- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

CRISPR-Cas9 Mediated Genome Editing in Drosophila

Published: Vol 9, Iss 2, Jan 20, 2019 DOI: 10.21769/BioProtoc.3141 Views: 11385
Reviewed by: Gal HaimovichAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Nov 2013
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
In recent years, great progress has been made in the research of genome editing systems, one of which is the CRISPR-Cas9 system, a powerful technology that is applied to edit animal genome. Here, we describe a CRISPR-Cas9 mediated mutation protocol for efficiently and specifically editing genes in Drosophila. In this optimized system, the mutant progeny can be generated by only injecting a DNA plasmid encoding synthetic guide RNA (sgRNA) under the control of the U6b promoter into transgenic fly embryos in which Cas9 is specifically expressed in the progenitor cells, thus the gene of interest can be edited by the CRISPR in germ cells, with high rate of heritable mutations and few side effects.
Keywords: Genome editingBackground
CRISPR-Cas9, an acquired immune system derived from bacteria, is made up of a single-stranded homing RNA and a Cas9 protein that has the properties of endonuclease activity (Barrangou et al., 2007; Gasiunas et al., 2012). These two elements can form a complex where the sgRNA guides the Cas9 protein to the specific DNA sequences, resulting in the double-stranded DNA break. Compared with the traditional gene editing system like ZFN and TALEN, the efficient CRISPR-Cas9 system is simpler to design and easier to manipulate, which greatly expands its applications.
Drosophila is one of the best model organisms favored by biomedical researchers in various fields that benefit from the advanced genetic tools. Here we describe how to apply the CRISPR-Cas9 system to generate gene-targeted mutations in Drosophila. The specificity of the target is determined by the 20 nt sequence within the sgRNA, which is transcribed under the control of U6b promoter