- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Monitoring Natural Killer Cell Function in Human Ovarian Cancer Cells of Ascitic Fluid
Published: Vol 8, Iss 24, Dec 20, 2018 DOI: 10.21769/BioProtoc.3124 Views: 6942
Reviewed by: HongLok LungAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2018

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Natural killer (NK) cells are the major effectors of the innate immune system when activated resulting in modulation of immune response of the host defense through target cell lysis and secretion of cytokines. Precise functions of NK cells are essential for the treatment outcome of different virus infections and malignant diseases. NK cells impart cytotoxic effect to the target cells lacking MHC class I molecules and thus the final readout of the activity is death of target cells. The NK cell function is evaluated by the 51Cr-release and/or flow cytometry-based assays. In the present protocol, we have determined the activation of NK cells by the liberation of IL-10 and IFNγ, and subsequently its function by enumerating the number of dead tumor cells originally isolated from the ascitic fluid of ovarian cancer patients. The entire assay is based on cells of the healthy donors and patients. Besides determining function, this method is able to demarcate between NK-cell sensitive and insensitive tumor cells. This technique enables researchers to study NK cell functions in healthy donors or in patients to reveal their impact on different malignancies and to further discover new therapeutic strategies.
Keywords: Ascitic fluidBackground
Being a part of innate immune system, natural killer (NK) cells involve in immune surveillance. They distinguish between healthy and abnormal cells (virus-infected or malignant) through a set of germline-encoded inhibitory and activating receptors (Marcus et al., 2014; Tognarelli et al., 2016). Under normal and healthy conditions, NK cells serve a role that is unlike other blood cells perform, until there is an infection. At that time natural killer cells assume their more recognized role in killing of targeted cells. NK cells exhibit direct cytotoxic effect via granzyme/perforin/death receptor pathways against the cells that lost the expression of HLA class I antigen (Geller and Miller, 2011; Paul and Lal, 2017). Further, NK cells are stimulated to produce IFNγ and TNF-α, which exert cytostatic/cytotoxic effects on specific targets. NK cell deficiency seems a part of larger immunological syndrome.
In general, the most common combination of surface markers used to identify the majority of NK cells is the absence of CD3, along with the expression of CD56 (i.e., CD3-CD56+). However, every NK cells of above phenotype may not be functionally active. It has been shown that Perforin and CD107a combination represent superior NK cells function (Rubin et al., 2017).
Several assays have been established for determination of the activity of NK and cytotoxic T cells, when they recognize the target cells. The 51Chromium release assay (CRA) was described for the first time (Brunner et al., 1968) to assay NK cells activity, which has been considered the ‘gold standard’ for measurement of cytotoxic effect. This assay indirectly determines dead cell population. Moreover, due to risks of handling and disposal of radioactive substance, several alternative methods have been developed time to time. Two fluorochromes based flow-cytometry assay is proposed for direct determination of dead target cells (Radosevic et al., 1990). In this case, fluorescently stained target cells, if lost viability, are counter stained with a DNA intercalating dye. Another fluorescence-based assay was proposed, which depends on the hydrolysis of Calcein AM (Lichtenfels et al., 1994) by intracellular esterases resulting in the formation of Calcein, a hydrophilic strongly fluorescent compound retained in the cytoplasm of the viable cells. In case of damaged ones, it releases in the medium producing an intense green signal measured by fluorimeter. The caveat of this assay is that the release calcein varies in its dynamic range for different tumor targets, and the calcein could retain within the apoptotic bodies and membrane fragments of the lysed cells results in incomplete release leading to underestimation. To overcome these limitations, a novel cytotoxicity assay using an image flow-cytometer was proposed, which is to be claimed as a simple and direct method (Srinivas et al., 2015). Later, two more assays based on LDH release (Konjevic et al., 1997) and bioluminescence (Karimi et al., 2014) have been proposed. While both of these methods provide indirect assessment, the former one is simple whereas the latter method is a complicated one. In this protocol, we have proposed a flow-cytometry based comprehensive method that not only provides direct value of percentage dead target cells but also analyzes sensitive cells in a heterogeneous population of ascitic or blood-derived cells. The earlier published methods were based on tumor cell lines, which is devoid of other cells present in the biological fluid in vivo. The scope of the present protocol is further extended to find out the efficacy of NK cells in cancer patients as compared to healthy persons. It is known that in cancer patients, depending upon the stages of the disease, the potency of NK cell immune surveillance is compromised (Levy et al., 2011). In an earlier paper, we discussed this protocol (Akhter et al., 2018), which clearly indicates that in ovarian cancer a subtype of tumor cells remains unaffected even in the presence of healthy donor NK cells. Thus, this protocol opens up the possibility to decide on a critical issue in NK cell immune therapy, that is how they should be generated for effective therapy.
Materials and Reagents
Note: The following materials were used to determine the activation and the function of NK cells.
