- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Genome-wide Estimation of Evolutionary Distance and Phylogenetic Analysis of Homologous Genes

Published: Vol 8, Iss 23, Dec 5, 2018 DOI: 10.21769/BioProtoc.3097 Views: 8010
Reviewed by: Chen ChenAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Dec 2017
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








Abstract
Homologous genes, including paralogs and orthologs, are genes that share sequence homologies within or between different species. Homologous genes originate from a common origin through speciation, genetic duplication or horizontal gene transfer. Estimation of the sequence divergence of homologous genes help us to understand divergence time, which makes it possible to understand the evolutionary patterns of speciation, gene duplication and gene transfer events. This protocol will provide a detailed bioinformatics pipeline on how to identify the homologous genes, compare their sequence divergence and phylogenetic relationships, focusing on homologous genes that show syntenic relationships using soybean (Glycine max) and common bean (Phaseolus vulgaris) as example species.
Keywords: Homologous genesBackground
Gene duplication, including whole genome duplication or polyploidy, segmental duplication, and tandem duplication, is a very important process that increases gene copy number and thus enhances genetic diversity in many organisms. Because of this, gene duplication is thought to be a major force in evolution (Ohno, 1970; Otto and Whitton, 2000; Blanc and Wolfe, 2004; Jiao et al., 2011). After duplication, duplicated genes are subject to a variety of changes, such as accumulation of point mutations, insertions and deletions, gene conversion and transposon insertions (Ilic et al., 2003; Gu et al., 2005; Sémon and Wolfe, 2007). Theoretically, two or multiple copies of the duplicated genes have undergone different levels of selective constraint, which makes the functional divergence of the duplicated genes. This can be reflected on the sequence divergence of the homologous genes, such as non-synonymous substitution (Ka) and synonymous substitution (Ks), the latter of which the produced amino acid sequence is not modified. Because it is neutral with respect to selection, Ks can be used to determine rough divergence time. The ratio of Ka/Ks can be used to estimate the selection pressure on genes. A Ka/Ks ratio equal to one indicates a lack of selection, as is observed in pseudogenes. The ratio of Ka/Ks higher and lower than one implies positive and purifying selection, respectively. The values of Ka/Ks for the vast majority of genes are < 1.0 due to purifying selection to maintain function (Makalowski and Boguski, 1998; Nekrutenko et al., 2002). When comparing duplicate genes, differences in Ka/Ks suggest different levels or kinds of selection.
In order to determine the evolutionary distance, we first need to identify the homologous genes within or between different species. Here, we will mainly focus on the homologous genes that show syntenic relationships between different species. Syntenic genes are those genes that retain an ancestral position on a given region of a chromosome. We refer to these syntenic homologous genes as “syntelogs” (Zhao et al., 2017). The advantage of focusing on syntelogs is that if they are the result of polyploidy, all syntelogs arose at the same time, so groups of syntenic gene pairs can be compared with high confidence. Here, we describe a detailed pipeline for the identification and comparison of the evolutionary distance of syntelogs by using soybean (Glycine max) and common bean (Phaseolus vulgaris) as example species (Figure 1). It has been proposed that soybean has experienced a recent whole genome duplication event roughly 5 to 13 million years ago (MYA, Schmutz et al., 2010), occurring after the split with its close relative common bean roughly 19 MYA (Lavin et al., 2005; McClean et al., 2010). In this protocol, we will estimate the evolutionary divergence of the duplicated gene pairs in soybean by comparing them with the orthologous genes in the common bean genome.
Equipment
- Linux/Unix cluster
In this study, we use the Purdue Halstead supercomputer, which contains 508 nodes in total. Each node contains 20 cores, two 10-Core Intel Xeon-E5 processors, and 128 GB memory. Please refer to the website for more information: https://www.rcac.purdue.edu/compute/halstead. - Personal computer for post data processing (Lenovo, T430s, Intel Core i5-3320M CPU, 4 GB RAM)
Software
- Blastall or Blast+ (Altschul et al., 1997)
https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE=BlastDocs&DOC_TYPE=Download
This program is developed and distributed by NCBI to do blast searches. - MCScanX (Wang et al., 2012), http://chibba.pgml.uga.edu/mcscan2/
This toolkit is for detection and evolutionary analysis of gene synteny and collinearity. This program also can generate tandemly duplicated genes. - Multiple Sequence Comparison by Log-Expectation (MUSCLE) (Edgar, 2004), https://www.drive5.com/muscle/
- ClustalW (Thompson et al., 1994), http://www.clustal.org/clustal2/
Both Muscle and ClustalW are software for multiple sequence alignment for nucleotide and protein sequences. - Phylogenetic Analysis by Maximum Likelihood (PAML) (Yang et al., 2007), http://abacus.gene.ucl.ac.uk/software/paml.html
PAML is a package of programs for phylogenetic analyses of DNA or protein sequences using maximum likelihood. - Molecular Evolutionary Genetics Analysis (MEGA X) (Kumar et al., 2018), https://www.megasoftware.net/
This software contains many sophisticated methods and tools for phylogenetic analysis, including constructing phylogenetic trees as well as evolutionary distance estimation. - Perl, https://www.perl.org/, or Python, https://www.python.org/, programming languages
These languages make it possible to post-process the primary data generated from some of the software used above. - SAS software, https://www.sas.com/en_us/home.html
SAS is a software for statistical analysis.