and then recruits the Cas9 protein which is driven by nanos regulator sequence (Ren et al., 2013). In this case, we only need to select sgRNA of the target gene and then clone it into the pU6B plasmid. After plasmid preparation and purification, we microinject it into the embryos of transgenic Cas9 flies, from whose offspring we can get heritable mutations.
Materials and Reagents
- 1.5 ml MaxyClear Microtubes (Axygen, catalog number: MCT-150-C)
- 0.2 ml Polypropylene PCR Tube (Axygen, catalog number: PCR-0208-C)
- Pipette tips (Corning, Axygen®)
- 0.22 μm Millipore filters (Millipore, catalog number: R7MA69670)
- Fly stocks (All Drosophila strains are stored in Tsinghua Fly Center)
- y,v; attp40{nos-Cas9} (TH00788)
- y,v;; attp2{nos-Cas9} (TH00787)
- y sc v (TB00077)
- y sc v; Gla Bc/CyO (TB00023)
- y sc v;; Dr,e/TM3,Sb (TB00139)
- Trans5α Chemically Competent Cell (Transgen Biotech, catalog number: CD201-01)
- PCR primers (Table 1), custom-synthesized
Table 1. The primers for colony PCR and DNA sequencing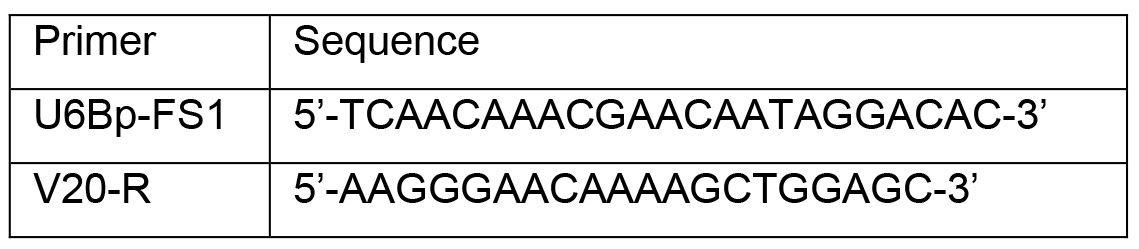
- pU6B (Ampicillin resistant, Figure 1)
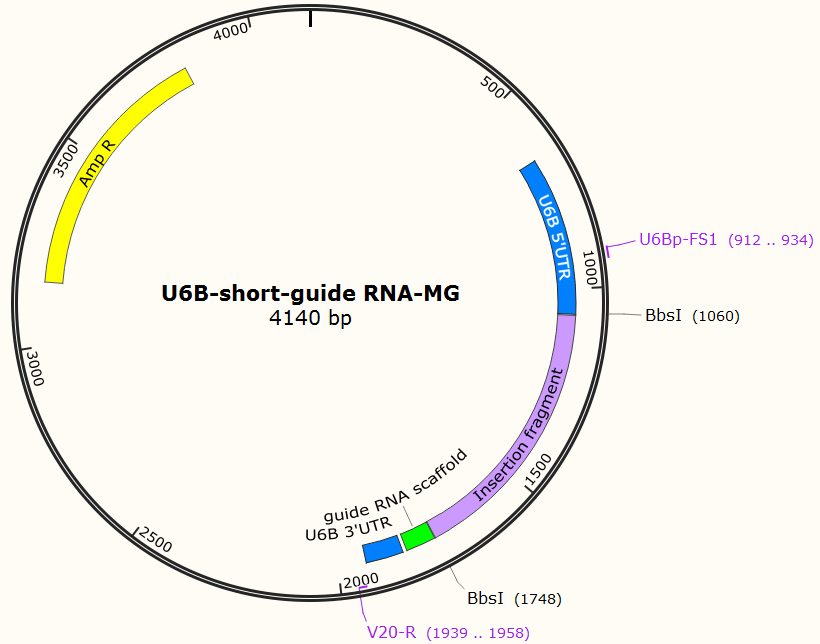
Figure 1. The construct map of pU6B. The insertion fragment between two BbsI sites is the marker to detect whether sgRNA is ligated with the vector. U6Bp-FS1 and V20-R are the primers for colony PCR and DNA sequencing. - BbsI (New England Biolabs, catalog number: R0539L)
- GoTag Green Master Mix, 2x (Promega, catalog number: 000179370)
- T4 DNA ligase (New England Biolabs, catalog number: M0202L)
- AxyPrep Plasmid Miniprep Kit (Corning, Axygen®, catalog number: AP-MN-P-250)
- AxyPrep DNA Gel Extraction Kit (Corning, Axygen®, catalog number: AP-GX-250)
- PurePlasmid Mini Kit (CWBiotech, catalog number: CW0500M)
- Binding Buffer PB (QIAGEN, catalog number: 154051779)
- Phenol-chloroform (Solarbio, catalog number: P1012)
- Ethanol absolute (Sigma, catalog number: SHBC3572V)
- Isopropyl alcohol (Sigma, catalog number: SZBC2260V)
- Sodium acetate (Amrosco, catalog number: 1123C206)
