codon (ATG), T7 terminator], fusion peptide tags (N-terminal 6x-His and C-terminal V5 tag) and the sequences complementary to the adaptor sequences common to the gene-specific primers:
Forward expression primer:
5’-gaaattaatacgactcactatagggagaccacaacggtttccctctagaaataattttgtttaagaaggagatatacatatgcatcatcatcatcatcatatgcaccaaacccaa-3’
Reverse expression primer:
5’-ctggaattcgcccttttattacgtagaatcgagaccgaggagagggttagggataggcttacccgcactggcatcatc-3'
- For the initial PCR, design gene-specific primers in a way that each primer pair is binding at the beginning and end of each ORF and is yielding an in-frame PCR-product. Primer length should be between 16 and 24 nucleotides (nt).
- Polymerase chain reaction
Perform two successive PCRs for each gene. The first PCR amplifies the gene of interest; the second PCR adds the elements needed for generating expression constructs. For the measurement of DNA concentrations, we use the NanoDrop spectrophotometer, since only small volumes are required. However, any other spectrophotometer could be used instead.
Note: Concerning the overall reaction, basically all PCR amplifications were successful up to a size of some 3 kb. For larger molecules, the yield varied although the polymerase used was suitable for synthesizing long fragments.- The first PCR is performed using genomic bacterial DNA as template.
Note: Genomic DNA can be obtained from the German Collection of Microorganisms and Cell Cultures (DSMZ), for example. As already mentioned in the background section above, also cDNA made from RNA preparations, existing ORF-libraries or any other molecule type that can be PCR-amplified could be used as templates. - Perform all PCR in 96-well PCR-plates–one well per gene–and group the genes based on the gene length and the melting temperatures of the respective primer pair.
- Use an annealing temperature (TA) that is 2 °C below the average melting temperature of all primers within each 96-well plate.
- Calculate the elongation time (timeE) as listed in the following table:

- Use a PCR thermal cycler to perform the amplification.
- Prepare the PCR-mixture for the first PCR as follows: 10.75 µl nuclease-free H2O, 5.0 µl Q5 High-Fidelity buffer, 5.0 µl Q5 High-Fidelity enhancer, 1.0 µl 10 mM gene-specific forward primer, 1.0 µl 10 mM gene-specific reverse primer, 1.0 µl 10 mM dNTP-mix, 1.0 µl genomic DNA template (2 ng/µl), 0.25 µl Q5 High-Fidelity polymerase.
- Use the PCR program listed below, followed by storage at 10 °C:

- Use 1 µl PCR-product of the first PCR as the template for the second PCR; perform the second amplification in 96-well PCR-plates as well.
- Store the remaining PCR-product of the first, gene-specific amplification frozen at -20 °C.
- Perform the second PCR with the elongation times used for the first PCR.
- Prepare the PCR-mixture for the second PCR as follows: 33.5 µl nuclease-free H2O, 5.0 µl 10x PCR buffer (with 10 mM MgCl2), 5.0 µl Q-Solution (QIAGEN), 2.0 µl 25 mM MgCl2, 1.0 µl 10 mM dNTP-mix, 1.0 µl 10 µM forward expression primer, 1.0 µl 10 µM reverse expression primer, 1.0 µl DNA template, 0.5 µl Taq DNA-polymerase (QIAGEN).
Note: Using a polymerase of low quality could result in incomplete on-chip protein expression and higher background signals at the spots (for example see Figure 1D). - Use the following PCR program: initial denaturation: 95 °C for 5 min; denaturation: 94 °C for 30 s; annealing: 52 °C for 30 s; elongation: 72 °C for calculated timeE; final elongation: 72 °C for 10 min. Repeat denaturation, annealing and elongation 35 times.
- Store the PCR-products as they are in the microtiter plate at -20 °C for long-term storage or at 4 °C for short-term storage.
- To check the success of all PCRs on a 1.3% agarose gel, dissolve 2.6 g of agarose in 200 ml 1x TAE buffer by heating, add 10 µl peqGreen and pour the gel.
