- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

In vitro Membrane Interaction and Liposome Fusion Assays Using Recombinant Hepatitis C Virus Envelope Protein E2
(*contributed equally to this work) Published: Vol 8, Iss 23, Dec 5, 2018 DOI: 10.21769/BioProtoc.3108 Views: 5845
Reviewed by: David PaulJan-Ulrik DahlAli Asghar Kermani
Original research article
The authors used this protocol in:

Mar 2018
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
In order to study the mechanism underlying the Hepatitis C Virus (HCV) fusion process we have performed assays using phospholipid liposomes and a truncated form of E2 protein, E2661 (amino acids 384-661 of the HCV polyprotein) lacking the transmembrane region. E2661 has been previously generated by using the baculovirus expression system. This form has been used in lipid-protein interaction studies with different model vesicles at different pHs, and monitored using a variety of fluorescent assays. After the analysis of the results, we observed that E2661 is able to insert into lipid bilayers and to induce vesicle aggregation, lipid mixing and liposome leakage, showing higher values of membrane destabilization for negatively charged phospholipids at acidic pH. This is indicative of the role of E2 glycoprotein in the HCV initial infective steps, interacting with the target membranes and producing their destabilization.
Keywords: Hepatitis C virusBackground
The fusion of the viral and cellular membranes is the first step of the infective cycle of an enveloped virus (Lemon et al., 2007; Lindenbach et al., 2007). This fusion can take place either at the plasma membrane or in an intracellular location after viral internalization by endocytosis mediated by binding to a receptor located at the plasma membrane. The Hepatitis C virus (HCV) genome is translated into a single large polyprotein, which is processed, among others, into the envelope proteins E1 and E2, which are involved in membrane fusion of this virus with the hepatocyte membranes. E1 (residues 192-383 of the polyprotein) and E2 (residues 384-746) are type-I transmembrane proteins, highly glycosylated, with an N-terminal, water soluble, ectodomain and a C-terminal hydrophobic domain anchoring these glycoproteins to the membrane. Sequence comparisons with fusion proteins of other viruses suggest that E2 could be the fusion protein (Cormier et al., 2004; Rocha-Perugini et al., 2008). However, several studies performed with synthetic peptides suggest the involvement of both E1 and E2 ectodomain sequences in the fusion process. To increase our knowledge of HCV biology two approaches have been developed: the use of HCV pseudoparticles (HCVpp), which consist of E1E2 glycoproteins assembled with retroviral core particles (Lavillette et al., 2006; Meertens et al., 2006), and a more recent cell culture model which enables the propagation of virus in cell culture (HCVcc) (Koutsoudakis et al., 2006). However, these systems are difficult to study fusion in vitro and the results obtained do not indicate which of the two glycoproteins has the fusogenic properties. To advance in the knowledge of HCV fusion mechanism, we have studied the membrane-perturbing properties of the ectodomain of E2, obtained using a baculovirus expression system, by employing a variety of fluorescent assays and different conditions (pH and phospholipid composition). We have previously shown that E2661 is folded as an independent unit and is recognized by HCV patient sera antibodies (Rodriguez-Rodriguez et al., 2009). Our study shows that E2661 is able to induce the essential steps required for fusion (Rodriguez-Rodriguez et al., 2018).
