- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Analysis of DNA Exchange Using Thymidine Analogs (ADExTA) in Trypanosoma cruzi


Published: Vol 8, Iss 24, Dec 20, 2018 DOI: 10.21769/BioProtoc.3125 Views: 4953
Reviewed by: Gal HaimovichNoelia LanderEmilia Krypotou
Original research article
The authors used this protocol in:

Sep 2018

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Trypanosoma cruzi is a protozoan parasite belonging to the Trypanosomatidae family. Although the trypanosomatids multiply predominantly by clonal generation, the presence of DNA exchange in some of them has been puzzling researchers over the years, mainly because it may represent a novel form that these organisms use to gain variability. Analysis of DNA Exchange using Thymidine Analogs (ADExTA) is a method that allows the in vitro detection and measurement of rates of DNA exchange, particularly in trypanosomatid cells, in a rapid and simple manner by indirect immunofluorescence assay (IFA). The method can be used to detect DNA exchange within one trypanosomatid lineage or among different lineages by paired analysis. The principle of this assay is based on the incorporation of two distinguishable halogenated thymidine analogs called 5′-chloro-2′-deoxyuridine (CldU) and 5′-iodo-2′-deoxyuridine (IdU) during DNA replication. After mixing the two cell cultures that had been previously incorporated with CldU and IdU separately, the presence of these unusual deoxynucleosides in the genome can be detected by specific antibodies. For this, a DNA denaturation step is required to expose the sites of thymidine analogs incorporated. Subsequently, a secondary reaction using fluorochrome-labeled antibodies will generate distinct signals under fluorescence analysis. By using this method, DNA exchange verification (i.e., the presence of both CldU and IdU in the same cell) is possible using a standard fluorescence microscope. It typically takes 2-3 days from the thymidine analogs incorporation to results. Of note, ADExTA is relatively cheap and does not require transfections or harsh genetic manipulation. These features represent an advantage when compared to other time-consuming protocols that demand DNA manipulation to introduce distinct drug-resistance markers in different cells for posterior selection.
Keywords: DNA exchangeBackground
Trypanosomatids parasites are single-celled eukaryotes within the supergroup Excavata (Adl et al., 2012). Among them, there are human pathogens responsible for causing several devastating diseases such as T. cruzi (etiological agent of Chagas disease, also called American trypanosomiasis), T. brucei (etiological agent of sleeping sickness, also called Human African trypanosomiasis), and Leishmania spp. (etiological agent of distinct forms of leishmaniasis). Altogether, these peculiar organisms are responsible for more than 50,000 deaths annually worldwide (Ponte-Sucre, 2016; Browne et al., 2017; da Silva et al., 2017b; Torres-Guerrero et al., 2017). Although trypanosomatids predominantly multiply by clonal generation through longitudinal binary fission, the presence of DNA exchange in these organisms, including T. cruzi and Leishmania spp., have been debated over the years (Gaunt et al., 2003; Alves et al., 2018). Due to the lack of practical and straightforward experiments and due to the difficulty of reproducing complex assays, DNA exchange still little explored, even though it may represent a novel and unknown form that trypanosomatids use to gain variability. However, a recent work developed by our group presented an elegant approach to detect DNA exchange in different strains of T. cruzi (Alves et al., 2018). In this study, we figured out higher rates of DNA exchange in naturally-occurring hybrid CL Brener strain (TcIV) relative to naturally-occurring non-hybrid Y strain (TcII). Also, this work pointed out that the recombinase Rad51 contributes significantly to the efficiency of this mechanism (Alves et al., 2018).
Here, we describe in detail the protocol used in this study to detect DNA exchange in trypanosomatids. We named it as Analysis of DNA Exchange using Thymidine Analogs (ADExTA). The principle of the method is based on the incorporation of two distinguishable halogenated thymidine analogs [e.g., 5′-chloro-2′-deoxyuridine (CldU) and 5′-iodo-2′-deoxyuridine (IdU)] into DNA during the S phase of the cell cycle. The presence of these unusual deoxynucleosides in the genome can be detected by specific antibodies after a DNA denaturation step using HCl, which will generate distinct signals after a secondary reaction using fluorochrome-labeled antibodies. This principle is the same used in the technique single-molecule analysis of DNA replication (Herrick and Bensimon, 1999), usually called DNA combing (Calderano et al., 2015; Stanojcic et al., 2016; da Silva et al., 2018). The method is rapid, relatively simple and provides a good overview of the presence of DNA exchange, as well as its rate. Of note, this protocol represents a considerable advantage when compared to other laborious approaches that require harsh genetic manipulation and is time-consuming, such as constructions using recombinant DNA to introduce distinct drug-resistance markers in different cells. By using this method, the presence of DNA exchange and its measurement can be carried out using a standard fluorescence microscope. Theoretically, this protocol can be applied to any organism that allows the incorporation of CldU/IdU, and it typically takes 2-3 days from the thymidine analogs incorporation to results. Of note, although thymidine analogs can be toxic for some cell types (Davidson and Kaufman, 1979; Hancock et al., 2009), in trypanosomatids they have been used apparently without showing any toxic effect (Elias et al., 2007; da Silva et al., 2013; Stanojcic et al., 2016; da Silva et al., 2017a).
Materials and Reagents
- Microtubes 1.5 ml (Axygen, Maxyclear, catalog number: MCT-150-C-S)
- Centrifuge tubes 15 ml (Corning, catalog number: CLS430791)
- Centrifuge tubes 50 ml (Corning, catalog number: CLS430290)
- Culture flasks 25 cm2 (Corning, canted neck, cap plug seal, catalog number: CLS430168)
- Syringe filter 0.22 μm (Sartori, Minisart Syringe filter, catalog number: 16534)
- Micropipette tips, 10 μl, 200 μl and 1,000 μl (Axygen, catalog numbers: T-300, T-200-Y and T-1000-B)
- Serological pipettes, 10 ml (Costar Sterile, catalog number: 4488)
- Microscope Slides (Knittel glass, non-color, catalog number: VA111101FKB.01)
- Coverslips (Knitell glass, 22 x 22 mm, catalog number: VD12222Y1A.01)
- Plastic coverslips (use a cut plastic pocket for binder)
- T. cruzi cell lines: Y strain (TcII) and CL Brener strain (TcIV)
- 5′-Iodo-2′-deoxyuridine (IdU) (Sigma-Aldrich, catalog number: I7125)
- 5′-Chloro-2′-deoxyuridine (CldU) (Abcam, catalog number: ab213715)
- Mouse α-IdU monoclonal antibody (BD, catalog number: 347580)
- Rat α-CldU monoclonal antibody (Accurate, catalog number: YSRTMCA2060GA)