- 10x PBS (Double Helix, catalog number: S0905A)
- 50x TAE (Double Helix, catalog number: P0309A)
- Agarose (Invitrogen, catalog number: 0000602315)
- LB powder (BD, catalog number: 2171199)
- KCl (AMRESCO, catalog number: 7447-40-7)
- Tris-HCl (AMRESCO, catalog number: 1185-53-1)
- EDTA (AMRESCO, catalog number: 60-00-4)
- NaCl (AMRESCO, catalog number: 7647-14-5)
- SDS (Beyotime, catalog number: ST628)
- Proteinase K (Roche, catalog number: P8601-100)
- Ampicillin (Solarbio, catalog number: A8180)
- Ethidium bromide (Sigma, catalog number: E8751)
- 0.2 M Sodium phosphate buffer stock solution (pH 6.8) (see Recipes)
- 0.2 M Na2HPO4 stock solution (see Recipes)
- 0.2 M NaH2PO4 stock solution (see Recipes)
- 10x Injection Buffer (see Recipes)
- 10x Annealing Buffer (see Recipes)
- 1x PBS (see Recipes)
- LB (Luria Bertani) medium (see Recipes)
- 1x TAE (see Recipes)
- Lysis Buffer (see Recipes)
- 1.5% agarose gel (see Recipes)
Equipment
- Pipette (Eppendorf)
- Microwave (SANYO, model: EM-2509EB1)
- Autoclave (SANYO, model: MLS-3780)
- PCR thermal cycler (Eppendorf, Mastercycler nexus GSX1)
- Refrigerated centrifuge (Eppendorf, model: 5417R)
- DNA electrophoresis apparatus (Bio-Rad)
- Microscope (Olympus, model: SZX10)
- NanoDrop 2000 Spectrophotometer (Thermo Scientific)
- Incubator (GINGKO)
- 4 °C refrigerator (Haier)
- -20 °C freezer (Haier)
Procedure
- Construction of plasmid expressing sgRNA targeting the gene of interest (see Figure 2 for an overview of CRISPR plasmid construction)
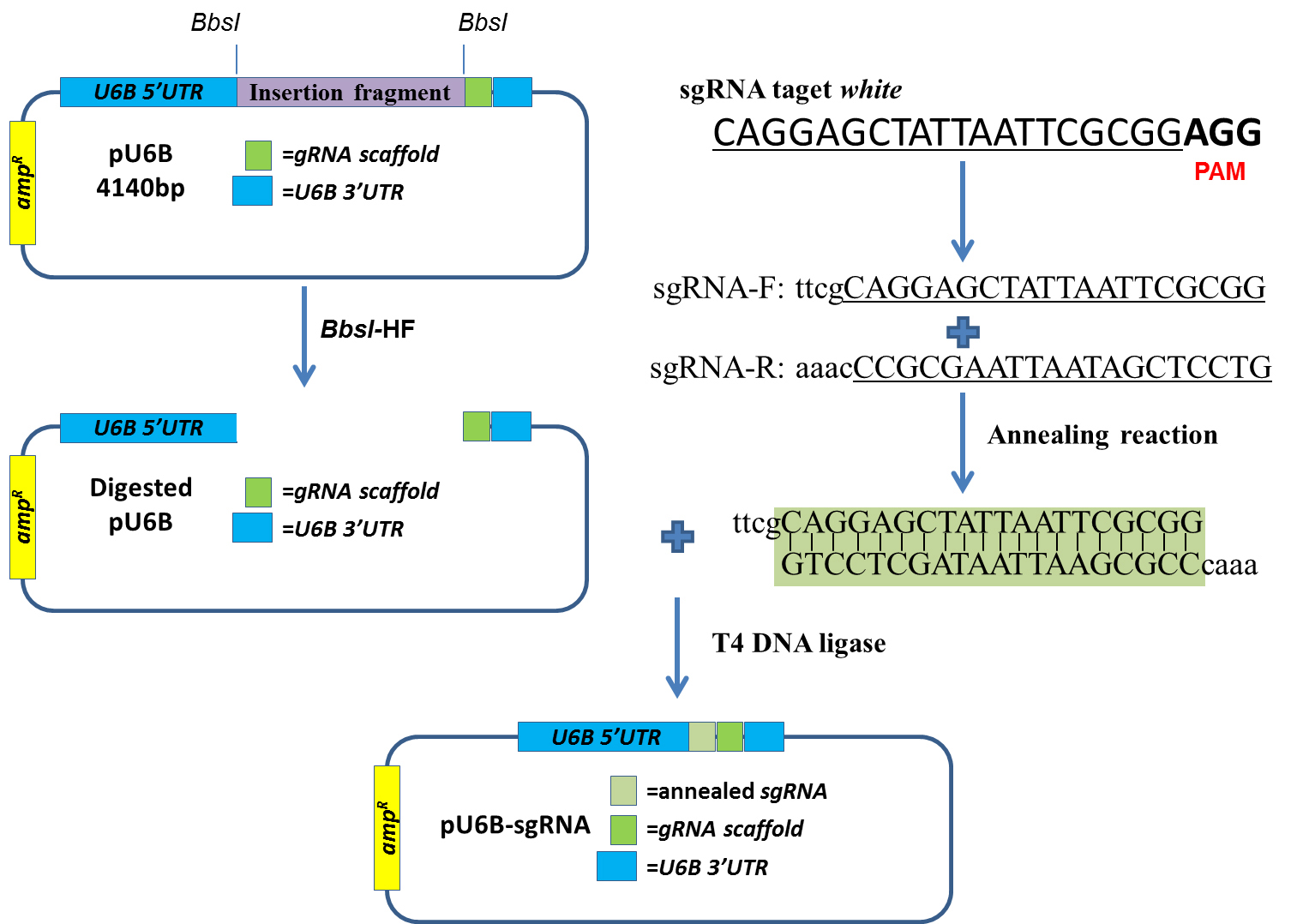
Figure 2. Schematic of the CRISPR plasmid construction. The sgRNA to target white is ligated with the BbsI digested pU6B plasmid.- Digestion of plasmid U6B-short-guide RNA-MG (pU6B) (Figure 1) with restriction endonuclease BbsI-HF
- pU6B is a construct consisting of the U6b regulatory sequence that directs transcription of sgRNA. Set up 50 μl restriction digest reaction:
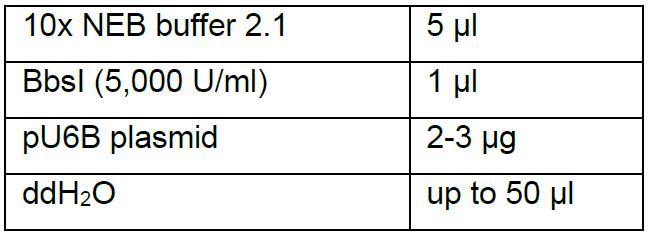