- Mix 5 µl of each PCR-product with 1 µl of 6x DNA loading dye and load the mixture onto the gel.
- Load at least one lane with the GeneRuler 1 kb ladder as a size standard.
- Run the gel with an electrophoresis power supply system at about 150 V and 500 mA for 30 min.
- Analyze the gel with UV-light using a gel documentation system. A single band per lane representing a DNA fragment of the expected size indicates a successful PCR.
- The first PCR is performed using genomic bacterial DNA as template.
- Generation of Ni-NTA slides
Note: All steps are performed in a dust-free environment of a sterile hood.- Place epoxysilane-coated slides into dust-free slide staining and storage systems.
- Prepare NTA-solution by dissolving sodium bicarbonate in sterile-filtered water to a concentration of 2.38 M and add NTA to 0.63 M.
- Stir the NTA-solution on a magnetic stirrer at 37 °C for 4 h.
- Soak the epoxysilane-coated slides in NTA-solution and incubate them on an orbital shaker set to 50 rpm at room temperature overnight.
- Discard the NTA-solution and wash the slides twice with sterile-filtered water for 5 min on an orbital shaker set to 50 rpm.
- Rinse slides in sterile-filtered water and dry them with compressed air.
- Incubate slides in 1% nickel sulfate solution at room temperature for 6 h.
- Rinse slides in sterile-filtered water.
- Incubate slides in Opti-solution containing 0.2 M acetic acid, 0.2 M CaCl2 and 0.1% Tween-20 at room temperature for 30 min.
- Rinse slides four times in sterile-filtered water and dry them with compressed air.
- Store slides in manufacturer’s storage boxes at 4 °C for a maximum of 3 months.
- Spotting of protein microarrays
Before each spotting procedure, the Nanoplotter 2.0 has to be washed twice with ddH2O for at least 20 min.- Transfer 45 µl each of all final PCR-products into 384-well plates.
- Add 5 µl 5 M betaine to each well and mix plates using a microplate mixer; centrifuge all plates at about 400 x g for 1 min to collect the entire volume at the bottom.
- Place up to 33 Ni-NTA-coated slides onto the slide tablet of the Nanoplotter.
- Design a work plate within the Nanoplotter software, containing information about the microarray pattern: number of rows and columns, distance between spots, spot distance to edges of the slide, number and distance of blocks as well as information about the type of the 384-well plate.
- Measure Z-values (vertical axis) for every slide individually to maintain proper spotting distance.
- Write a transfer file compatible to the work plate in Excel and transfer it to Nanoplotter Notepad.
- Transfer per spot 0.6 nl of each PCR-product onto the Ni-NTA slides.
Note: Confirm correct sample delivery by using the automated detection with a stroboscope system that is part of the Nanoplotter. - Prepare S30 T7 High-Yield Protein Expression Kit by mixing immediately before use the ingredients provided by the manufacturer: 20 µl S30 Premix Plus, 18 µl T7 S30 Extract and 12 µl nuclease-free water.
- Transfer the 50 µl expression mixture to a new well of a 384-well microplate.
Note: Avoid air bubbles while handling the expression mixture. - Dispense with the Nanoplotter 2.4 nl expression mixture directly on top of each spot of a expression construct.
Note: Transfer the expression mixture slide by slide. One piezo element of the spotter can produce a maximum of 800 spots using one sample uptake. If necessary, use multiple piezo elements for spotting the expression mixture or multiple sample uptakes per slide. - Place the slide immediately into a microarray hybridization cassette containing 30 µl of nuclease-free water in each reservoir.
- Incubate the hybridization cassette in a preheated box containing wet tissue paper at 37 °C for 1 h.
- Transfer the cassettes into a ventilated oven and incubate at 30 °C overnight.
- Store slides without loss of reactivity at -20 °C for a maximum of 3 months.
- Determination of on-chip protein expression by antibody staining
Note: To avoid drying, perform all subsequent steps immediately after one another. All washing and incubation steps are performed at room temperature unless stated otherwise on an orbital shaker set to 50 rpm.- Take slides from the freezer and place them into ProPlate Slide Modules.