- Goat α-mouse IgG1 secondary antibody, Alexa Fluor 568 (Thermo Scientific, catalog number: A-21124)
- Goat α-rat IgG (H + L) secondary antibody, Alexa Fluor 488 (Thermo Scientific, catalog number: A-11006)
- Paraformaldehyde (Sigma-Aldrich, catalog number: 158127)
- Poly-L-lysine hydrochloride (Sigma-Aldrich, catalog number: P2658)
- Bovine serum albumin (Sigma-Aldrich, catalog number: 05470)
- Triton X-100 (Sigma-Aldrich, catalog number: T8787)
- NaCl (Sigma-Aldrich, catalog number: S9888)
- KCl (Sigma-Aldrich, catalog number: P9541)
- Na2HPO4 (Sigma-Aldrich, catalog number: 255793)
- KH2PO4 (Merck, catalog number: 104873)
- D-Glucose (Sigma-Aldrich, catalog number: G8270)
- Liver Infusion broth (BD, catalog number: 226920)
- Tryptose (Sigma-Aldrich, catalog number: 70937)
- Hemin (Sigma-Aldrich, catalog number: H9039)
- Triethanolamine (Sigma-Aldrich, catalog number: 90279)
- Fetal Bovine Serum (Fisher Scientific, catalog number: 10500056)
- Penicillin G sodium salt (Sigma-Aldrich, catalog number: 13752)
- Streptomycin sulfate salt (Sigma-Aldrich, catalog number: S9137)
- Boric Acid (Sigma-Aldrich, catalog number: B6768)
- Sodium tetraborate (Sigma-Aldrich, catalog number: 221732)
- NaOH (Merck, catalog number: 1064980500)
- Vectashield Mounting Medium with DAPI (Vector Labs, catalog number: H-1200)
- Hydrochloric acid fuming 37% (Merck, catalog number: 100317)
- Nail varnish (any brand, preferably colorless)
- Alkalinized water (see Recipes)
- Phosphate buffered saline (1x PBS) (see Recipes)
- Liver Infusion Tryptose (LIT) medium (see Recipes)
- 5′-chloro-2′-deoxyuridine solution (CldU-S) (see Recipes)
- 5′-iodo-2′-deoxyuridine solution (IdU-S) (see Recipes)
- Fixation buffer (FB) (see Recipes)
- Poly-L-lysine solution (PLS) (see Recipes)
- Permeabilization solution (PS) (see Recipes)
- Denaturation buffer (DB) (see Recipes)
- Neutralization buffer (NB) (see Recipes)
- Blocking solution (BS) (see Recipes)
Equipment
- Microcentrifuge (Eppendorf, model: 5424 R)
- Motorized pipet dispenser (Fisher Scientific, Fisherbrand, catalog number: 03-692-172)
- Water bath (Cientec, model: CT-226)
- Incubator BOD (Vitrex, model: NI1705)
- Fluorescence Microscope [Olympus, model: BX51, coupled to an XM10 digital camera. Filters specifications: U-MWU2 (excitation = 330-385 nm; emission = 420 nm), U-MWIBA3 (excitation = 460-495 nm; emission = 510-550 nm), and U-MWG2 (excitation = 510-550 nm; emission = 590 nm)]
- Micropipettes (Gilson, models: Pipetman P10, P20, P200 and P1000)
- Centrifuge (Eppendorf, model: 5810 R), equipped with 4 x 250 ml Swing-Bucket Rotor
- Neubauer chamber with cover glass (Sigma-Aldrich, model: Bright-LineTM Hemacytometer)
- Biosafety Class II A2 cabinet (Pachane, model: PA 700)
- pH meter (Gehaka, model: PG1800)
- Autoclave
Software
- Olympus Cell F software (Olympus, version 5.1.2640)
- ImageJ (NIH, version 1.47t)
- Microsoft Excel (Microsoft Office-any version) or GraphPad Prism (GraphPad software, Inc.)
Procedure
-Before starting with this protocol, we recommend reading it completely, especially the Notes section at the end of this article.
-Cells should be cultured in medium and growth conditions that are appropriate for the given cell type. To achieve a suitable number of labeled cells during data analysis, they must be in the exponential phase.
-Although the thymidine analogs can be toxic when used in high dosages for long periods of incorporation, we did not detect any alterations regarding toxicity in our T. cruzi cell lines during our analysis.
-Important: If the antibodies that will be used in this protocol have not been previously tested for specificity, we strongly recommend that Procedure B (controls for checking the specificity of the antibodies) be performed first.
- Analysis of the DNA exchange
- Incubate T. cruzi cells (28 °C) until they reach exponential phase (~1 x 107 cells/ml) in 10 ml of culture. The parasites density varies according to the lineage used.
Note: In our assays, we used two different lineages: T. cruzi Y strain (TcII), and T. cruzi CL Brener strain (TcIV), both in epimastigote forms. The exponential phase may vary according to each strain. - Split the exponential T. cruzi culture into two culture flasks (25 cm2) adding 5 ml per flask.
- Thaw both solutions of thymidine analogs CldU-S and IdU-S at room temperature.
- Add one of the thymidine analogs (e.g., CldU) to a final concentration of 100 μM in one flask, and the other one (e.g., IdU) in the remaining flask, also to a final concentration of 100 μM.
- Incubate each flask containing the different thymidine analogs for 12 h at 28 °C.
Note: The incubation time, as well as the temperature, may vary according to each parasite strain or cell type. In our protocol, the incubation time (period of thymidine analogs incorporation) was determined empirically after a long period of testing. We managed to get good results using a period corresponding to the half of the doubling time of the lineage used (T. cruzi doubling time is 24 h). - Warm an aliquot of 200 ml of Liver Infusion Tryptose (LIT) medium at 28 °C.
Note: If it is available, use a water bath. - Collect the 5 ml of T. cruzi parasites CldU- or IdU-incorporated by centrifugation at 800 x g for 5 min (from Step A6).
- Remove the medium from cells carefully and replace it with 10 ml of LIT at 28 °C.
- Suspend the pellet carefully and repeat the Steps A7-A8 twice for a total of three washes with LIT. For the last wash, suspend the pellet carefully into 5 ml LIT.
- Mix the both CldU- and IdU-incorporated T. cruzi cultures into one single flask, totalizing a volume of 10 ml.
- Incubate for 24 h at 28 °C.
Note: The incubation time, as well as the temperature, may vary according to each parasite strain or cell type. - Chill the 1x PBS to 4 °C.
- Harvest the cells by centrifugation at 800 x g for 5 min at 4 °C and wash twice each using 5 ml of cold 1x PBS.
- Remove the 1x PBS from cells carefully to preserve the pellet.
- Suspend the pellet in 1 ml of cold Fixation buffer (FB) and transfer to a 1.5 ml microcentrifuge tube.
- Incubate at 4 °C for 7 min and wash three times using 5 ml of cold 1x PBS (centrifuging at 800 x g for 5 min in each wash) each. For the last wash, suspend the pellet carefully into 500 μl of 1x PBS. If the pellet is too small (i.e., almost invisible to the naked eye), decrease the volume of the last wash to 100 μl of 1x PBS.
- Prepare the slides to receive the T. cruzi cells by spreading 2.5 μl of Poly-L-lysine solution (PLS) onto slide surface until PLS dry out. Prepare three slides for each sample from Step A15.
Note: Use a coverslip to help spreading the PLS onto slide (see Figure 1 for more detail).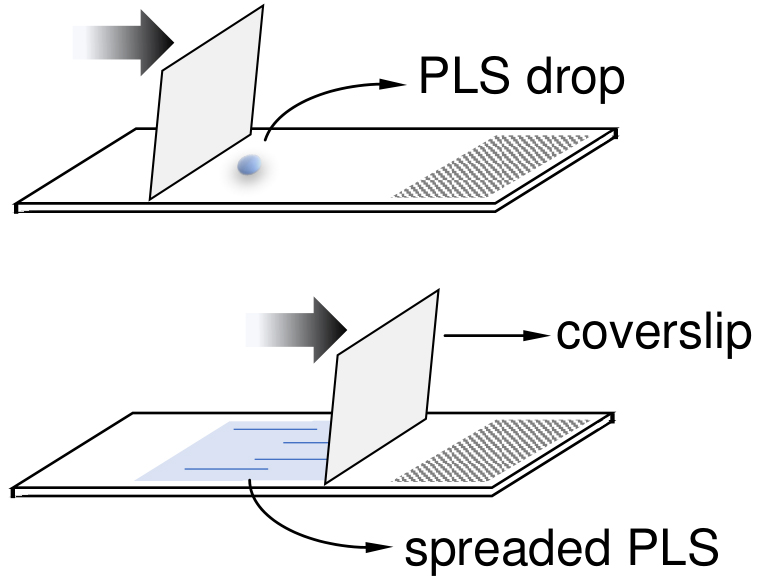
Figure 1. Illustration to clarify the Step A17. A coverslip can be used to help to spread out the PLS drop (2.5 μl) onto slides. - Spread the suspended-pellet (from Step A16) carefully in each of the three slides. Use 25-30 μl and save the remaining suspended-pellet volume in case you need to remake some slides.