- Incubate the reaction at 37 °C for 1.5 h, followed by 1.5% agarose gel extraction of the 3.5 kb band with AxyPrep DNA Gel Extraction Kit.
- Elute the product with 50 μl ddH2O (the final concentration should be around 50 ng/μl).
- pU6B is a construct consisting of the U6b regulatory sequence that directs transcription of sgRNA. Set up 50 μl restriction digest reaction:
- Preparation of the 20 nt sgRNA targeting the gene of interest
- 20 nt upstream of NGG in the exon of the target gene can be selected as the 20 nt sgRNA. Also, the website (https://www.flyrnai.org/crispr/) can help to select the proper sgRNA sequence. After entering the gene ID, symbol, or chromosome–location in the specified block and clicking the 'Submit' button, a series of sgRNA sequence marked by green points can be selected. According to the efficiency, frameshift and seedScore, optimal sequence can be obtained. Previous study showed that sgRNAs with three or fewer GCs in the six nucleotides closest to the PAM rarely reach a 60% heritable mutation rate, but sgRNAs with at least 4 GCs in that region nearly always have a heritable mutation rate over 60% (Ren et al., 2014).
Note: It is better to select the sgRNA close to the 5’ terminus of the target gene or located in the essential functional domains. In addition, if a restriction site is found in the 20 nt sgRNA sequence, it is likely to be destroyed by CRISPR-Cas9 mediated heritable mutagenesis, providing a convenient way for screening mutant progeny. - Two complementary 24 nt primers with a 20 nt sgRNA sequence should be designed to generate a double-strand DNA with 4 bp appropriate overhangs on both ends and cloned into BbsI digested pU6B. Forward primer consists of the 5’ insert TTCG followed by the 20 nt sgRNA sequence specific to the target gene while reverse primer consists of the 5’ insert AAAC followed by the 20 nt reverse complement.
- Preparation of DNA annealing:

- Denature at 95 °C for 5 min in a thermocycler, then stop heating and let it cool down to room temperature slowly.