Procedure
- Identification of Syntenic Genes among Closely Related Species
- Retrieve gene sequence
For given plant genomes of which the genome annotation is available, download the coding sequences (CDS) and protein sequences of all the protein-encoding genes from corresponding databases. Some species have their respective websites, such as Arabidopsis TAIR (https://www.arabidopsis.org/), soybean SoyBase (https://soybase.org/), and maize MaizeGDB (https://www.maizegdb.org/), etc. The sequences of many of other species were deposited at NCBI (https://www.ncbi.nlm.nih.gov/), CoGe (https://genomevolution.org/coge/), Phytozome (https://phytozome.jgi.doe.gov/pz/portal.html) or other relevant databases. We downloaded all the genome sequences and gene annotation of soybean (v1.1, Schmutz et al., 2010) and common bean (Schmutz et al., 2014) from Phytozome. - Removal of transposon-related and hypothetical proteins
In order to detect highly confident syntelogs among closely related species, transposon-related and hypothetical proteins were first removed using BLAST (Altschul et al., 1997). Taking soybean (v1.1) as an example, in total 53,927 annotated genes from the 20 soybean chromosomes were BLASTN queried against the soybean transposon database SoyTEdb (Du et al., 2010, https://www.soybase.org/soytedb/). Any genes over the 80% of the total length matching the transposon-related sequences with the sequence similarities of greater than 80% were removed. Genes annotated as hypothetical proteins were excluded as well (Figure 1). Here is a typical setting for doing local blast.
formatdb -i SoyBase_TE_Fasta.txt -p F -o T
blastall -p blastn -i Soybean_gene_cds.fa -d SoyBase_TE_Fasta.txt -m 8 -a 8 -o Soybean_gene_cds_blast_TEs
Note: Hypothetical proteins were identified based on the gene annotation file. The genes annotated as hypothetical proteins were not included in the analysis. Here formatdb is to format the nucleotide source database “SoyBase_TE_Fasta.txt” before it can be searched using blastall. blastall is used to compare the gene sequences in the file “Soybean_gene_cds.fa” with the database “SoyBase_TE_Fasta.txt”. A detailed description of each parameter can be found in the software manual.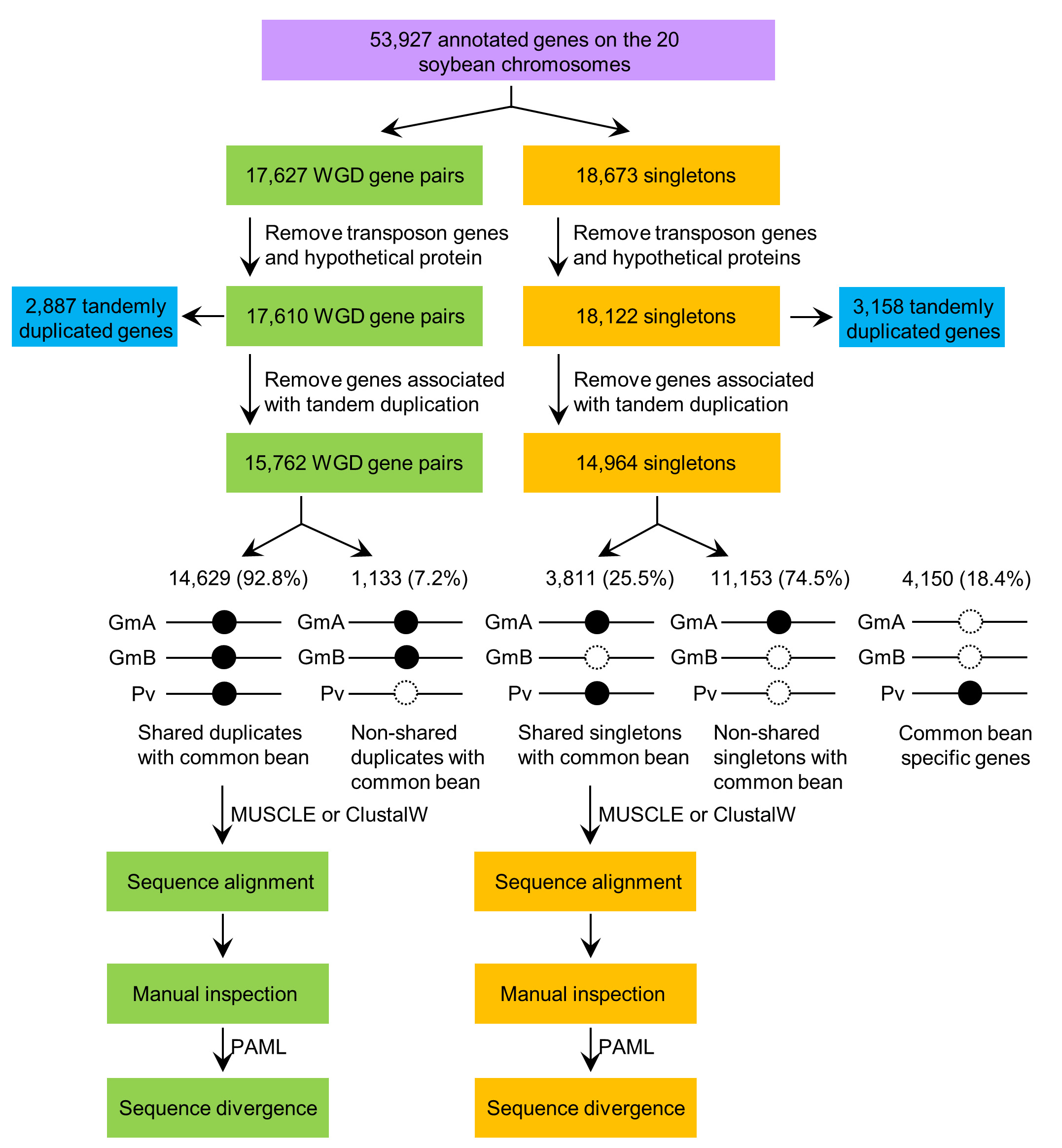