- Pipette tips
- 10 ml Dispovan syringe (BD, catalog number: 301001)
- 10 ml Vacutainer Heparin Tube (BD, catalog number: 367880)
- Nylon mesh cell strainer 100 micron (BD, Pharmingen, catalog number: 352360)
- Nylon mesh cell strainer 40 micron (BD, Pharmingen, catalog number: 352340)
- 15 ml centrifuge tubes (BD, Pharmingen, catalog number: 352096)
- 50 ml centrifuge tubes (BD, Pharmingen, catalog number: 352070)
- 48-well cell culture cluster (Corning, catalog number: 3548)
- 75 cm2 cell culture flask (Corning, catalog number: 430720)
- 5 ml round bottom tube (BD, Pharmingen, catalog number: 352054)
- Ficoll-Hypaque, ρ= 1.077 g/ml (Sigma-Aldrich, catalog number: 10771)
- Phosphate-buffered saline (PBS) 1x (HiMedia Laboratories, catalog number: 023)
- Trypan blue solution (Sigma-Aldrich, catalog number: T8154)
- CD3/PE-Cy7 1:100 (BD, Pharmingen, catalog number: 563423)
- CD56/V450 1:200 (BD, Pharmingen, catalog number: 560360)
- CD45/APC 1:100 (BD, Pharmingen, catalog number: 555485)
- EpCAM antibody 1:50 (Abcam, catalog number: ab46714)
- HLA class I/FITC 1:100 (BD, Pharmingen, catalog number: 555555)
- HLA class II/FITC 1:100 (BD, Pharmingen, catalog number: 565558)
- RPMI-1640 medium (Thermo Fisher Scientific, catalog number: 11875093)
- ITS Liquid Media Supplement (100x) (Sigma-Aldrich, catalog number: 13146)
- Human Serum Albumin (HSA) 10 mg/ml (Sigma-Aldrich, catalog number: 12055-091)
- Epidermal Growth Factor (EGF) 20 mg/ml (Sigma-Aldrich, catalog number: E9644-2MG)
- IL10 ELISA kit (Pepro Tech Worldwide, catalog number: 900-K21)
- IFNγ ELISA (Pepro Tech Worldwide, catalog number: 900-K27)
- Propidium Iodide (PI) 5μg/100 ml (Sigma-Aldrich, catalog number: P4864-10ML)
- Citrate phosphate dextrose (CPD) 0.25 mM (Sigma-Aldrich, catalog number: C7165)
- 99.9% ethanol (Analytical CS Reagent, catalog number: 1170)
- Complete medium (see Recipes)
Equipment
- Pipettes
- Flow cytometer (FACSAriaIII, BD Sciences, San Jones, CA)
- ELISA reader (BioTEK, model: PowerWave Elite XS)
- CO2 incubator (NuAire, model: Nu4750)
- Refrigerated centrifuge (Eppendorf, model: 5810R)
- Biosafety cabinet (ESCO, model: Class II, Type A2)
- Inverted Microscope (Olympus, model: CK2)
- Hemacytometer (Sigma-Aldrich, catalog number: Z359629)
- Pipets 10 μl, 100 μl, 200 μl and 1,000 μl (Eppendorf)
- -80 °C Freezer (Thermo Scientific, model: FormaTM 900 Series)
- 4 °C Refrigerator (Whirlpool, model: FF-350 Elite)
Software
- BD FACSDivaTM Software (BD, Pharmingen, San Jones, CA) (has been used to analyze data of flow-cytometry based experiments)
- All statistical analyses are based on GraphPad Prism 5.01 (GraphPad Software, La Jolla, CA)
Procedure
The entire protocol is divided into 5 parts: (A) Preparation of NK cells of healthy donor; (B) Preparation of purified tumor cells from ascitic fluid of ovarian cancer patient; (C) Co-culture of NK and tumor cells for cytotoxicity assay; (D) Determination of HLA antigens in the tumor cells; and (E) Cytokine analysis of the culture supernatant.
- Preparation of NK cells of healthy donor
- Withdraw 10 ml of peripheral blood from the vein of a healthy donor and place on a 10 ml heparin vial. The blood is diluted 1:1 with RPMI-1640 medium.
- Slowly overlay heparinized diluted blood over 10 ml of Ficoll-Hypaque in a 50 ml plastic tube. Separate the mononuclear cells by density-gradient centrifugation at 600 x g for 25 min without a break.
- Density-gradient centrifugation fractionates the whole blood into three constituents: erythrocytes/granulocytes, plasma and buffy coat. The buffy coat, a thin layer (~1% of original blood volume) is sandwiched between the heavy erythrocytes/granulocytes and platelet-rich plasma, yet contains the majority of the mononuclear cells (MNCs). Figure 1 shows the location of three layers after centrifugation. Remove as much plasma layer as possible; collect the buffy coat material with a disposable transfer pipette without disturbing the bottom layer. Transfer the content into a new sterile tube and wash the sample twice in 10 ml of medium and enumerate viable cell count using trypan blue exclusion method.
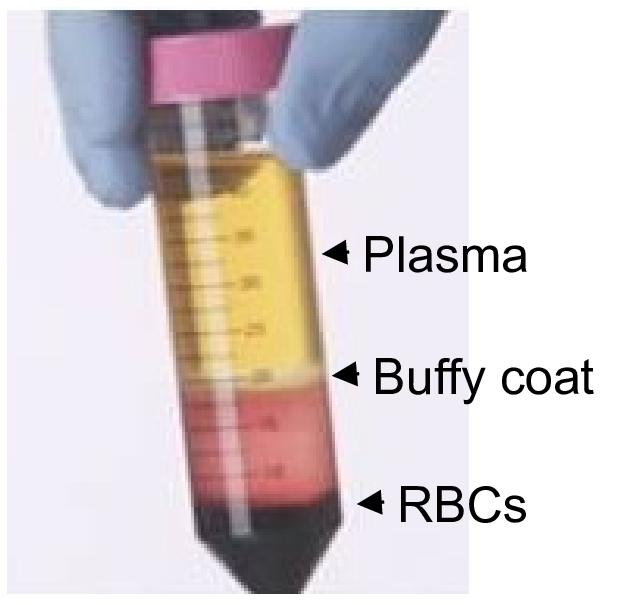
Figure 1. Three layers of peripheral blood after density gradient centrifugation. Top plasma layer contains most of the platelets, middle (buffy coat) consists of mononuclear cells, and bottom Ficoll layer consists of RBCs and granulocytes. - Take nine million mononuclear cells in a 3 ml plastic tube, in which 5 μl each of 1:10 diluted CD3/PE-Cy7 and CD56/V450 antibody mixture is added. Mix the content and incubate at 4 °C for 30 min.
- Wash the cell pellet 1x each with 3 ml of PBS and medium containing 2% human serum by centrifugation at 400 x g for 5 min and finally resuspend in 3 ml of medium containing 2% human serum and store in an ice bath under darkness.
- Perform cell sorting using FACSAriaIII with a 70 μm nozzle. The dot-plot of side scatter (SSC) and forward scatter (FSC) properties of the cells are shown in the density plots (Figure 2A), which gives an estimation of the size and granularity, respectively. Gate potential mononuclear cells after excluding dead cells, cell fragments and cells with high granularity. Subject this gated population in two-stage doublet discrimination (Figures 2B and 2C). The gated population in Figure 1C is expected to be free from any doublet or higher aggregate.
- Acquire data in a dot plot as a combination of CD3/PE-Cy7 and CD56/Horizon V450, which gives a pre-sort result (Figure 2D). The pre-sort analysis shows that CD3-CD56+ cells are about 18.5%.