- 20 nt upstream of NGG in the exon of the target gene can be selected as the 20 nt sgRNA. Also, the website (https://www.flyrnai.org/crispr/) can help to select the proper sgRNA sequence. After entering the gene ID, symbol, or chromosome–location in the specified block and clicking the 'Submit' button, a series of sgRNA sequence marked by green points can be selected. According to the efficiency, frameshift and seedScore, optimal sequence can be obtained. Previous study showed that sgRNAs with three or fewer GCs in the six nucleotides closest to the PAM rarely reach a 60% heritable mutation rate, but sgRNAs with at least 4 GCs in that region nearly always have a heritable mutation rate over 60% (Ren et al., 2014).
- Ligation
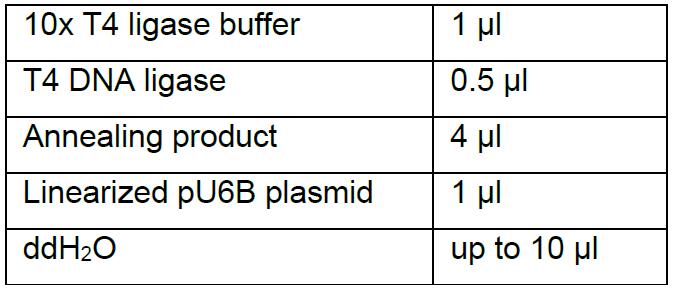
Incubate the reaction at room temperature for half an hour, transform 10 μl Trans5α Chemically Competent Cells (Transgene) with 5 μl ligation product according to the standard transfection procedure and then plate on the LB solid medium with 100 μg/ml ampicillin. Incubate the plate upside-down at 37 °C overnight. - Selection of the correct clone by PCR and verification of the insertion by DNA sequencing
- Perform colony PCR with primer U6Bp-FS1 and primer V20-R. Mark and pick the colonies with 20 μl tips into 10 μl PCR reaction mix:
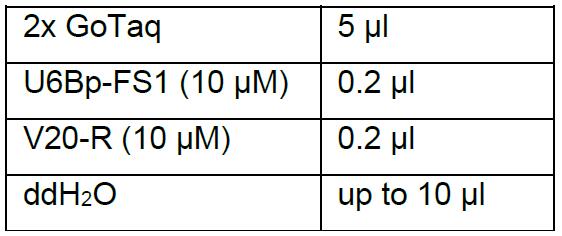
Thermal cycling condition is: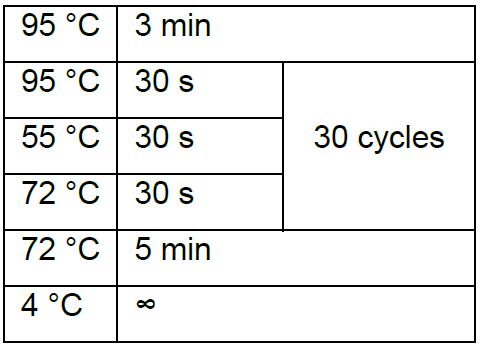
- The correct clone will give around 300 bp PCR product, while no band or around 1 kb band is incorrect. Then inoculate a correct clone into the liquid LB culture and extract plasmids with AxyPrep Plasmid Miniprep Kit following standard procedure. Confirm the correct insertion by Sanger sequencing with primer U6Bp-FS1.
- Perform colony PCR with primer U6Bp-FS1 and primer V20-R. Mark and pick the colonies with 20 μl tips into 10 μl PCR reaction mix:
- Digestion of plasmid U6B-short-guide RNA-MG (pU6B) (Figure 1) with restriction endonuclease BbsI-HF
- Plasmid pU6B-sgRNA purification and injection
- Add Binding Buffer PB, isopropanol and sodium acetate to 10 mg plasmid pU6B-sgRNA DNA and invert to mix thoroughly. Transfer the mixture to mini prep column to purify the plasmids. Elute the column with 1x Injection Buffer. Adjust the final concentration to approximately 75 ng/μl (Ren et al., 2014).
Note: Before injection, we recommend centrifuging the purified plasmid DNA dissolved in the 1x injection buffer at 4 °C at 12,000 x g for more than 20 min to allow some insoluble material to precipitate. - Inject the plasmids in 1x Injection Buffer (~75 ng/μl) into the embryos of transgenic Cas9 flies collected within one hour. The microinjection follows the standard procedure as described in (Ni et al., 2011). The injected embryos should be kept at 25 °C and 60% humidity to adulthood (G0).