Materials and Reagents
- Pipette tips
- Pyrex borosilicate glass tubes (Corning, catalog numbers: 99445-10 [10 x 75 mm] and 99445-13 [13 x 100 mm])
- 100 nm polycarbonate filters (Whatman, catalog number: 800309)
- Absorption and fluorescence cuvettes (Hellma, absorption cuvette 1 x 0.2, catalog number: QS 104-10-40; fluorescence cuvette 1 x 0.2, catalog number: 104002F-10-40; fluorescence cuvette 1 x 0.4, catalog number: 104F-10-40)
- Glass column manually made by faculty glass blower facility
- Sephadex G-75 (Pharmacia Biotech, catalog number: 17-0051-01)
- Chloroform (Scharlau, catalog number: CL02001000)
- Methanol (Merck, catalog number: 106002)
- Dimyristoylphosphatidylcholine (DMPC) (Avanti Polar Lipids, catalog number: 850345)
- Dimyristoylphosphatidylglycerol (DMPG) (Avanti Polar Lipids, catalog number: 840445)
- Egg phosphatidylcholine (PC) (Avanti Polar Lipids, catalog number: 840051)
- Egg yolk phosphatidylglycerol (PG) (Sigma-Aldrich, catalog number: 50445)
- Nitrogen Gas (Carburos metálicos, catalog number: UN1066)
- Tris-Base (Sigma-Aldrich, catalog number: T1503)
- NaCl (Merck, catalog number: 106404)
- 2-Morpholinoethanesulfonic acid monohydrate buffer substance MES (Merck, catalog number: 106126)
- Sodium citrate (Merck, catalog number: 106448)
- EDTA (Sigma-Aldrich, catalog number: 03695)
- N-(7-nitro-2,1,3-benzoxadiazol-4-yl)dimyristoylphosphatidylethanolamine (NBD-PE) (Avanti Polar Lipids, catalog number: 810143)
- N-(lissaminerhodamine B sulfonyl)diacylphosphatidyl-ethanolamine (Rh-PE) (Avanti Polar Lipids, catalog number: 810157)
- 8-aminonaphthalene-1,3,6-trisulfonic acid (ANTS) (Thermo Fisher Scientific, InvitrogenTM, catalog number: A350)
- p-xylenebis-(pyridinium) bromide (DPX) (Thermo Fisher Scientific, InvitrogenTM, catalog number: X1525)
- Liquid nitrogen (Air Liquide, Spain)
- Triton X-100 (USB, catalog number: 22686)
- Tetrahydrofuran (Sigma-Aldrich, catalog number: 401757)
- 1,6-diphenyl-1,3,5-hexatriene (DPH) (Sigma-Aldrich, catalog number: D208000)
- 1-(4-trimethylammoniumphenyl)-6-phenyl-1,3,5-hexatriene (TMA-DPH) (Sigma-Aldrich, catalog number: T0775)
- Medium buffer (see Recipes)
Equipment
- Pipettes: volume ranges of 2-20 μl, 20-200 μl and 200-1,000 μl
- Speed Vac (Uniequip, model: Univapo 100 H)
- Water bath Incubator (Selecta, model: unitronic 320)
- Bath sonicator (Bransonic, model: Branson 1200)
- Extruder apparatus (Avestin, model: LiposoFastTM-Basic)
- Spectrophotometer (Beckman Coulter, model: Beckman DU-640)
- Spectrofluorometer, equipped with a thermostated cell holder in which the temperature of the cuvette is maintained by a circulating water bath (SLM Aminco, model: SLM Aminco 8000C)
Procedure
- Vesicle preparation
- Dissolve the lipids in chloroform/methanol (2:1) at a concentration of 10 mg/ml. Prepare aliquots of 100 μl in 13 x 100 mm glass tubes and dry under nitrogen current to obtain a lipid film. Keep the film under vacuum in a speed-vac for 6-8 h.
- Make a suspension of phospholipids at a concentration of 1 mg/ml in 1 ml of medium buffer at the appropriate pH value.
- Incubate at 37 °C for 1 h in a water bath incubator without shaking and vortex vigorously every 10 min.
- Sonicate the suspension for 10 min in a bath sonicator at a frequency of 40 kHz and subject to 15 cycles of extrusion in an extruder apparatus using 100 nm polycarbonate filters. For a successful extrusion process, avoid the formation of bubbles in the lipid suspension when inserting it into the syringe and squeeze the syringe plunger carefully and slowly.
It is recommended to prepare the lipid vesicles just before carrying out an experiment, although they may be stored for 1-2 days at 4 °C.
After vesicle preparation, the exact phospholipid concentration is calculated based on the phosphorus content determined according to Bartlett (1959).
- Aggregation studies
- Add different amounts (between 10 and 100 μl) of a 15-20 μM stock solution of the E2661 recombinant protein (see Gómez-Gutiérrez et al., 2018) to 50 μl of recently prepared lipid vesicles and the needed amount of medium buffer at the appropriate pH to reach a total volume of 500 μl. After gently mixing the solutions in 10 x 75 mm glass tubes, the final phospholipid and protein concentrations must be 0.14 mM and in the range of 0.2-3.5 μM, respectively. Use controls of lipid vesicles in the absence of protein and of protein in the absence of lipid.
- Incubate at 37 °C for 1 h in a water bath incubator without shaking.
- Measure the optical density at 360 nm (OD360) on a spectrophotometer, using a 1 x 0.2 cm absorption cuvette.