- Sort the blue gated cells (CD3-CD56+) using the same machine and collect into a new tube. Re-analyze post-sorted cells in the same settings of the flow-cytometer for checking the purity of the NK cells. As shown in Figure 2E, the fraction of the cells falling within the same gated region like in pre-short represents 94.4% pure. The remaining fraction seems dead cells and/or fragments.
- Wash the sorted cells in PBS and enumerate the viable count by trypan blue exclusion method. The viability of the cells is about 95%.
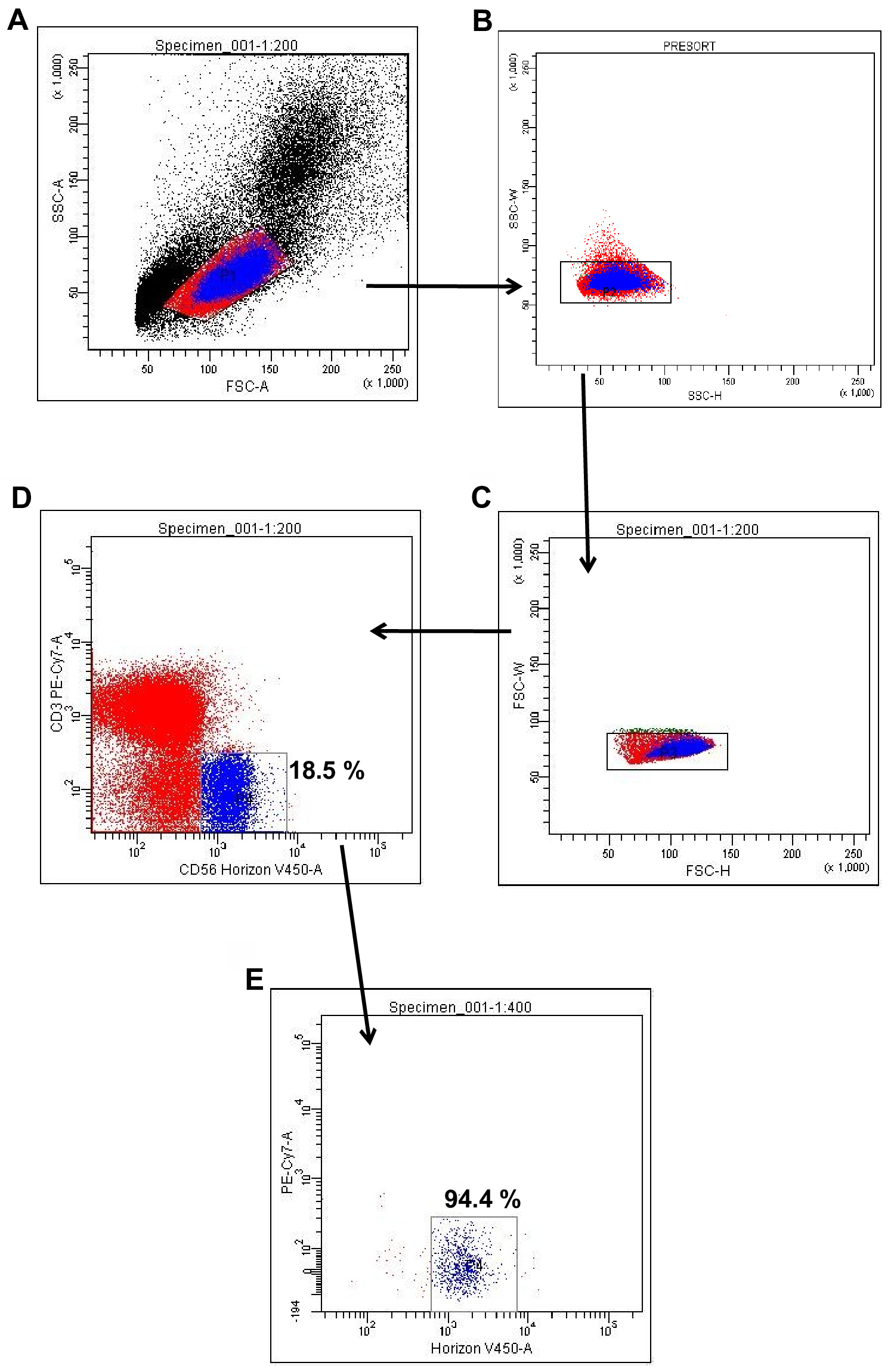
Figure 2. Sorting of NK cells. Sorting strategy and sort purity of NK cells are shown. The representative dot-plots show pre- and post-sort purity of CD3-CD56+ cells. A. Side scatter (SSC) and forward scatter (FSC) properties of mononuclear cells. B. Doublet discrimination based on cells’ granularity. C. Doublet discrimination based on cells’ size. D. Pre-sort analysis of NK cells. E. Post-sort analysis of NK cells.
- Preparation of purified tumor cells from ascitic fluid of ovarian cancer patient
- Take about 200 ml of ascitic fluids of serous epithelial ovarian cancer patient in a sterile container containing CPD. Pellet-down the cells by centrifuging at 600 x g for 20 min. Resuspend cells in 10 ml of medium and follow Step A2. Remove the buffy coat containing mononuclear cells, wash and then enumerate the viable count following Step A3.
- Take about 5 x 106 cells in a 3 ml plastic tube and stain with primary antibodies (EpCAM and CD45/APC) by incubating in an ice bath for 30 min. In the case of EpCAM, a second incubation step is followed after adding fluorochrome-labeled secondary antibody (anti-rabbit/PECy5). Wash the cells and resuspend in the medium as per Step A5.
- Sort cells in two different phenotypes: EpCAM+CD45+ and EpCAM+CD45- following the Step A6. Dot-plots of presort-sorted and post-sorted cells are shown in Figure 3. Both cellular fractions are found to be more than 90% pure and viable.

Figure 3. Analysis of tumor cells and sort purity. Pre-sort dot-plot shows different phenotypes of mononuclear cells present in the ascitic fluid of the ovarian cancer patient. Representative dot-plots (right) show highly purified EpCAM+CD45+ and EpCAM+CD45- cells.