Note: The transgenic Cas9 flies were created through integrating nos-Cas9 plasmid (Ren et al., 2013) into attP40 landing site on the second chromosome or attP2 landing site on the third chromosome by phiC31 integrase, followed by screening for the vermillion+ marker present in the nos-Cas9 plasmid. If we want to mutate a gene on the third chromosome, we should choose TH00788 (y,v; attp40{nos-Cas9}) to microinject, while a gene on the second chromosome to choose TH00787 (y,v;; attp2{nos-Cas9}). Also, both TH00788 and TH00787 can be selected to microinject to mutate a gene on the X or the fourth chromosome. Please note that nos-Cas9 and target gene should be expressed on different chromosomes in order to separate the chromosome with Cas9 in next step.
- Add Binding Buffer PB, isopropanol and sodium acetate to 10 mg plasmid pU6B-sgRNA DNA and invert to mix thoroughly. Transfer the mixture to mini prep column to purify the plasmids. Elute the column with 1x Injection Buffer. Adjust the final concentration to approximately 75 ng/μl (Ren et al., 2014).
- Screening of mutations (see Figure 3 for an overview of screening procedure)
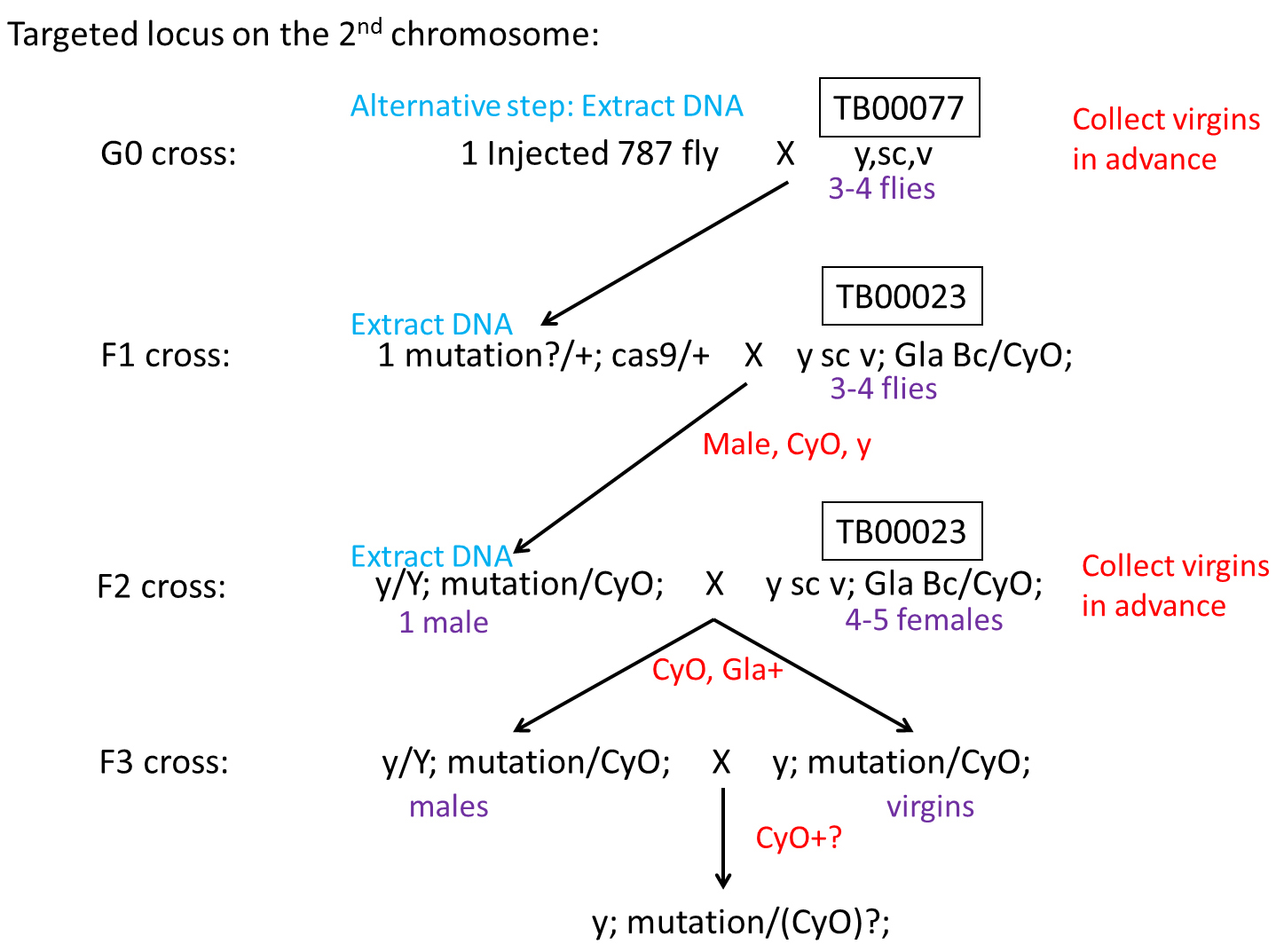
Figure 3. Overview of the procedure of screening CRISPR/Cas9-induced mutations on the 2nd chromosome- First cross all G0 adult flies hatched from injected embryos with TB00077 individually in order to increase the number of the offspring.
- Cross each F1 progeny with flies carrying an appropriate balancer (e.g., for chromosome II targets, use y sc v; Gla Bc/CyO or similar), making the offspring with a chromosome mutation more stable. After spawning, pick a single F1 and extract genomic DNA for PCR amplification of target loci via phenol-chloroform extraction.
- Homogenize each one in 400 μl of Lysis Buffer. Incubate the lysate at 55 °C for 1 h, followed by extraction in 400 μl of phenol-chloroform.
- Centrifuge the mixture is at 12,000 x g for 20 min at 4 °C. Then transfer the supernatant is to a new Eppendorf tube and add an equal volume of isopropanol and 10 μl sodium acetate.