- Block each slide with 2 ml BSA-blocking-buffer for 1 h.
- Dilute fluorophore-conjugated antibodies directed against the protein tags (Penta-His Alexa Fluor 647 conjugate; monoclonal Anti-V5-Cy3 antibody) 1:1,000 in BSA-blocking-buffer and incubate the mixture at 4 °C for 1 h.
- Discard blocking buffer and wash slides twice with 2 ml PBST each for 5 min.
- Incubate each slide with 1 ml of the antibody solution for 1 h.
- Remove slides from the ProPlate Slide Modules, place them into quadriPerm chambers and wash three times with 5 ml PBST each for 10 min.
- Rinse slides in sterile-filtered water and air-dry them in a ventilated oven at 30 °C by using slide staining and storage systems.
- Scan slides in a Power Scanner at excitation wavelengths of 532 nm and 635 nm.
- Proteome immunoassay
Note: To avoid drying, perform all subsequent steps immediately after one another. All washing and incubation steps are performed at room temperature unless stated otherwise on an orbital shaker set to 50 rpm.- Take slides from freezer and place them into ProPlate Slide Modules.
- Block each slide with 2 ml SuperBlock blocking buffer for 1 h.
- Prepare patient serum samples by diluting sera 1:33 in SuperBlock blocking buffer also containing 1 µg/µl E. coli wild-type lysate and incubate mixture at 4 °C for 1 h.
- Discard blocking buffer and wash each slide twice with 2 ml PBST for 5 min.
- Discard PBST and incubate slide with 1.5 ml serum dilution for 1 h.
- Prepare a 1:350 dilution of the secondary antibody (Alexa Fluor 647-conjugated goat anti-human IgA, IgG, IgM) in SuperBlock blocking buffer and incubate the dilution at 4 °C for 1 h.
- Wash each slide twice with 2 ml PBST for 5 min
- Incubate slide with 1.5 ml of the 1:350 dilution of the secondary antibody for 1 h.
- Remove slide from the ProPlate Slide Modules, place it into a quadriPerm chamber and wash three times with 5 ml PBST each for 10 min.
- Rinse slide in sterile-filtered water and air-dry it in a ventilated oven at 30 °C by using slide staining and storage systems.
- Scan slide with a Power Scanner at 635 nm excitation wavelength (Figure 1).
Note: E. coli lysate can be produced using the following protocol.- Inoculate 200 ml LB-medium with E. coli BL21 wild-type and incubate the medium at 37 °C overnight on an orbital shaker set to 150 rpm.
- Centrifuge bacteria suspension at 3,000 x g at 4 °C for 10 min.
- Discard the supernatant and resuspend pellet in 10 ml PBS (this suspension can be stored at -20 °C).
- Add 2 mM DTT and 500 µl Complete Protease inhibitor to the 10 ml bacteria suspension.
- Lyse bacteria with a homogenizer.
- Centrifuge lysate at 22,000 x g at 4 °C for 30 min.
- Take supernatant and determine protein concentration.
- Mix cleared lysate with water-free glycerol at a ratio of 1:1.
- Store lysate at -20 °C.
Data analysis
The acquisition and analysis software GenePix Pro 6.0 is used to generate from the scanner images gpr-files containing the mean fluorescence intensity (MFI) values for each processed slide. The gpr-files are analyzed using the statistical programming language R.
For determination of the degree of on-chip protein expression by antibodies targeting the terminal 6xHis- or V5-tag, respectively, the acquired gpr-files are imported to R and each protein spot’s MFI is compared to the MFI values of the negative control spots (made from control PCR lacking DNA-template). A protein is considered to be expressed if its final MFI (either 6xHis or V5 signal) exceeds the mean MFI value of the control spots plus five standard deviations.