Notes:- Use the same surface where PLS was previous spread.
- Each slide is one technical replicate.
- Wait for the cells to precipitate and settle on slide for 10-15 min at room temperature. Ensure that the cells do not dry out.
- Permeabilize the cells by adding 50 μl of Permeabilization solution (PS) for 10 min at room temperature.
- Wash the slide containing cells three times using 1x PBS.
Note: Use a P1000 micropipette to spread (by sneezing) 1x PBS (1 ml) onto slide three times. - Denature the DNA of the cells by adding 50 μl of Denaturation buffer (DB) for 20 min at room temperature.
Note: Use a plastic coverslip to help DB spread out and avoid dry out. - Remove the plastic coverslip and wash the slide containing cells once using 1x PBS.
- Neutralize the reaction by adding 50 μl of Neutralization buffer (NB) for 10 min at room temperature.
Note: Use a plastic coverslip to help NB spread out. - Remove the plastic coverslip and wash the slide containing cells three times using 1x PBS.
- Dilute both primary antibodies [i.e., rat α-CldU monoclonal antibody (Accurate) and mouse α-IdU monoclonal antibody (BD)] 1:300 in Blocking solution (BS).
- Spread out 50 μl of the diluted primary antibodies on the slide-surface containing cells (from Step A25). Incubate for 2 h at room temperature.
Note: Use a plastic coverslip to help primary antibodies spread out and avoid dry out. - Remove the plastic coverslip and wash the slide containing cells three times using 1x PBS.
- Dilute both secondary antibodies [i.e., Alexa Fluor 568-conjugated goat α-mouse (Thermo Scientific) and Alexa Fluor 488-conjugated goat α-rat (Thermo Scientific)] 1:300 in BS.
Note: From this step onwards, do not expose the slides to light. - Spread out 50 μl of the diluted secondary antibodies on the slide-surface containing cells (from Step A28). Incubate for 2 h at room temperature.
Note: Use a plastic coverslip to help primary antibodies spread out and avoid dry out. - Remove the plastic coverslip and wash the slide containing cells three times using 1x PBS.
- Ensure remove of all the liquid from the slide surface containing cells and add 4 μl of Vectashield mounting medium containing DAPI.
Note: This solution is used as the anti-fade mounting solution and to stain organelles containing DNA. - Add a glass coverslip and seal using colorless nail varnish. Wait the varnish dry out for 5 min. The slide can be analyzed under a fluorescence microscope immediately or stored at 4 °C up to one month. Figure 2 shows a scheme containing the main steps of this protocol section.
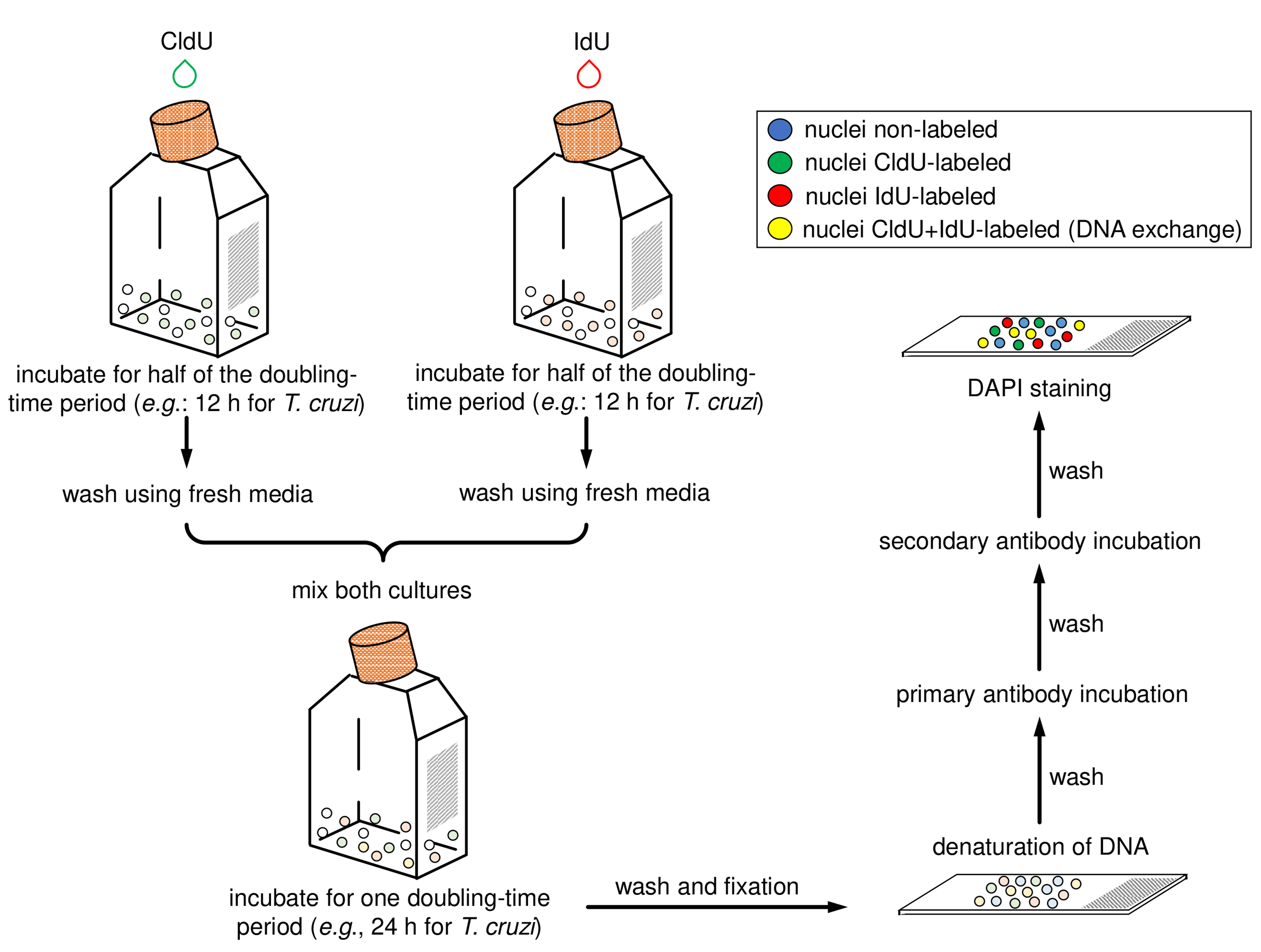
Figure 2. Schematic diagram representing the main steps of the application of the protocol for CL Brener and Y strains (epimastigote forms). The halogenated thymidine analogs (CldU and IdU) are added separately in each culture for 12 h. After that, parasites must be washed with fresh media, mixed, and incubated for 24 h. Then samples must be washed, fixed, and added onto slides. Next, the parasite cells must have their DNA denatured and should be then processed for the detection of the thymidine analogs using primary antibodies (α-CldU and α-IdU) and the corresponding secondary antibodies (Alexa Fluor 488 α-rat and Alexa Fluor 568 α-mouse). Finally, mounting medium with DAPI must be added and the slides sealed.