- The well-mixed liquid is kept at -20 °C for at least 1 h, followed by centrifugation at 12,000 x g for 20 min at 4 °C. Remove the supernatant with a pipette, and wash the pellet with 500 μl of 75% ethanol, followed by centrifugation at 12,000 x g for 20 min at 4 °C.
- Finally, dissolve the pellet in 30 μl of ddH2O.
- Design the gene-specific primers to amplify a region surrounding the target locus, and make sure that the GC content of the primers is about 60% so that they can be annealed at 55 °C. Using 0.6 μl of the genomic DNA extract as a template, amplify the DNA sequence surrounding the target loci by PCR for 35 cycles using the following reaction system:

- Further confirm the mutagenesis events by sequencing the PCR-amplified product.
- The F2 progeny are then screened for yellow- to separate the chromosome with nos-Cas9.
- Half of the F2 progenies now carry a single set of mutagenized chromosomes and thus can be tested for the presence of the mutation by mating males singly to several virgin females. After spawning, we pick a single F2 and extract genome DNA for PCR amplification of target loci via phenol-chloroform extraction. Further identify the mutants we want through enzyme digestion or DNA sequencing as described in Step C2.
Note: Since female flies are prone to homologous recombination to repair the mutant chromosomes, we recommend using male flies for mating. - Finally, F3 flies with the same mutation results are crossed individually, homozygote mutant would be generated in the offspring. Keep the mutant lines as balanced heterozygotes if the homozygous flies are unhealthy or lethal.
Data analysis
PCR products amplified from the genome DNA of individual F1 and F2 flies will be confirmed by DNA sequencing. The sequence of the mutation will be detected instead of the wild-type sequence through the overlapped double peaks. The homozygous progenies in F3 will only have single peaks of the mutated gDNA sequence.