- Lipid mixing assay
Note: This protocol is adapted from that described in Struck et al. (1981). This method measures the decrease in efficiency of the fluorescence energy transfer between a donor and an acceptor incorporated into liposomes, as a consequence of lipid mixing.- Prepare two different kinds of phospholipid vesicles: (1a) labeled vesicles containing 1 mol% NBD-PE and 0.6 mol% Rh-PE and (1b) unlabeled vesicles. An example of labeled vesicle (1a) preparation would be as follows: dissolve NBD-PE and Rh-PE in chloroform/methanol (2:1) at a concentration of 1 mg/ml in separate glass tubes; mix in a glass tube 10 μl and 6 μl of, respectively, NBD-PE and Rh-PE solutions with 100 μl of a solution of unlabeled phospholipid in chloroform/methanol (2:1) at a concentration of 10 mg/ml and operate according to Procedure A. For (1b), directly operate according to Procedure A. All the vesicle suspensions are prepared at a final lipid concentration of 1 mg/ml in medium buffer at the appropriate pH value.
- Mix labeled (1a) and unlabeled (1b) liposomes in a 1:9 molar proportion, maintaining the final lipid concentration of 1 mg/ml in a total volume of 1 ml. This mixed vesicle preparation is diluted ten times (100 μl + 900 μl of medium buffer) to a final concentration of 0.14 mM for lipid mixing assays.
- Add different amounts (between 5 and 100 μl) of protein from a 15-30 μM stock solutions in 20 mM Tris pH 7.5, NaCl 100 mM. The final protein concentration must be in the range of 0.1-3 μM.
- Incubate at 37 °C for 1 h in a water bath incubator without shaking.
- Record the emission spectra (wavelength range 480-620 nm) in a 1 x 0.4 cm fluorescence cuvette with the excitation wavelength set at 450 nm, in an SLM Aminco 8000C spectrofluorometer. Set the excitation and emission slits at 4 nm. In order to minimize dispersive interference, keep constant the excitation and emission polarizers at 90° and 0° respectively.
- Use the ratio of the emission intensities at 530 and 590 nm to calculate the efficiency of FRET (%FRET) together with the appropriate calibration curve. This curve is obtained by using labeled vesicles containing 1 mol% NBD-PE and variable concentrations of Rh-PE (0, 0.1, 0.2, 0.3, 0.5, 0.7, 1 and 1.3 mol%), according to the equation:
%FRET = [1 - (F/F0)] x 100
where,
F is the fluorescence intensity at 530 nm of vesicles containing 1% NBD-PE and 0-1% Rh-PE.
F0 is the fluorescence intensity at 530 nm of vesicles containing 1% NBD-PE and 0% Rh-PE.
- Release of aqueous contents
Note: This protocol is adapted from that described in Ellens et al. (1985). The assay is based on the dequenching of ANTS fluorescence caused by its dilution upon release of the aqueous contents of a vesicle preparation containing both ANTS and DPX.- The fluorophore ANTS and the quencher DPX must be co-encapsulated at a concentration of 12.5 and 45 mM respectively in either PG or PC phospholipid vesicles. For that purpose, weigh 13.4 mg of ANTS and 47.5 mg of DPX and dissolve in 2.5 ml of medium buffer at the appropriate pH value.
- Add 2 ml of this solution to a lipid film (prepared according to Procedure A1) containing 2 mg of the appropriate phospholipid. Hydrate the film at 37 °C for 1 h in a water bath incubator without shaking, and sonicate for 30 min in a bath sonicator.
- Subject the vesicles to five cycles of freeze-thawing in liquid nitrogen.
- Extrude the vesicles at least 15 times through 100 nm polycarbonate filters (Avestin) by using an extruder apparatus.
- Remove the non-encapsulated material by passing the sample through a Sephadex G-75 column using medium buffer at the appropriate pH value for elution. A typical column with 12-15 ml bed volume is prepared by swelling 1 g of dry Sephadex G75 in 20 ml of medium buffer for 24 h. After swelling is complete, decant supernatant, add buffer and degas the suspension. For packing, pour the complete slurry into a glass column of 1-1.5 cm diameter. Run chromatography with a 1 ml/min flow rate, and collect 1 ml fractions. The encapsulated material elutes in the void volume in fractions with high turbidity, and therefore is detected by measuring the optical density at 360 nm of each fraction in a spectrophotometer, whereas the non-encapsulated free dyes elute in the column volume. Dilute the encapsulated vesicles to a final phospholipid concentration in the assay of 0.14 mM.
- Add different amounts of a stock solution (15-20 μM) of E2661 recombinant protein to 0.8 ml of the diluted phospholipid vesicle suspension. The final protein concentration must be in the range of 0.1-3 μM.