- Incubate T. cruzi cells (28 °C) until they reach exponential phase (~1 x 107 cells/ml) in 10 ml of culture. The parasites density varies according to the lineage used.
- Controls (checking the specificity of the antibodies)
- Prepare a new set of T. cruzi culture. Choose one strain if you carried out the previous analysis with two strains. Incubate T. cruzi cells (28 °C) until they reach exponential phase (~1 x 107 cells/ml) in 10 ml of culture. The parasites density varies according to the lineage used.
Note: In our assays to check the specificity of the antibodies, we used only the T. cruzi Y strain in epimastigote forms. - Follow the Steps A2-A5 exactly as previously described.
- Chill the 1x PBS to 4 °C.
Note: Do not mix both cultures as previously done. - After the incubation time, harvest separately the CldU- and IdU-incorporated cells by centrifugation at 800 x g for 5 min at
4 °C and wash each sample twice using 5 ml of cold 1x PBS. - Remove the 1x PBS from each sample carefully and suspend each pellet in 1 ml of cold FB. Transfer to a 1.5 ml microcentrifuge tube.
- Incubate each pellet at 4 °C for 7 min and wash them three times using 5 ml of cold 1x PBS (centrifuging at 800 x g for 5 min in each wash). For the last wash, suspend each pellet carefully into 500 μl of 1x PBS. If any of them is too small (i.e., almost invisible to the naked eye), decrease the volume to 100 μl of 1x PBS.
- Prepare four slides to receive the CldU-incorporated cells and four to receive the IdU-incorporated (totalizing eight slides). Spread 2.5 μl of PLS in each slide and let PLS dry out.
Note: Use a coverslip to help spreading the PLS onto slide. - Spread the suspended-pellet from each sample (i.e., CldU- and IdU-incorporated cells) (from Step B7) carefully in the slides (four slides for each thymidine analog group). Use 25 μl and save the remaining suspended-pellet volume in case you need to remake some slides.
Note: Use the same surface where PLS was previous spread. - Wait for the cells to settle on the slides for 10-15 min at room temperature. Ensure that the cells do not dry out.
- Permeabilize the cells by adding 50 μl of PS for 10 min at room temperature.
- Wash each slide containing cells three times using 1x PBS.
Note: Use a P1000 micropipette to spread (by sneezing) 1x PBS (1 ml) onto slide three times. - Denature the DNA of the cells by adding 50 μl of DB for 20 min at room temperature.
Note: Use a plastic coverslip to help DB spread out and avoid dry out. - Remove the plastic coverslip and wash each slide containing cells once using 1x PBS.
- Neutralize the reaction by adding 50 μl of NB for 10 min at room temperature.
Note: Use a plastic coverslip. - Remove the plastic coverslip and wash each slide containing cells three times using 1x PBS.
- Dilute both primary antibodies [i.e., rat α-CldU monoclonal antibody (Accurate) and mouse α-IdU monoclonal antibody (BD)] individually using two separated microtubes. Dilute each one 1:300 in BS.
- Spread out 50 μl of the diluted primary antibody rat α-CldU on the surface of two slides containing CldU-incorporated cells (from Step B15) and on the surface of two slides containing IdU-incorporated cells (from Step B15). Repeat the same procedure for the four remaining slides using the other diluted primary antibody, i.e., mouse α-IdU. Incubate for 2 h at room temperature.
Note: Use a plastic coverslip to help primary antibodies spread out and avoid dry out. - Remove the plastic coverslip and wash the slide containing cells three times using 1x PBS.
- Dilute both secondary antibodies [i.e., Alexa Fluor 568-conjugated α-mouse (Thermo Scientific) and Alexa Fluor 488-conjugated α-rat (Thermo Scientific)] individually using two separated microtubes. Dilute each one 1:300 in BS.
Note: From this step onwards, do not expose the slides to light. - Spread out 50 μl of the diluted secondary antibody Alexa Fluor 488 α-rat on the surface of the following slides containing cells (from Step B19): CldU-incorporated + primary antibody rat α-CldU, CldU-incorporated + primary antibody mouse α-IdU, IdU-incorporated + primary antibody rat α-CldU, and IdU-incorporated + primary antibody mouse α-IdU. Repeat the same procedure for the four remaining slides (from Step B19) using the other diluted secondary antibody Alexa Fluor 568 α-mouse, i.e., CldU-incorporated + primary antibody rat α-CldU, CldU-incorporated + primary antibody mouse α-IdU, IdU-incorporated + primary antibody rat α-CldU, and IdU-incorporated + primary antibody mouse α-IdU. Incubate for 2 h at room temperature.
Note: Use a plastic coverslip to help secondary antibodies spread out and avoid dry out. - Remove the plastic coverslip and wash each slide three times using 1x PBS.
- Ensure removing all the liquid from each and add 4 μl of Vectashield mounting medium containing DAPI.
Note: This solution is used as the anti-fade mounting solution and to stain organelles containing DNA. - Add a glass coverslip and seal each slide using colorless nail varnish. Wait the varnish dry out for 5 min. The slide can be analyzed under a fluorescence microscope immediately or stored at 4 °C up to one month. Figure 3 shows a scheme containing the main steps of this control protocol.