Recipes
- 10x Injection Buffer
1 mM sodium phosphate buffer (pH 6.8)
50 mM KCl
Prepare in sterile distilled water and filter through 0.22 μm Millipore filters
Store at -20 °C - 0.2 M Sodium phosphate buffer (pH 6.8) (stock solution)
51 ml 0.2 M NaH2PO4
49 ml 0.2 M Na2HPO4
Store at room temperature - 0.2 M Na2HPO4 (stock solution)
Dissolve 71.6 g Na2HPO4•12H2O in 1 L of ddH2O, mix thoroughly
Store at room temperature - 0.2 M NaH2PO4 (stock solution)
Dissolve 31.2 g Na2HPO4•2H2O in 1 L of ddH2O, mix thoroughly
Store at room temperature - 10x Annealing Buffer
100 mM Tris-HCl (pH 7.5)
10 mM EDTA
1 M NaCl
Store at room temperature - 1x PBS
100 ml 10x PBS
900 ml ddH2O
Store at room temperature - LB (Luria Bertani) medium
Dissolve 25 g of LB powder in 1 L of ddH2O, mix thoroughly
Autoclave at 121 °C for 15 min - 1x TAE
20 ml 50x TAE
980 ml ddH2O
Store at room temperature - Lysis Buffer
50 ml 1x PBS
0.2% SDS
200 μg/ml Proteinase K
Store at 4 °C - 1.5% agarose gel
1.5 g agarose
100 ml 1x TAE
Heat solution to dissolve agarose in a microwave
Add ethidium bromide to a final concentration of 0.2 μg/ml
Acknowledgments
This work was supported by NIH Grants R01GM084947 and R24OD021997 and the NIH’s Ruth L. Kirschstein National Research Service Award F32GM113395 from the NIH General Medical Sciences Division. This work was supported by the National Key Technology Research and Development Program of the Ministry of Science and Technology of the People's Republic of China (2015BAI09B03, 2016YFE0113700), and the National Natural Science Foundation of China (31571320).
Competing interests
The authors declare no competing financial interests.
References
- Barrangou, R., Fremaux, C., Deveau, H., Richards, M., Boyaval, P., Moineau, S., Romero, D. A. and Horvath, P. (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315(5819): 1709-1712.
- Gasiunas, G., Barrangou, R., Horvath, P. and Siksnys, V. (2012). Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci U S A 109(39): E2579-E2586.
- Ni, J. Q., Zhou, R., Czech, B., Liu, L. P., Holderbaum, L., Yang-Zhou, D., Shim, H. S., Tao, R., Handler, D., Karpowicz, P., Binari, R., Booker, M., Brennecke, J., Perkins, L. A., Hannon, G. J. and Perrimon, N. (2011). A genome-scale shRNA resource for transgenic RNAi in Drosophila. Nat Methods 8(5): 405-407.
- Ren, X., Sun, J., Housden, B. E., Hu, Y., Roesel, C., Lin, S., Liu, L. P., Yang, Z., Mao, D., Sun, L., Wu, Q., Ji, J. Y., Xi, J., Mohr, S. E., Xu, J., Perrimon, N. and Ni, J. Q. (2013). Optimized gene editing technology for Drosophila melanogaster using germ line-specific Cas9. Proc Natl Acad Sci U S A 110(47): 19012-19017.
- Ren, X., Yang, Z., Xu, J., Sun, J., Mao, D., Hu, Y., Yang, S. J., Qiao, H. H., Wang, X., Hu, Q., Deng, P., Liu, L. P., Ji, J. Y., Li, J. B. and Ni, J. Q. (2014). Enhanced specificity and efficiency of the CRISPR/Cas9 system with optimized sgRNA parameters in Drosophila. Cell Rep 9(3): 1151-1162.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Peng, P., Wang, X., Shen, D., Sun, J., Jia, Y., Xu, R., Zhu, L. and Ni, J. (2019). CRISPR-Cas9 Mediated Genome Editing in Drosophila. Bio-protocol 9(2): e3141. DOI: 10.21769/BioProtoc.3141.
Category
Molecular Biology > DNA > DNA modification
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.