- Use a 1 x 0.4 cm fluorescence cuvette to record the increase of the fluorescence emission at 520 nm upon excitation at 385 nm in a thermostated cell holder at 37 °C. Set the excitation and emission slits at 4 nm. In order to minimize dispersive interference, keep constant the excitation and emission polarizers at 90° and 0° respectively.
- Set the fluorescence scale to 100% by addition of 40 μl of 10% Triton X-100 (final concentration: 0.5% Triton) to 0.8 ml of the diluted phospholipid vesicle suspension and to 0% by measuring the fluorescence of 0.8 ml of the control, diluted, phospholipid vesicles. For each protein concentration, the parameter % Fmax is calculated according to the following equation:
% Fmax = [(Fprot - F0)/( Fmax - F0)] x 100
where,
-Fprot is the fluorescence intensity at 520 nm after the addition of E2 protein.
-F0 is the initial fluorescence intensity at 520 nm, before addition of the protein (0%).
-Fmax is the fluorescence intensity at 520 nm after addition of Triton X-100 (100%).
- Fluorescence polarization measurements
- Prepare DMPG or DMPC phospholipid vesicles labeled with the fluorescent probes DPH or TMA-DPH. The probe/phospholipid weight ratio is 1:500 for DPH and 1:100 for TMA-DPH. An example of labeled vesicle preparation would be as follows: use two glass tubes to dissolve DPH in tetrahydrofuran and TMA-DPH in methanol; at a concentration of 1 mg/ml; mix in glass tubes 2 μl of DPH (or 10 μl of TMA-DPH) solution with 100 μl of a solution of the synthetic phospholipid (DMPG or DMPC) in chloroform/methanol (2:1) at a concentration of 10 mg/ml and operate according to Procedure A. All the vesicle suspensions are prepared at a final lipid concentration of 1 mg/ml in medium buffer at the appropriate pH value.
- Incubate labeled vesicles in the absence or in the presence of E2661 recombinant protein at 37 °C for 30 min in a water bath incubator without shaking at a final lipid concentration of 0.14 mM and protein/lipid molar ratios of 1:20, 1:50 or 1:100. As an example, for a total volume of 400 μl, pipette 40 μl of labeled vesicles suspension, 20-100 μl of a 28 μM stock solution of E2 protein, and the needed amount of medium buffer at the appropriate pH to reach a total volume of 400 μl. After this incubation, cool the samples to 10 ºC.
- Adjust the excitation and emission slit widths of the spectrofluorometer at 4 nm.
- Set the excitation wavelength at 365 nm and measure the emission at 425 nm after equilibration of the samples for 10 min at the required temperature. The temperature in the 1 x 0.2 cm fluorescence cuvette is maintained in the thermostated cell holder with a circulating water bath. The fluorescence polarization value is directly provided by the apparatus and is calculated using the spectrofluorometer software according to the following equation:
P (polarization) = (IVV – IVH)/(IVV + IVH)
where, IVV and IVH are, respectively, the vertical and horizontal fluorescence intensities when the sample is excited with the excitation polarizer oriented vertically.
Data analysis
- Aggregation studies
The ability of E2661 to induce liposome aggregation was studied by measuring the increase of the optical density at 360 nm (OD360) due to scattering of a phospholipid suspension. The results obtained at pH 5.0 and pH 7.0 for both PC and PG vesicles at different phospholipid/protein ratios are depicted in Figure 1. The analysis of the initial rate of these processes indicates that aggregation was almost instantaneous. In all cases, the OD360 values were dependent on the E2661 concentration and became saturated as the protein concentration increased.