Figure 1. Bioinformatics pipeline for identification and comparison of the syntenic homologous genes in soybean and common bean. The solid and the dotted circles indicate the presence and absence of the genes in the corresponding genomes, respectively. GmA and GmB represent either of the duplicated genes in soybean. Gm, Glycine max, soybean; Pv, Phaseolus vulgaris, common bean. This figure was modified from Zhao et al., 2017. - Detection of candidate syntelogs
The remaining protein-encoding genes in soybean and common bean were used to do an all-against-all BLASTP search using default parameters, with the E-value cutoff 10-10 (Altschul et al., 1997). For each pair of genes, BLAST hits were loaded to the software MCScanX (Wang et al., 2012) to scan the syntelog homologous gene pairs.
blastall -p blastp -i Soybean_gene_pep.fa -d Commonbean_gene_pep.fa -m 8 -a 8 -F F -o Soybean _blast_Commonbean
MCScanX Soybean_CommonBean
Note: The setting of E-value is based on the rough divergence time. You can leave it as default setting if the divergence time is unknown. Please prepare the required gff file for all gene locations to MCScanX. Syntenic gene pairs between soybean and common bean were identified with MCScanX’s default settings (Match Score = 50, Match Size = 5, Gap Penalty = -1, Overlap Window = 5, Max Gaps = 25 and an E-value cutoff 10-10). - Post process the candidate syntelogous genes
Since soybean has undergone a whole genome duplication after its split with common bean, the genes in the common bean genomes were corresponding to one or two copies in the soybean genome, depending on the duplication status of the genes retained in soybean. MCScanX provided all the homologous gene pairs in addition to syntelogous gene pairs. In order to remove the false positive syntelogous genes, the duplicated block information from the reference genome was incorporated (Schmutz et al., 2014) to keep the homologous gene pairs detected in the duplicated regions and showing syntenic relationship between soybean and common bean.
Example of the final list of candidate syntelogous genes was shown in Table 1. Genes involved in tandem duplication have an ambiguous retention status and thus were put aside for separate analysis.