- Co-culture of NK and tumor cells for cytotoxicity assay
- Take a 48-well flat bottom culture plate. In multiple wells, acquire 100,000 cells of each type of cancer cells from Step B3 in 200 μl volume of complete medium. Pour freshly isolated NK cells from Step A9 in the proportion of 1:5 (1 part tumor and 5 parts NK cells) in 100 μl of the same medium in each well. Keep two sets of 3 wells each containing tumor cells as control, which do not receive NK cells. Similarly, one set of well contains only NK cells.
- Incubate the culture plate in a 5% CO2 incubator at 37 °C for 24 h. Transfer cell suspension from each well to the corresponding marked tube and centrifuge at 300 x g for 5 min. Remove the culture supernatants and store at -80 °C, wash the cell pellets 2x with 400 μl PBS.
- Resuspend cell pellets in 400 μl PBS, add 1 μl PI into each tube and gently mix the content prior to incubation for 1-2 min in the dark.
Note: Mixing of cells is carried out gently by tapping with fingers. Cell analysis should be carried out within 5 min of addition of PI. - Adjust the setting of the flow-cytometer in a way such that on the basis of the physical properties (size and granularity), tumor cells can be demarcated from NK cells. Thus two classes of cell form two major clusters. Due to low granularity, NK cells remain in the bottom part of the FSC-SSC dot-plot (Figure 4A). Subject this gated population in two-stage doublet discrimination and then analyze for viability on the basis of PI- cells. The histogram plot of SSC versus PI intensity shows only 2% cells are dead (Figure 4D).
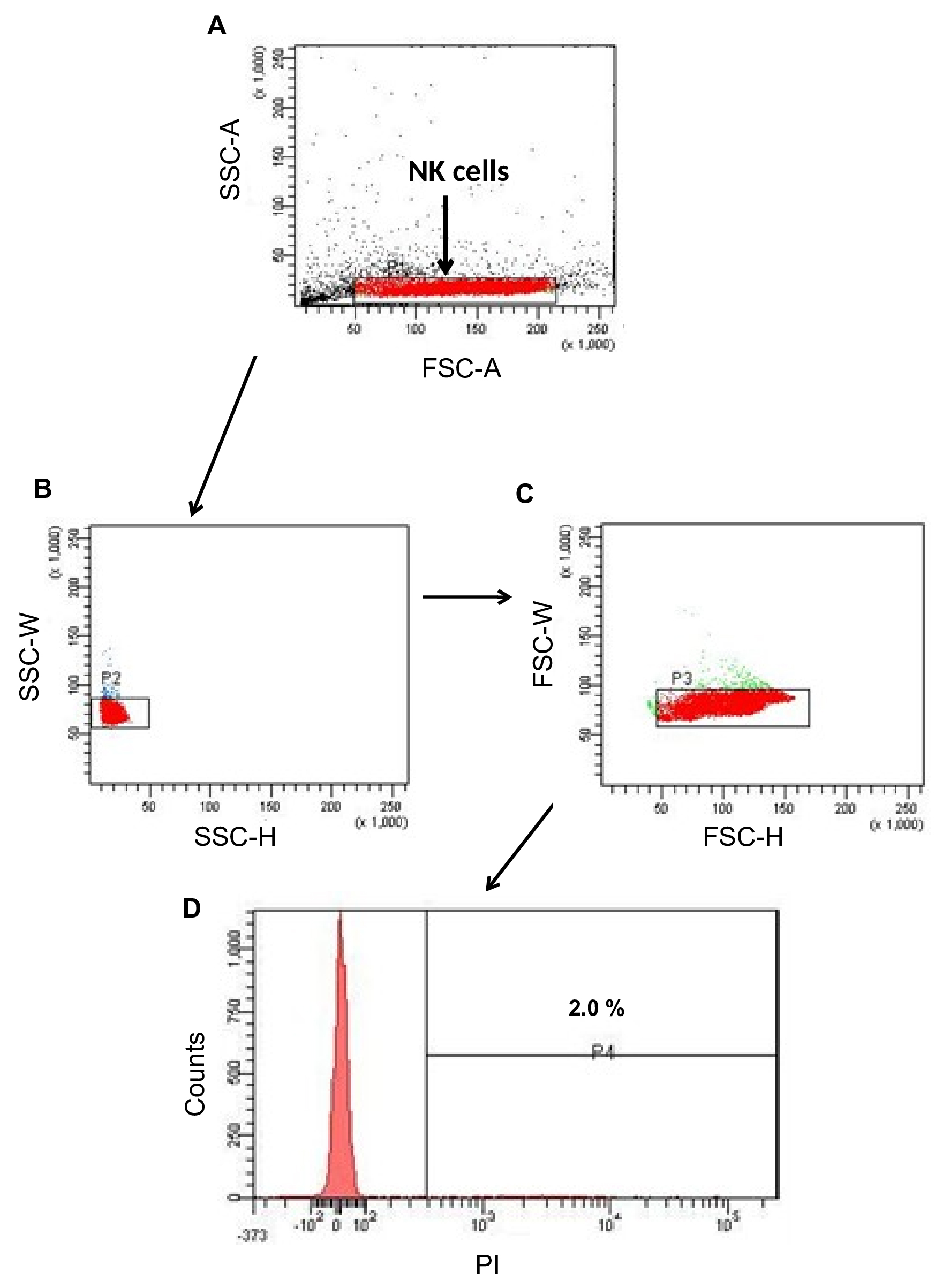
Figure 4. Post-incubation viability of NK cells. The representative dot-plot shows the strategy of gating NK cells and histogram profile for PI staining after 24 h incubation. A. Side scatter (SSC) and forward scatter (FSC) properties of NK cells. B. Doublet discrimination based on cells’ granularity. C. Doublet discrimination based on cells’ size. D. Histogram analysis of PI-stained dead cells. - Similar analysis in case of EpCAM+CD45+ cells alone (control) shows cluster of NK cells at different location than other cells/cell fragments (Figures 5A and 5C). Following doublet discrimination, the viability of the control cells is 6.8% (Figure 5B). In co-culture with NK cells, the viability of EpCAM+CD45+ cells is marginally increased to 9.8% (Figure 5D).