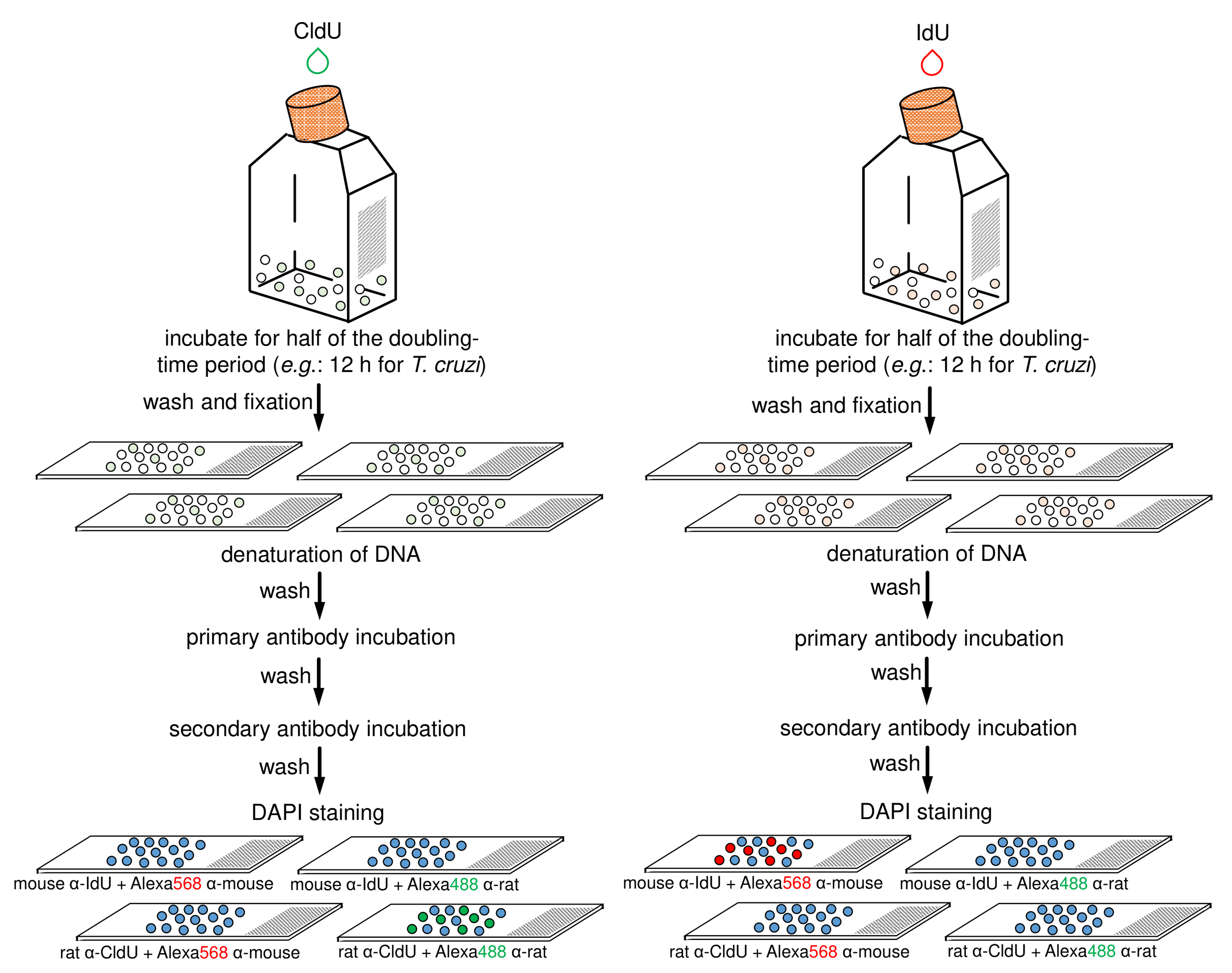
Figure 3. Schematic diagram representing the main steps of antibodies specificity control. This control assay can be applied to any of the two previously strains (i.e., CL Brener and Y). The halogenated thymidine analogs (CldU and IdU) are added separately in each culture for 12 h. After that, each parasite-group (CldU-incorporated and IdU-incorporated) must be washed with 1x PBS, fixed, and aliquots distributed onto four slides (totalizing eight slides for the both groups). Next, each slide containing parasite cells must have their DNA denatured and should be then processed for the detection of the thymidine analogs using primary and secondary antibodies according to the specifications (on the scheme, see the specifications on the bottom of slides) (for details see previous Step B21). Finally, mounting medium with DAPI must be added in each slide and then sealed.
- Prepare a new set of T. cruzi culture. Choose one strain if you carried out the previous analysis with two strains. Incubate T. cruzi cells (28 °C) until they reach exponential phase (~1 x 107 cells/ml) in 10 ml of culture. The parasites density varies according to the lineage used.
Data analysis
- Analyze each slide under a fluorescence microscope (see Equipment for specifications). Capture images using the differential interference contrast (DIC) (if available) or phase contrast. Also, capture images in the fields corresponding to the fluorescence emission blue (DAPI), green (CldU), and red (IdU). Capture images with the same exposure time, especially for CldU and IdU fields.
Notes:- In our analysis, we used an objective lens 100x. Also, the exposure time used to capture the images can vary according to the microscope, filters, and software used. In our case, the images were captured using an exposure time ranging from 200-800 ms.
- Ensure a minimum of one hundred T. cruzi cells is captured per experiment. Each slide of the analysis of DNA exchange (Procedure A) is one technical replicate. This protocol must be prepared in biological triplicate from independent experiments.
- To avoid bias, a double-blind approach should be applied, especially for paired analysis using two different strains.
- In our analysis, we used the Olympus Cell F software (version 5.1.2640).