For analysis of proteome immunoassays, the acquired gpr-files of all relevant microarray experiments are imported into R, and a threshold is defined as mean MFI of all negative controls plus five standard deviations. Before moving on to marker selection, visual quality control is done to all scanned slides for identification of possible artifacts. In addition, each slide’s MFI values are plotted according to their position on the slide. This allows the recognition of technical effects on individual slides, like gradients of the background or of the MFI values. All antigens showing MFI values above threshold are considered to be reactive with antibodies of the respective patient serum sample. In order to allow comparisons between different slides, an MFI signal is calculated by:
MFI signal = (MFI of antigen)/(MFI of threshold)
An MFI signal > 1 indicates a significant reactivity with a certain antigen.
Recipes
- BSA-blocking buffer
2% BSA in PBST - NTA-solution
90 g nitrilotriacetic acid
150 g NaHCO3
Add ddH2O to 750 ml - Nickel-solution
Dissolve 10 g NiSO4 in 1,000 ml ddH2O - LB-medium
10 g Tryptone
5 g NaCl
5 g Yeast extract
Add 800 ml of ddH2O
Adjust pH to 7.4 with NaOH
Add ddH2O to 1,000 ml and autoclave - Opti-Solution
22.4 g CaCl2
12 ml glacial acetic acid
1 ml Tween-20
Add ddH2O to 1,000 ml - 10x PBS
80 g NaCl
2 g KH2PO4
11.1 g Na2HPO4
2 g KCl
Add ddH2O to 1,000 ml - PBST
100 ml 10x PBS
5 ml 10% Tween-20
Add ddH2O to 1,000 ml - TAE (50x)
242 g Tris base
57.1 ml glacial acetic acid
100 ml 0.5 M EDTA
Add ddH2O to 1,000 ml
Dilute to 1x before use
Acknowledgments
During early stages, work was supported by the European Union, as part of the Affinomics consortium and the PaCaNet project funded by the German Federal Ministry of Education and Research (BMBF).
Competing interests
The authors declare no conflicts of interest or competing interests.
References
- Angenendt, P., Kreutzberger, J., Glokler, J. and Hoheisel, J. D. (2006). Generation of high density protein microarrays by cell-free in situ expression of unpurified PCR products. Mol Cell Proteomics 5(9): 1658-1666.
- He, M. and Taussig, M. J. (2001). Single step generation of protein arrays from DNA by cell-free expression and in situ immobilisation (PISA method). Nucleic Acids Res 29(15): E73-73.
- He, M., Stoevesandt, O., Palmer, E. A., Khan, F., Ericsson, O. and Taussig, M. J. (2008). Printing protein arrays from DNA arrays. Nat Methods 5(2): 175-177.
- Hufnagel, K., Lueong, S., Willhauck-Fleckenstein, M., Hotz-Wagenblatt, A., Miao, B., Bauer, A., Michel, A., Butt, J., Pawlita, M., Hoheisel, J. D. and Waterboer, T. (2018). Immunoprofiling of Chlamydia trachomatis using whole-proteome microarrays generated by on-chip in situ expression. Sci Rep 8(1): 7503.
- Ramachandran, N., Hainsworth, E., Bhullar, B., Eisenstein, S., Rosen, B., Lau, A. Y., Walter, J. C. and LaBaer, J. (2004). Self-assembling protein microarrays. Science 305(5680): 86-90.
- Syafrizayanti, Lueong, S. S., Di, C., Schaefer, J. V., Pluckthun, A. and Hoheisel, J. D. (2017). Personalised proteome analysis by means of protein microarrays made from individual patient samples. Sci Rep 7: 39756.
- The ORFeome Collaboration. (2016). The ORFeome Collaboration: a genome-scale human ORF-clone resource. Nat Methods 13(3): 191-192.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Hufnagel, K., Reininger, D., Ng, S. W., Gassert, N., Rohland, J. K., Shahryarhesami, S., Bauer, A. S., Waterboer, T. and Hoheisel, J. D. (2019). In situ, Cell-free Protein Expression on Microarrays and Their Use for the Detection of Immune Responses. Bio-protocol 9(3): e3152. DOI: 10.21769/BioProtoc.3152.
Category
Cancer Biology > Tumor immunology > Immunological assays > Protein analysis
Microbiology > Microbe-host interactions > Bacterium
Molecular Biology > Protein > Protein-protein interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.