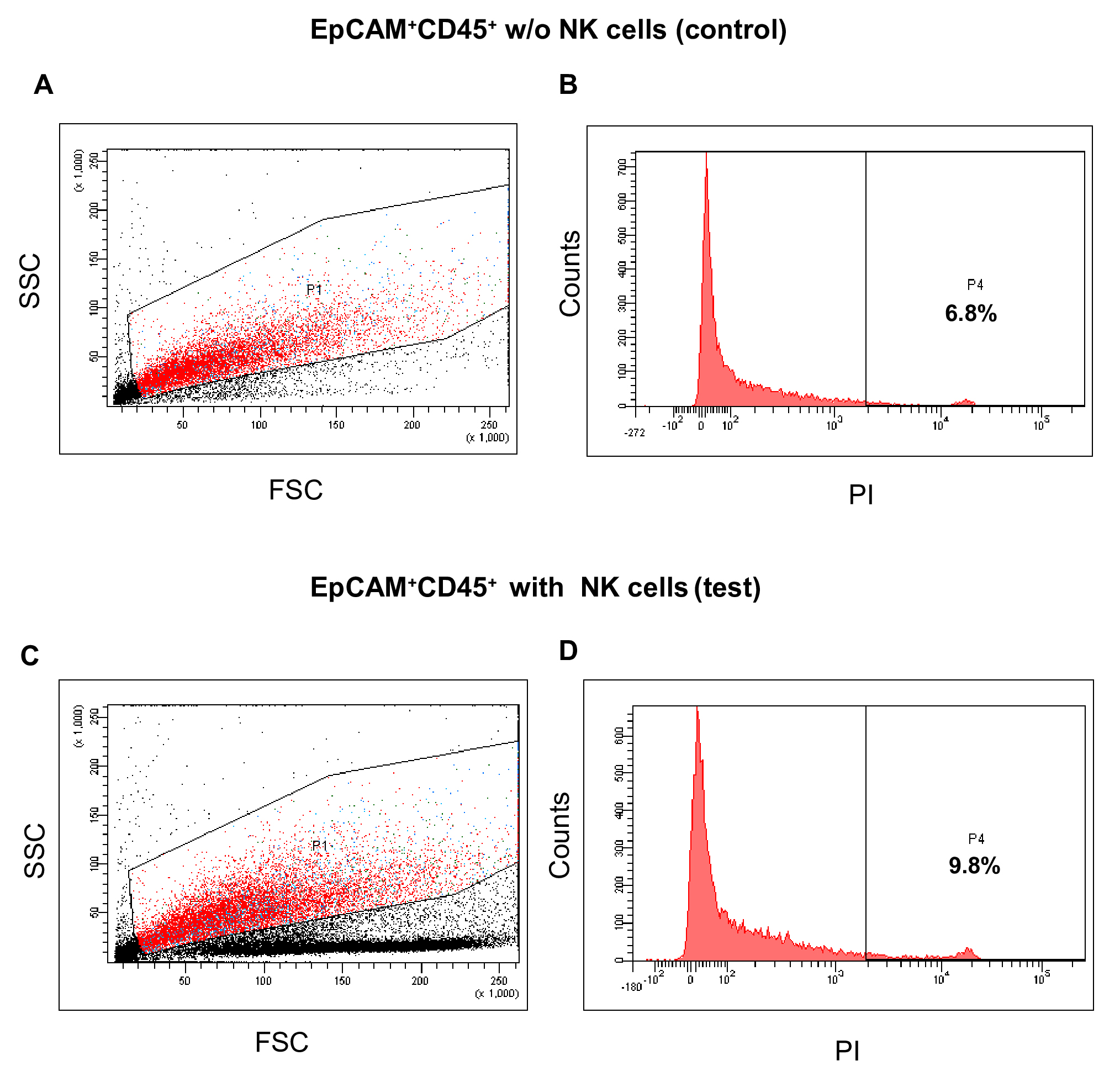
Figure 5. NK cells mediated death of EpCAM+CD45+ tumor cells. The representative histograms show no increase of death of EpCAM+CD45+ cells in the test sample as compared with control (without NK cells). A. Side scatter (SSC) and forward scatter (FSC) properties of NK (control) and tumor cells mixture. B. Histogram analysis of PI-stained dead cells. C. Side scatter (SSC) and forward scatter (FSC) properties of NK (test) and tumor cells mixture. D. Histogram analysis of PI-stained dead cells. - EpCAM+CD45- cells when co-cultured with NK cells under identical condition show a steep increase of the dead cells from 4.8% to 26.8% (Figures 6B and 6D).
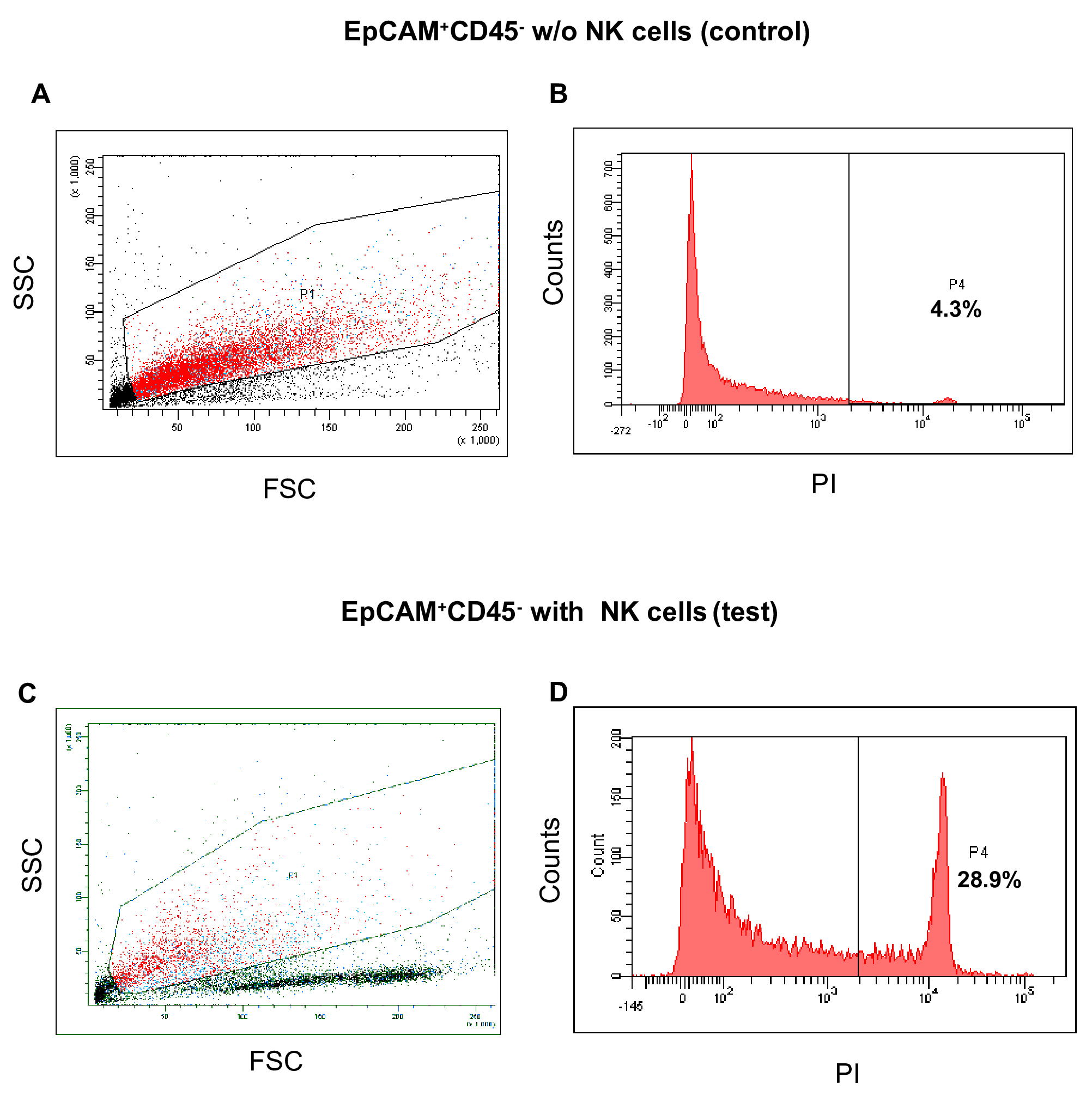