- Merge the images captured using ImageJ software (or other software that allows this approach). Figures 4 and 5 show representative images of cells containing, respectively: DNA exchange captured in each strain analyzed (i.e., CL Brener and Y), and antibodies specificity control.
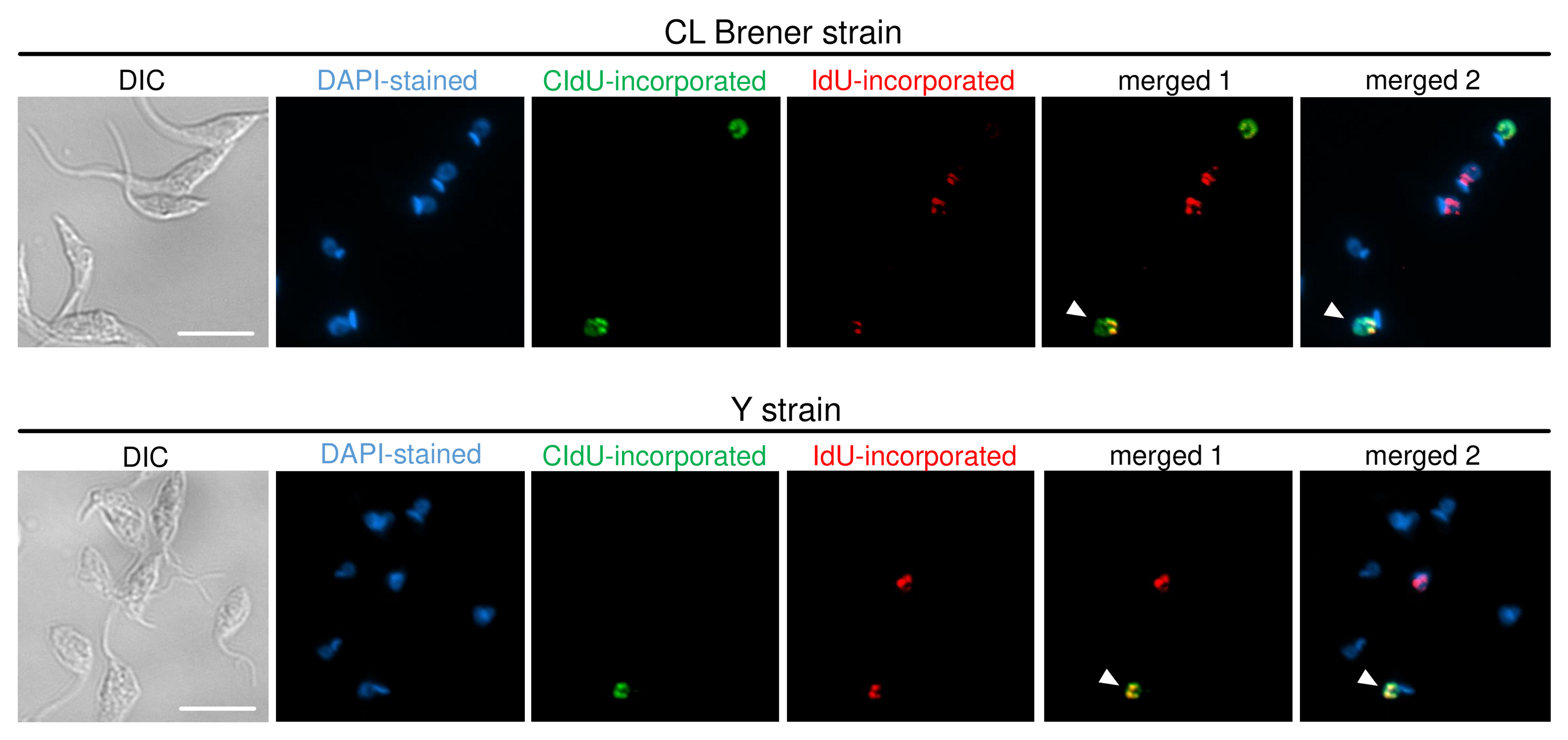
Figure 4. ADExTA applied in CL Brener and Y strains. Representative images are organized in six columns: DIC (to see morphology of the cells), DAPI (staining organelles containing DNA), CldU-incorporated cells (green), IdU-incorporated cells (red), merged 1 (CldU + IdU overlay), and merged 2 (DAPI + CldU + IdU overlay). The white arrows indicate cells that suffered DNA exchange, i.e., they have a nucleus containing CldU and IdU incorporated. Scale bars = 10 μm.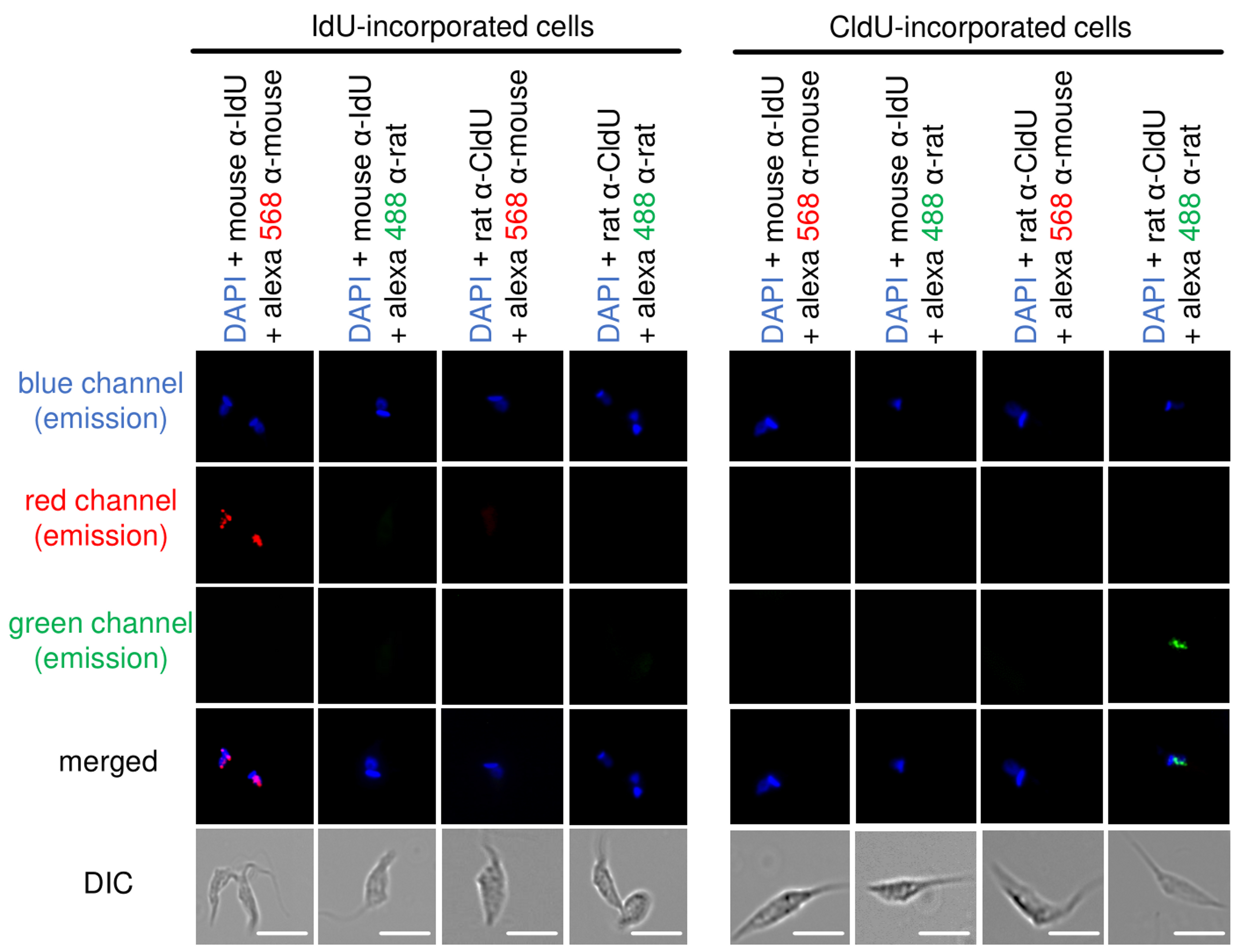
Figure 5. Antibodies specificity control. Representative images of CL Brener strain show that the recognition of thymidine analogs is specific. CldU- and IdU-incorporated epimastigotes were added onto slides and processed for detection using mouse α-IdU + Alexa Fluor 568 (mouse), mouse α-IdU + Alexa Fluor 488 (rat), rat α-CldU + Alexa Fluor 568 (mouse), and rat α-CldU + Alexa Fluor 488 (rat). We can observe complete absence of cross-reactions between primary and secondary antibodies in each fluorescence channel analyzed (blue, red and green). Images were captured randomly. This figure was adapted from Alves et al. (2018). Scale bars = 10 μm. - Count the number of total cells, cells CldU-labeled (green), cells IdU-labeled (red), cells CldU + IdU-labeled (yellow when merged), and cells non-labeled (blue-DAPI).
Note: The number of total cells must be higher than one hundred to allow to obtain reliable results. - Use the Excel software (Microsoft Office) to establish a table (or graphic) containing the percentage of cell CldU- and IdU-labeled, as well as double-labeled cells (i.e., CldU + IdU-labeled).
Note: Other software can be used in place of Excel, for example, GraphPad Prism (GraphPad software). - If you are comparing the DNA exchange between two different strains (e.g., CL Brener and Y strains), perform statistical analysis using Student’s t-test (two-tailed, unpaired t-test with Welch’s correction) to establish a P-value. Applying this test, you will find out if the difference observed is statistically significant or not. Figure 6 shows graphs with the percentage of DNA exchange (yellow column), as well as the P-value estimated for CL Brener and Y strains.