Figure 1. Aggregation of PC (- - -) and PG (—) phospholipid vesicles induced by E2661 at different concentrations. The optical density at 360 nm (OD360) was measured after incubation at 37 °C for 1 h of vesicles (0.14 mM) in medium buffer at pH 7.0 (▲) and pH 5.0 (●) with aliquots of protein at different concentrations. The final phospholipid concentration was 0.14 mM. The results shown are the averages and standard deviations of three independent experiments. - Lipid mixing assay
Lipid mixing was followed by the resonance energy transfer (FRET) assay between the fluorescent probes NBD-PE and Rh-PE (Struck et al., 1981). In Figure 2 are depicted the results obtained with PG at pH 7.0 and 5.0 and PC vesicles at pH 5.0. These results indicate that E2661 is able to induce a considerable fusion of labeled and unlabeled vesicles. In the case of PC vesicles, no decrease of energy transfer was observed at pH 5.0. Thus, lipid mixing is specifically induced by E2 glycoprotein on vesicles containing lipids with acidic head groups in an almost pH-independent manner and occurred in the absence of receptors on the target liposomes.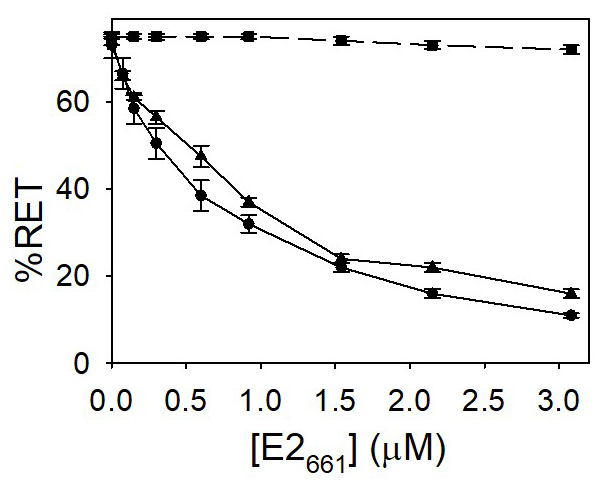
Figure 2. Lipid mixing induced by E2661. Vesicles containing both the donor and acceptor molecules were mixed with unlabeled ones in a 1:9 (mol:mol) ratio. Increasing concentrations of the E2661 were added to the mixture. The concentrations of NBD-PE and Rh-PE were 1 mol% and 0.6 mol%, respectively. PG (●, ▲) or PC (■) vesicles hydrated in medium buffer at pH 7.0 (●) and 5.0 (■, ▲). The final phospholipid concentration was 0.14 mM. The results shown are the averages and standard deviations of three independent experiments. - Release of aqueous content
The ability to cause the release of the aqueous contents of phospholipid vesicles was also used to check the destabilization properties of E2661. Liposome leakage was monitored by the dilution of the co-encapsulated fluorophore ANTS and its collisional quencher DPX, which leads to a rapid increase in ANTS fluorescence at 520 nm (Ellens et al., 1985). The leakage induced by the protein added to PG or PC phospholipid suspensions at the two pH values tested is shown in Figure 3. This process induced on neutral phospholipid vesicles was lower than that observed for PG vesicles suggesting the importance of the electrostatic component on the protein-lipid interactions.
Figure 3. Leakage of ANTS/DPX induced by E2661. Increasing concentrations of E2 peptides were added to vesicles of PC (- - -) and PG (—) phospholipid vesicles loaded with ANTS and DPX in medium buffer at pH 7.0 (■) and 5.0 (●). Leakage was initiated by addition of the protein and the increase of the fluorescence emission at 520 nm upon excitation at 385 nm was continuously recorded. Fmax was obtained upon addition of 0.5% Triton X-100. The measurements were performed at 37 °C and the final phospholipid concentration in the assay was 0.14 mM. The results shown are the averages and standard deviations of three independent experiments. - Fluorescence polarization measurements
E2 ectodomain was able to interact with phospholipid vesicles altering their thermotropic behavior. This was evidenced by measuring the fluorescence depolarization of the fluorescent probe DPH, incorporated into phospholipid vesicles. This molecule provides clues on how the interaction with the protein alters the deeper environment of the acyl chains (Lentz et al., 1976; Prendergast et al., 1981) (Figure 4).
Figure 4. Effect of E2661 on the temperature dependence of fluorescence polarization of DPH-labeled DMPG (A, B) and DMPC (C, D) at pH 5.0 (A, C) and 7.0 (B, D) liposomes. The protein:lipid molar ratios were: (●) 0.0 (No protein), (♦) 1:100, (■) 1:50, (▲) 1:20. The results shown are the averages and standard deviations of three independent experiments.
Recipes
- Medium buffer
Tris-HCl 5 mM
NaCl 100 mM
MES 5 mM
Sodium citrate 5 mM
EDTA 1 mM
Acknowledgments
This work was supported by grant BFU 2006-13033 from the Ministerio de Educación y Ciencia, Spain and the research project SANTANDER/COMPLUTENSE PR26/16-20271.
Competing interests
Authors declare that there are no conflicts of interest or competing interests.
References
- Bartlett, G. R. (1959). Phosphorus assay in column chromatography. J Biol Chem 234(3): 466-468.