Figure 6. NK cells mediated death of EpCAM+CD45- tumor cells. The representative histograms show substantial death of EpCAM+CD45- cells in the test sample as compared to control (without NK cells). A. Side scatter (SSC) and forward scatter (FSC) properties of NK (control) and tumor cells mixture. B. Histogram analysis of PI-stained dead cells. C. Side scatter (SSC) and forward scatter (FSC) properties of NK (test) and tumor cells mixture. D. Histogram analysis of PI-stained dead cells. - The comparative killing effect of NK cells towards both fractions of tumor cells is shown in Figure 7.
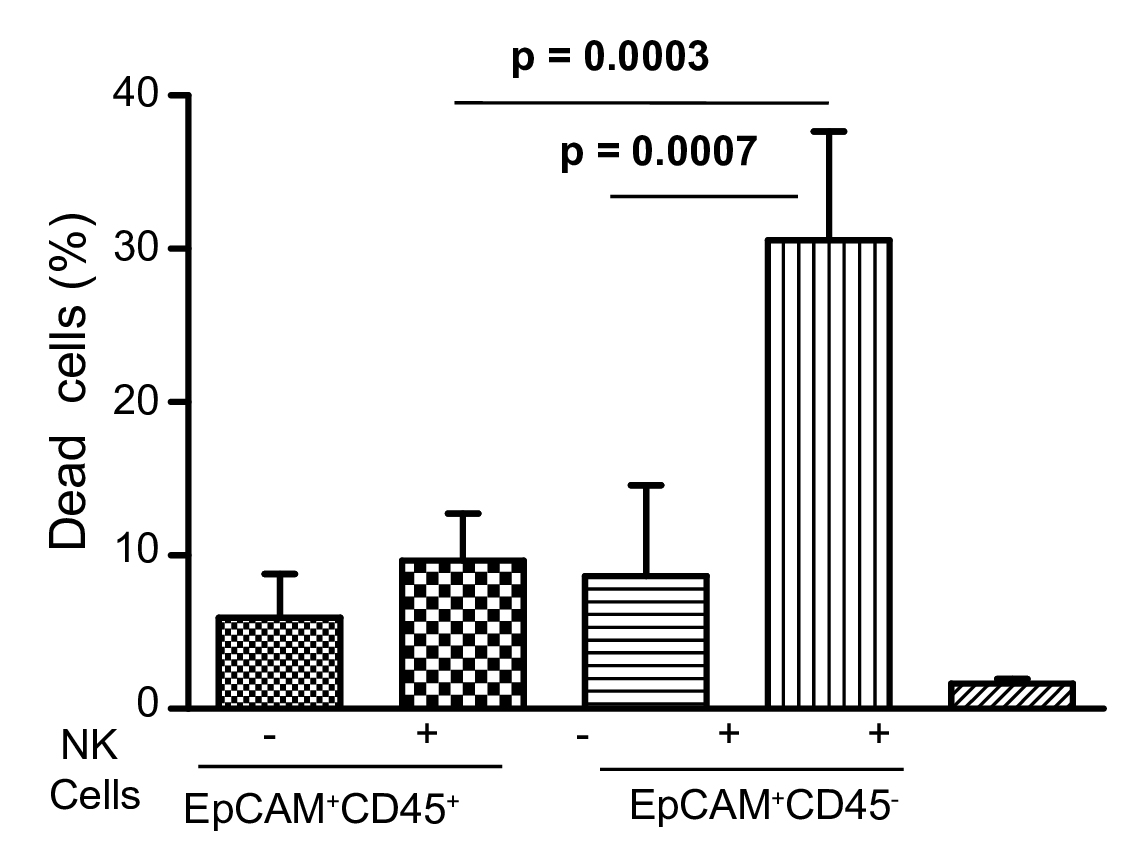
Figure 7. Statistical analysis of NK cell-mediated death of tumor cells. NK cells mediated death of tumor cells are shown. Bar diagrams show comparative values of dead cells. A significantly high cell death is seen in case of sorted EpCAM+CD45- cells. The error bars represent means ± SD (n = 5). Significance are calculated based on Student’s t-test.