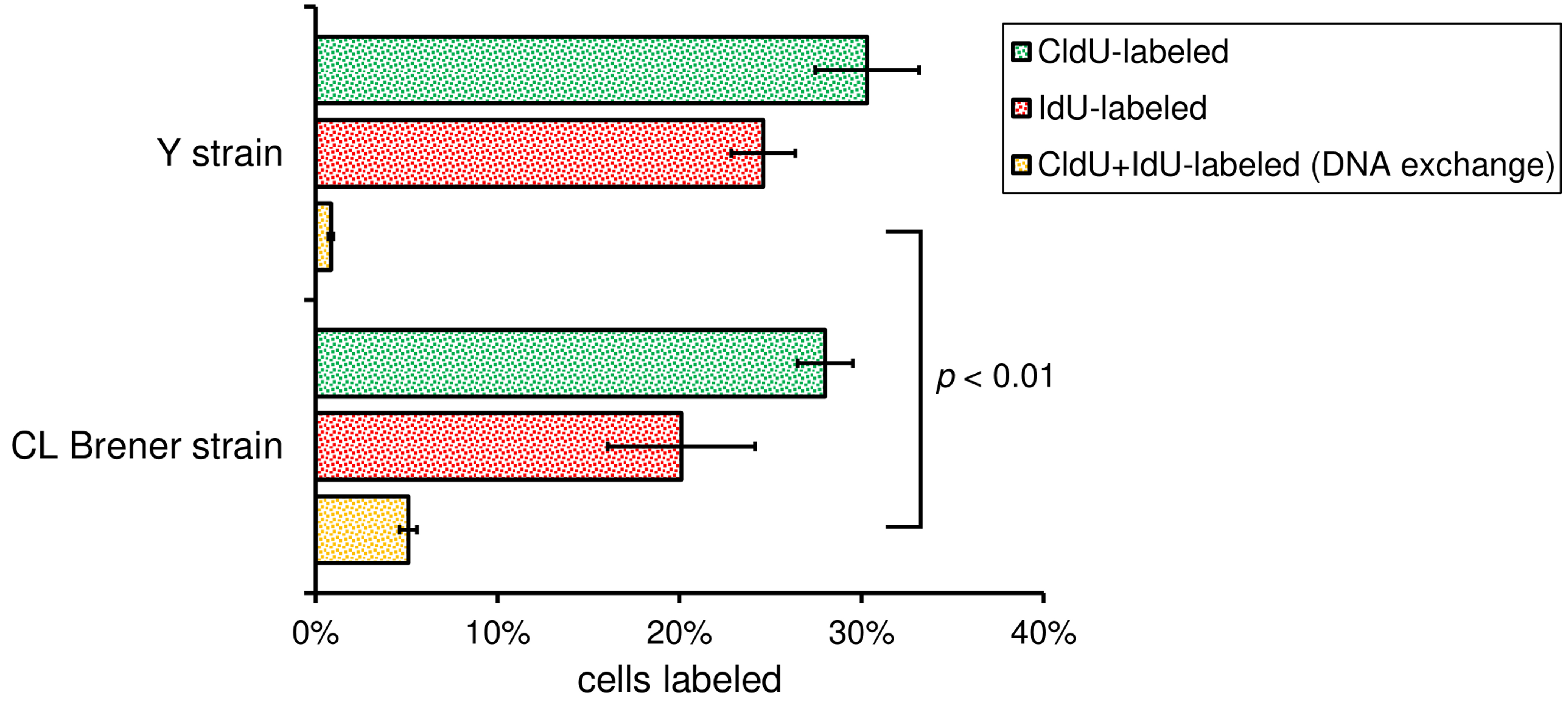
Figure 6. Bar graphs showing the DNA exchange measurement. Bars represent the percentage of cells CldU-labeled (green), IdU-labeled (red), and CldU + IdU-labeled (yellow), which represents DNA exchange, for the two strains analyzed (Cl Brener and Y). More than 200 cells of each strain were analyzed per biological replicate. Error bars indicate SD of the triplicates. P-value was obtained using Student’s t-test. The numerical analysis presented in this figure was originally published in Alves et al. (2018).
Notes
- General notes
- This protocol should be carried out in biological triplicate from three independent experiments. Each experiment generates three technical replicates (three slides).
- Although the most companies that sell the halogenated thymidine analogs (BrdU, CldU or IdU) recommend a DNA denaturation step of 2 M HCl to allow their detection (e.g., Abcam, Sigma-Aldrich), we decided to use 2.5 M HCl to increase the number of sites containing the incorporated thymidine analogs. This approach improves the efficiency of detection, but impair DAPI staining, as evidenced in a recent study (da Silva et al., 2017a).
- Technical tips
- The incubation time of the thymidine analogs varies according to the doubling-time, and it can be optimized for other cell types. The same is valid for the incubation time after cell mix.
- Perform this protocol in the dark from the step that requires secondary antibodies incubation onwards.
- To avoid false positives, do not overexpose fluorescence images during capturing step.
Recipes
- Alkalinized water
Prepare the alkalinized water by adjusting the pH of Milli-Q water to 9.5 (using 5 M NaOH with the aid of a pH meter) - Phosphate buffered saline (1x PBS)
137 mM NaCl
2.7 mM KCl
10 mM Na2HPO4
2 mM KH2PO4- Prepare the buffer by adding 8 g of NaCl, 0.2 g of KCl, 1.44 g of Na2HPO4, and 0.24 g of KH2PO4
- Add 800 ml of water and adjust the pH to 7.4 with HCl
- Complete the volume to 1 L
- Dispense the solution into aliquots (e.g., 250 ml) and sterilize by autoclaving (20 min, 120 °C, liquid cycle)
- Store at room temperature for up to six months
- Check the pH after prolonged use
- Liver Infusion Tryptose (LIT) medium
68 mM NaCl
5.3 mM KCl
56 mM Na2HPO4
0.2% (w/v) glucose
0.5% (w/v) liver infusion broth
0.5% (w/v) tryptose
10 mg/L hemin
10% (v/v) fetal bovine serum
133 mg/L streptomycin sulfate salt
59 mg/L penicillin G sodium salt- Prepare the medium by adding 4 g of NaCl, 0.4 g of KCl, 8 g of Na2HPO4, 5 g of tryptose, 5 g of Liver infusion broth
- Add 800 ml of water, adjust pH to 7.3 (using 5 M NaOH with the aid of a pH meter), complete the volume to 880 ml, and sterilize by autoclaving (20 min, 120 °C, liquid cycle)
- Before utilization, add 1 ml of a hemin solution (10 mg/ml in 0.1 M of triethanolamine), 100 ml of fetal bovine serum, 20 ml of 20% (w/v) glucose, 133 mg of streptomycin sulfate, and 59 mg of penicillin
- Sterilize again by filtering (using 0.22 μm filter)
- Store at -20 °C for up to six months
- 5′-chloro-2′-deoxyuridine solution (CldU-S)
10 mM of CldU diluted in water- Prepare the buffer by adding 26.26 mg of 5′-chloro-2′-deoxyuridine in 9 ml of water
- Solubilizes and complete the volume to 10 ml
- Sterilize by filtering (using 0.22 μm syringe filter) and dispense the solution into aliquots (e.g., 1 ml)
- Store at -20 °C for up to six months
- 5′-iodo-2′-deoxyuridine solution (IdU-S)
10 mM of IdU diluted in water alkalinized
Note: IdU is not readily soluble at physiological pH.- Prepare the buffer by adding 35.4 mg of 5′-iodo-2′-deoxyuridine in 5 ml of alkalinized Milli-Q water (adjust the Milli-Q water pH to 9.5 using 5 M NaOH with the aid of a pH meter)
- Solubilize and complete the volume to 10 ml with Milli-Q water
- Sterilize by filtering (using 0.22 μm syringe filter) and dispense the solution into aliquots (e.g., 1 ml)
- Store at -20 °C for up to six months
- Fixation buffer (FB)
4% (w/v) Paraformaldehyde in 1x PBS- Prepare the buffer by adding 2.1 g of Paraformaldehyde (powder, 95%) in 40 ml 1x PBS
- Solubilize and complete the volume to 50 ml with 1x PBS
- Store at 4 °C for up to one month
- Poly-L-lysine solution (PLS)
0.1% (w/v) Poly-L-Lysine hydrochloride diluted in water- Prepare the buffer by adding 10 mg of Poly-L-Lysine hydrochloride in 9 ml of water
- Solubilize and complete the volume to 10 ml with water
- Sterilize by filtering (using 0.22 μm syringe filter) and dispense the solution into aliquots (e.g., 1 ml)
- Store at 4 °C for up to six months
- Permeabilization solution (PS)
0.1% (v/v) Triton X-100 diluted in 1x PBS- Prepare the buffer by adding 10 μl of Triton X-100 in 9,990 μl of 1x PBS
- Mix well, sterilize by filtering (using 0.22 μm syringe filter), and dispense the solution into aliquots (e.g., 1 ml)
- Store at 4 °C for up to six months
- Denaturation buffer (DB)
2.5 M HCl- Prepare the buffer by adding 2.1 ml of HCl (concentrated 37%) in 7.9 ml of water
- Mix carefully because this solution is irritant
- This solution cannot be stored and must be immediately made prior to use
- Neutralization buffer (NB)
100 mM Boric Acid
75 mM NaCl
25 mM Sodium Tetraborate- Prepare the buffer by adding 620 mg of Boric Acid, 440 mg of NaCl, and 950 mg of Sodium Tetraborate
- Add 80 ml of water and adjust the pH to 8.4 with NaOH
- Complete the volume to 100 ml and sterilize by filtering (using 0.22 μm syringe filter)
- Store at room temperature for up to six months
- Blocking solution (BS)
4% (w/v) Bovine serum albumin dilute in 1x PBS- Prepare the buffer by adding 400 mg of Bovine serum albumin (powder, ≥ 98%) in 9 ml 1x PBS
- Solubilize and complete the volume to 10 ml with 1x PBS
- Sterilize by filtering (using 0.22 μm syringe filter)
- Store at 4 °C for up to one month
Acknowledgments
We would like to thank all the members from the laboratories involved in developing this protocol. We also thank the support of the funding agencies: São Paulo Research Foundation (FAPESP) - Center of Toxins, Immune Response and Cell Signaling (CeTICS) (grants numbers 2013/07467-1, 2014/24170-5, 2017/18719-2), the National Council for Scientific and Technological Development (CNPq) (projects numbers: 870219/1997-9, 304329/2015-0), Minas Gerais Research Support Foundation (FAPEMIG), Coordination for the Improvement of Higher Education Personnel (CAPES), and The Wellcome Centre for Molecular Parasitology.