Table 1. Example of syntenic homologous genes identified in the common bean and soybean genomes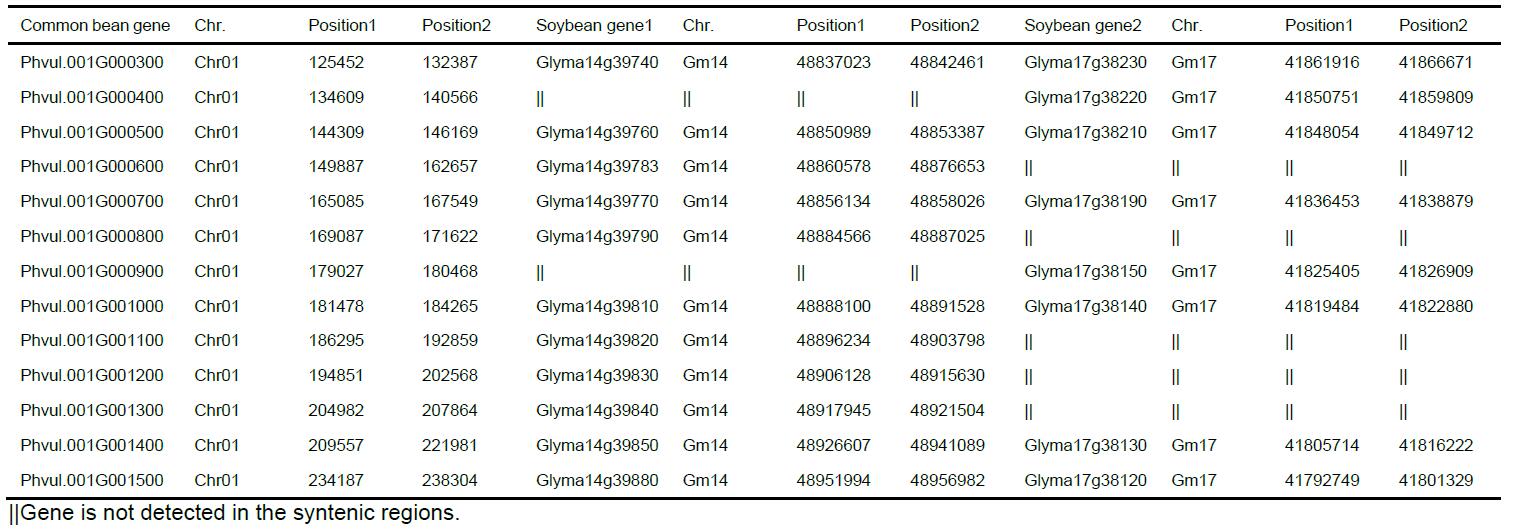
- Retrieve gene sequence
- Estimation of Evolutionary Distance for Syntelogous Genes
- Sequence alignment
Although many genes have several alternative transcripts, only the primary transcripts of the genes based on the gene annotation were used to estimate the sequence divergence between different syntenic genes of soybean and common bean. The nucleotide sequences of the syntenic genes were aligned using the MUSCLE program (Edgar, 2004) or ClustalW (Thompson et al., 1994) using default parameters. The alignment can be viewed by Jalview (Figure 2A).
muscle -in input -out output or clustalw input
Note: The primary transcripts of the genes were determined based on the gene annotation file which shows the primary transcripts of the genes. MUSCLE or ClustalW can only run one group of syntelogs at a time. For whole genome level analysis, we recommend that the authors write a Perl or Python script to automatically load each pair of sequences to MUSCLE or ClustalW to do the alignment. At this step, we used MUSCLE to run the alignment first, and then performed ClustalW for the remained gene pairs of which the nucleotide alignments were not integer multiples of three after MUSCLE alignment. - Manual inspection
The output alignment was manually inspected to modify incorrectly aligned nucleotides. This step is very important although it may not be practical if there is a very large amount of data to verify (Figure 2B).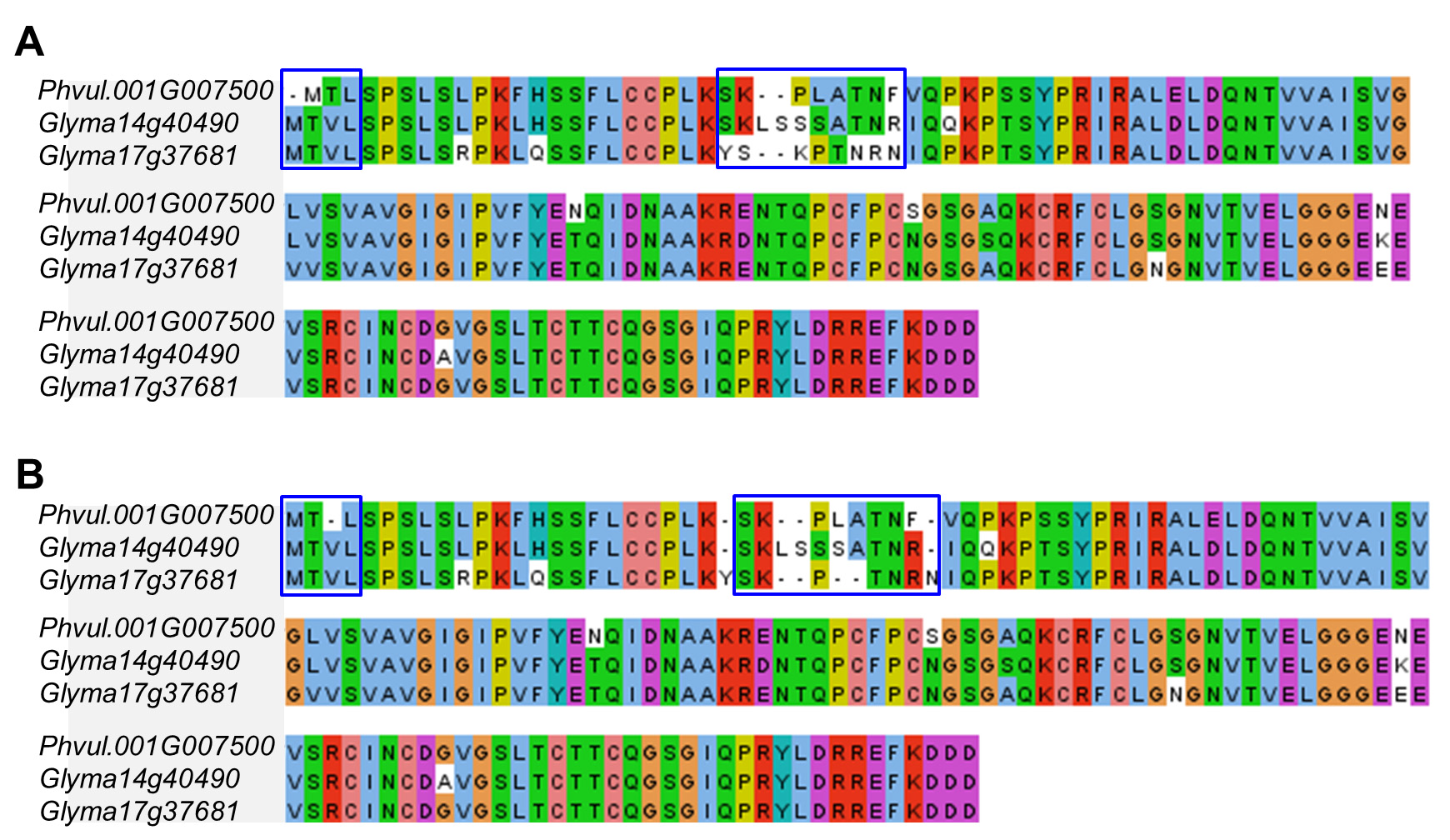
Figure 2. An example of sequence alignments of syntenic homologous genes in soybean and common bean. A. Original alignment generated by MUSCLE (Edgar, 2004). B. Manually modified alignment. Blue boxes indicate modified regions. - Sequence divergence