- Determination of HLA antigens in the tumor cells
In order to confirm that NK cells sensitive fraction of the tumor cells (EpCAM+CD45-) in fact down-regulate the expression of HLA class I antigen and not the insensitive cells (EpCAM+CD45+), separately stain both the fractions of cells with HLA class I and II antigens following Step B3. The expression of both HLA antigens is significantly declined in EpCAM+CD45- cells, keeping high-level expression in EpCAM+CD45+ cells (Figure 8).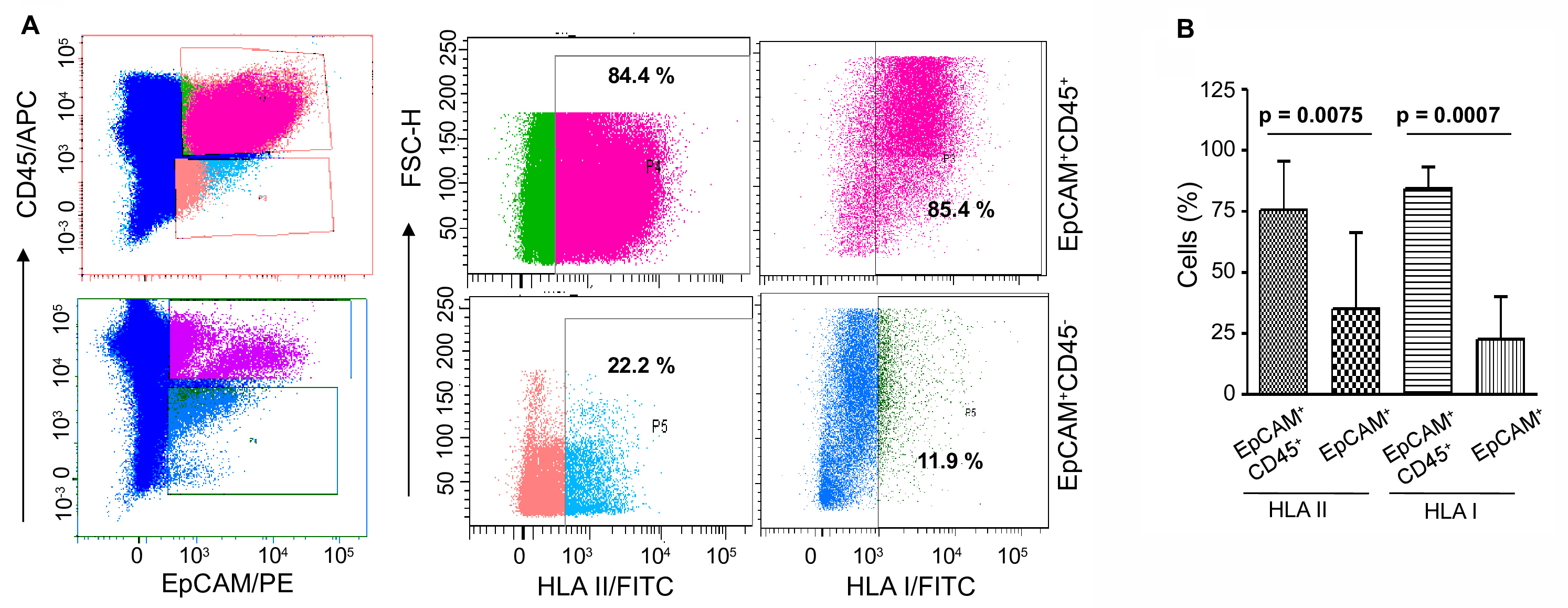
Figure 8. Comparative expressions of HLA class I and II antigens in EpCAM+CD45+ and EpCAM+CD45- cells. A. The representative dot-plots show both classes of antigens are highly expressed in EpCAM+CD45+ cells, whereas they are significantly declined in EpCAM+CD45- compartment. B. Statistical analysis of the expression of HLA class I and II antigens in tumor cells. Bar diagrams show comparative values in both cell fractions. The error bars represent means ± SD (n = 5). The statistical significance is calculated based on Student’s t-test. - Cytokine analysis of the culture supernatant
Examine the culture supernatants, as stored in Step C2, by ELISA using standard protocol as described in the kit. Assay supernatants at different dilutions (1:1, 1:5, 1:10) for the presence of IFNγ and IL-10. The results confirm the presence of both cytokines (use the dilutions at which concentrations fall within the linearity of the calibration plot) in the culture supernatants when EpCAM+CD45- and NK cells are incubated together (Figures 9A and 9B).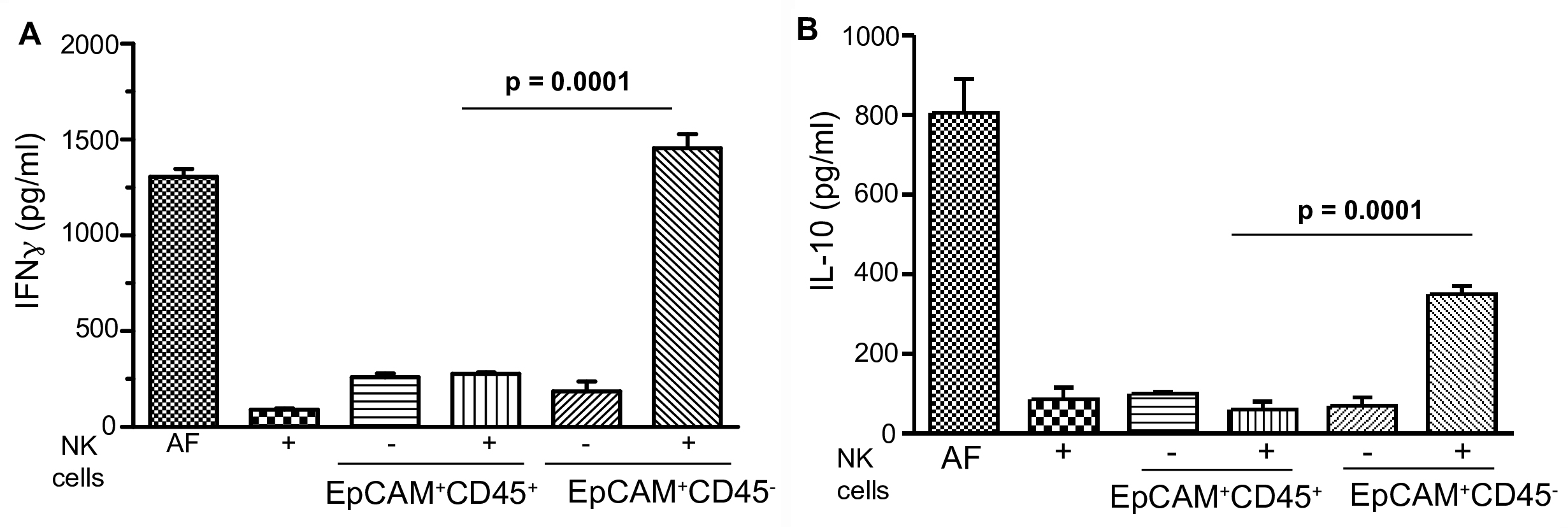
Figure 9. Activation of NK cells. A. Concentration of IFNγ in ascitic fluid (AF) and in co-culture media of Step C2. B. Concentration of IL-10 in ascitic fluid (AF) and in co-culture media of Step C2. The error bars represent means ± SD (n = 4). The statistical significance is calculated based on Student’s t-test.