Competing interests
The authors declare that there is no conflict of interest regarding the publication of this article.
References
- Adl, S. M., Simpson, A. G., Lane, C. E., Lukeš, J., Bass, D., Bowser, S. S., Brown, M. W., Burki, F., Dunthorn, M., Hampl, V., Heiss, A., Hoppenrath, M., Lara, E., Le Gall, L., Lynn, D. H., McManus, H., Mitchell, E. A., Mozley-Stanridge, S. E., Parfrey, L. W., Pawlowski, J., Rueckert, S., Shadwick, L., Schoch, C. L., Smirnov, A. and Spiegel, F. W. (2012). The revised classification of eukaryotes. J Eukaryot Microbiol 59(5): 429-493.
- Alves, C. L., Repolês, B. M., da Silva, M. S., Mendes, I. C., Marin, P. A., Aguiar, P. H. N., Santos, S. D. S., Franco, G. R., Macedo, A. M., Pena, S. D. J., Andrade, L. O., Guarneri, A. A., Tahara, E. B., Elias, M. C. and Machado, C. R. (2018). The recombinase Rad51 plays a key role in events of genetic exchange in Trypanosoma cruzi. Sci Rep 8(1): 13335.
- Browne, A. J., Guerra, C. A., Alves, R. V., da Costa, V. M., Wilson, A. L., Pigott, D. M., Hay, S. I., Lindsay, S. W., Golding, N. and Moyes, C. L. (2017). The contemporary distribution of Trypanosoma cruzi infection in humans, alternative hosts and vectors. Sci Data 4: 170050.
- Calderano, S. G., Drosopoulos, W. C., Quaresma, M. M., Marques, C. A., Kosiyatrakul, S., McCulloch, R., Schildkraut, C. L. and Elias, M. C. (2015). Single molecule analysis of Trypanosoma brucei DNA replication dynamics. Nucleic Acids Res 43(5): 2655-2665.
- da Silva, M. S., Cayres-Silva, G. R., Vitarelli, M. O., Marin, P. A., Hiraiwa, P. M., Araújo, C. B., Avila, A. R., Reis, M. S. and Elias, M. C. (2018). Transcription activity contributes to the firing of non-constitutive origins in Trypanosoma brucei. bioRxiv. doi: https://doi.org/10.1101/398016.
- da Silva, M. S., Monteiro, J. P., Nunes, V. S., Vasconcelos, E. J., Perez, A. M., Freitas-Junior Lde, H., Elias, M. C. and Cano, M. I. (2013). Leishmania amazonensis promastigotes present two distinct modes of nucleus and kinetoplast segregation during cell cycle. PLoS One 8(11): e81397.
- da Silva, M. S., Muñoz, P. A. M., Armelin, H. A. and Elias, M. C. (2017a). Differences in the detection of BrdU/EdU incorporation assays alter the calculation for G1, S, and G2 phases of the cell cycle in trypanosomatids. J Eukaryot Microbiol 64(6): 756-770.
- da Silva, M. S., Pavani, R. S., Damasceno, J. D., Marques, C. A., McCulloch, R., Tosi, L. R. O. and Elias, M. C. (2017b). Nuclear DNA replication in trypanosomatids: there are no easy methods for solving difficult problems. Trends Parasitol 33(11): 858-874.
- Davidson, R. L. and Kaufman, E. R. (1979). Resistance to bromodeoxyuridine mutagenesis and toxicity in mammalian cells selected for resistance to hydroxyurea. Somatic Cell Genet 5(6): 873-885.
- Elias, M. C., da Cunha, J. P., de Faria, F. P., Mortara, R. A., Freymuller, E. and Schenkman, S. (2007). Morphological events during the Trypanosoma cruzi cell cycle. Protist 158(2): 147-157.
- Gaunt, M. W., Yeo, M., Frame, I. A., Stothard, J. R., Carrasco, H. J., Taylor, M. C., Mena, S. S., Veazey, P., Miles, G. A., Acosta, N., de Arias, A. R. and Miles, M. A. (2003). Mechanism of genetic exchange in American trypanosomes. Nature 421(6926): 936-939.
- Hancock, A., Priester, C., Kidder, E. and Keith, J. R. (2009). Does 5-bromo-2'-deoxyuridine (BrdU) disrupt cell proliferation and neuronal maturation in the adult rat hippocampus in vivo? Behav Brain Res 199(2): 218-221.
- Herrick, J. and Bensimon, A. (1999). Single molecule analysis of DNA replication. Biochimie 81(8-9): 859-871.
- Ponte-Sucre, A. (2016). An overview of Trypanosoma brucei infections: an intense host-parasite interaction. Front Microbiol 7: 2126.
- Stanojcic, S., Sollelis, L., Kuk, N., Crobu, L., Balard, Y., Schwob, E., Bastien, P., Pages, M. and Sterkers, Y. (2016). Single-molecule analysis of DNA replication reveals novel features in the divergent eukaryotes Leishmania and Trypanosoma brucei versus mammalian cells. Sci Rep 6: 23142.
- Torres-Guerrero, E., Quintanilla-Cedillo, M. R., Ruiz-Esmenjaud, J. and Arenas, R. (2017). Leishmaniasis: a review. F1000Res 6: 750.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
da Silva, M. S., Marin, P. A., Repolês, B. M., Elias, M. C. and Machado, C. R. (2018). Analysis of DNA Exchange Using Thymidine Analogs (ADExTA) in Trypanosoma cruzi. Bio-protocol 8(24): e3125. DOI: 10.21769/BioProtoc.3125.
Category
Microbiology > Microbial biochemistry > DNA
Molecular Biology > DNA > DNA recombination
Molecular Biology > DNA > DNA labeling
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.