- Cormier, E. G., Durso, R. J., Tsamis, F., Boussemart, L., Manix, C., Olson, W. C., Gardner, J. P. and Dragic, T. (2004). L-SIGN (CD209L) and DC-SIGN (0209) mediate transinfection of liver cells by hepatitis C virus. Proc Natl Acad Sci 101: 14067-14072.
- Ellens, H., Bentz, J. and Szoka, F. C. (1985). H+- and Ca2+-induced fusion and destabilization of liposomes. Biochemistry 24(13): 3099-3106.
- Gómez-Gutiérrez, J., Rodríguez-Rodríguez, M., Gavilanes, F. and Yélamos, B (2018) Expression and Ni-NTA-Agarose purification of recombinant hepatitis C virus E2 ectodomain produced in a baculovirus expression system. Bio Protoc 8(19): e3030.
- Koutsoudakis, G., Kaul, A., Steinmann, E., Kallis, S., Lohmann, V., Pietschmann, T. and Bartenschlager, R. (2006). Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J Virol 80(11): 5308-5320.
- Lavillette, D., Bartosch, B., Nourrisson, D., Verney, G., Cosset, F. L., Penin, F. and Pecheur, E. I. (2006). Hepatitis C virus glycoproteins mediate low pH-dependent membrane fusion with liposomes. J Biol Chem 281(7): 3909-3917.
- Lemon, S. M., Walker, C. M., Alter, M. J. and Yi, M. (2007). Hepatitis C virus. In: Knipe, D. M., Howley, P. M. (Eds). Fields Virology. Lippincott Williams & Wilkins, Philadelphia. pp: 1253-1304.
- Lentz, B. R., Barenholz, Y. and Thompson, T. E. (1976). Fluorescence depolarization studies of phase transitions and fluidity in phospholipid bilayers. 2 Two-component phosphatidylcholine liposomes. Biochemistry 15(20): 4529-4537.
- Lindenbach, B. D., Thiel, H. J. and Rice, C. M. (2007). Flaviviridae: The viruses and their replication. In: Knipe, D. M. and Howley, P. M. (Eds). Fields Virology. 5th ed. Lippincott Williams & Wilkins, Philadelphia. pp: 1101-1152.
- Meertens, L., Bertaux, C. and Dragic, T. (2006). Hepatitis C virus entry requires a critical postinternalization step and delivery to early endosomes via clathrin-coated vesicles. J Virol 80(23): 11571-11578.
- Prendergast, F. G., Haugland, R. P. and Callahan, P. J. (1981). 1-[4-(Trimethylamino)phenyl]-6-phenylhexa-1,3,5-triene: synthesis, fluorescence properties, and use as a fluorescence probe of lipid bilayers. Biochemistry 20(26): 7333-7338.
- Rocha-Perugini, V., Montpellier, C., Delgrange, D., Wychowski, C., Helle, F., Pillez, A., Drobecq, H., Le Naour, F., Charrin, S., Levy, S., Rubinstein, E., Dubuisson, J. and Cocquerel, L. (2008). The CD81 partner EWI-2wint inhibits hepatitis C virus entry. PLoS One 3(4): e1866.
- Rodriguez-Rodriguez, M., Tello, D., Gomez-Gutierrez, J., Peterson, D. L., Gavilanes, F. and Yelamos, B. (2018). Fusogenic properties of the Ectodomain of HCV E2 envelope protein. Biochim Biophys Acta Biomembr 1860(3): 728-736.
- Rodriguez-Rodriguez, M., Tello, D., Yelamos, B., Gomez-Gutierrez, J., Pacheco, B., Ortega, S., Serrano, A. G., Peterson, D. L. and Gavilanes, F. (2009). Structural properties of the ectodomain of hepatitis C virus E2 envelope protein. Virus Res 139(1): 91-99.
- Struck, D. K., Hoekstra, D. and Pagano, R. E. (1981). Use of resonance energy transfer to monitor membrane fusion. Biochemistry 20(14): 4093-4099.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Yélamos, B., Rodríguez-Rodríguez, M., Gavilanes, F. and Gómez-Gutiérrez, J. (2018). In vitro Membrane Interaction and Liposome Fusion Assays Using Recombinant Hepatitis C Virus Envelope Protein E2. Bio-protocol 8(23): e3108. DOI: 10.21769/BioProtoc.3108.
Category
Biochemistry > Protein > Fluorescence
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.