Data analysis
The notion of this protocol is to demonstrate a comprehensive method and to confirm that NK cells of healthy donor recognize tumor cells that down-regulate HLA class I antigen, in turn self-activated and kill the recognized cells (Akhter et al., 2018). As shown in Figure 6, EpCAM+CD45- cells suffer high death (30.54 ± 3.18%, P < 0.0007) within 24 h of culture as compared to controls (Figure 7). Further, this protocol ensures that upon recognition of tumor cells, NK cells are activated and secreted high level of IFNγ (1,487 ± 68 pg/ml, P < 0.0001) and IL-10 (345 ± 28 pg/ml, P < 0.0001) as compared to the controls (Figures 9A and 9B). Thus the protocol provides a non-radioactive, flow-cytometry based method that can be easily adopted in the laboratory to generate numerous information: a) functional evaluation of NK cells of patients and healthy donor’s origin, b) to confirm its specific recognition to tumor cells, and c) determine self-activation of NK cells.
Recipes
- Complete medium
RPMI-1640 supplemented with 10 mg HSA/ml, 1x ITS and 20 ng/ml EGF
Acknowledgments
We are thankful to Department of Biotechnology (DBT), Government of India for generous support in the Center for Molecular Medicine Program at the institute.
Competing interests
Authors have no conflict of interest.
Ethics
Samples were obtained from Department of Medical Oncology, All India Institute of Medical Sciences, (AIIMS), New Delhi, India on patient’s informed consent basis during tapping. This project was pursued with due approval of Ethical Committees from AIIMS (IEC/NP-277/01-08-2014) and NII (HEC/XX/2014). The samples were handled as per the approved procedures.
References
- Akhter, M. Z., Sharawat, S. K., Kumar, V., Kochat, V., Equbal, Z., Ramakrishnan, M., Kumar, U., Mathur, S., Kumar, L. and Mukhopadhyay, A. (2018). Aggressive serous epithelial ovarian cancer is potentially propagated by EpCAM+CD45+ phenotype. Oncogene 37(16): 2089-2103.
- Brunner, K. T., Mauel, J., Cerottini, J. C. and Chapuis, B. (1968). Quantitative assay of the lytic action of immune lymphoid cells on 51-Cr-labelled allogeneic target cells in vitro; inhibition by isoantibody and by drugs. Immunology 14(2): 181-196.
- Geller, M. A. and Miller, J. S. (2011). Use of allogeneic NK cells for cancer immunotherapy. Immunotherapy 3(12):1445-1459.
- Karimi, M. A., Lee, E., Bachmann, M. H., Salicioni, A. M., Behrens, E. M., Kambayashi, T. and Baldwin, C. L. (2014). Measuring cytotoxicity by bioluminescence imaging outperforms the standard chromium-51 release assay. PLoS One 9(2): e89357.
- Konjevic, G., Jurisic, V. and Spuzic, I. (1997). Corrections to the original lactate dehydrogenase (LDH) release assay for the evaluation of NK cell cytotoxicity. J Immunol Methods 200(1-2): 199-201.
- Levy, E. M., Roberti, M. P. and Mordoh, J. (2011). Natural killer cells in human cancer: from biological functions to clinical applications. J Biomed Biotechnol 2011: 676198.
- Lichtenfels, R., Biddison, W. E., Schulz, H., Vogt, A. B. and Martin, R. (1994). CARE-LASS (calcein-release-assay), an improved fluorescence-based test system to measure cytotoxic T lymphocyte activity. J Immunol Methods 172(2): 227-239.
- Marcus, A., Gowen, B. G., Thompson, T. W., Iannello, A., Ardolino, M., Deng, W., Wang, L., Shifrin, N. and Raulet, D. H. (2014). Recognition of tumors by the innate immune system and natural killer cells. AdvImmunol 122: 91-128.
- Paul, S. and Lal, G. (2017). The molecular mechanism of natural killer cells function and its importance in cancer immunotherapy. Front Immunol 8: 1124.
- Radosevic, K., Garritsen, H. S., Van Graft, M., De Grooth, B. G. and Greve, J. (1990). A simple and sensitive flow cytometric assay for the determination of the cytotoxic activity of human natural killer cells. J Immunol Methods 135(1-2): 81-89.
- Rubin, T. S., Zhang, K., Gifford, C., Lane, A. Choo, S., Bleesing, J. J. and Marsh, R. A. (2017). Perforin and CD107a testing is superior to NK cell function testing for screening patients for genetic HLH. Blood 129(22): 2993-2999.
- Srinivas, S. S., McCulley, K. J., Somanchi, A., Chan, L. L. and Lee, D. A. (2015). A novel method for assessment of natural killer cell cytotoxicity using image cytometry. Plos One 10(10): e0141074.
- Tognarelli, S., Jacobs, B., Staiger, N. and Ullrich, E. (2016). Flow cytometry-based assay for the monitoring of NK cell functions. J Vis Exp (116). doi: 10.3791/54615.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Kumar, V. and Mukhopadhyay, A. (2018). Monitoring Natural Killer Cell Function in Human Ovarian Cancer Cells of Ascitic Fluid. Bio-protocol 8(24): e3124. DOI: 10.21769/BioProtoc.3124.
Category
Immunology > Immune cell function > Cytotoxicity
Cancer Biology > Cell death > Immunological assays
Cell Biology > Cell viability > Cell death
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.