All pairwise alignments of the syntenic genes were prepared into the required format by the PAML software using Perl or Python programming in order to calculate non-synonymous (Ka) and synonymous (Ks) substitution using the yn00 and baseml modules with the default parameters except model was set to 1 instead of 0 (Yang, 2007). Please refer to the manual for more information about running programs in the PAML package.
- Sequence alignment
- Phylogenetic analysis
Phylogenetic trees are used to tell the phylogenetic relationship among homologous genes.- Sequence Alignment
The nucleotide sequences or protein sequences of the syntenic genes were aligned using the MUSCLE program (Edgar, 2004) or ClustalW (Thompson et al., 1994) using default parameters.
muscle -in input -out output or clustalw input - Phylogenetic Tree Construction
The sequence alignments of the homologous genes were transferred to MEGA software to construct the phylogenetic trees using the neighbor-joining maximum composite likelihood model integrated for nucleotide sequences and Poisson correction for protein sequences with pairwise deletions (Kumar et al., 2018). Bootstrap values were calculated from 1,000 replicates.
- Sequence Alignment
Data analysis
Student’s t-test was performed to compare the evolutionary distance between duplicates and singletons using the SAS software. The Bonferroni correction was further performed to correct the P values. P < 0.05 was considered to be significant, and P < 0.0001 was considered to be significant under the Bonferroni correction. Experimental values are reported as mean ± standard deviation or in a box plot (Figure 3).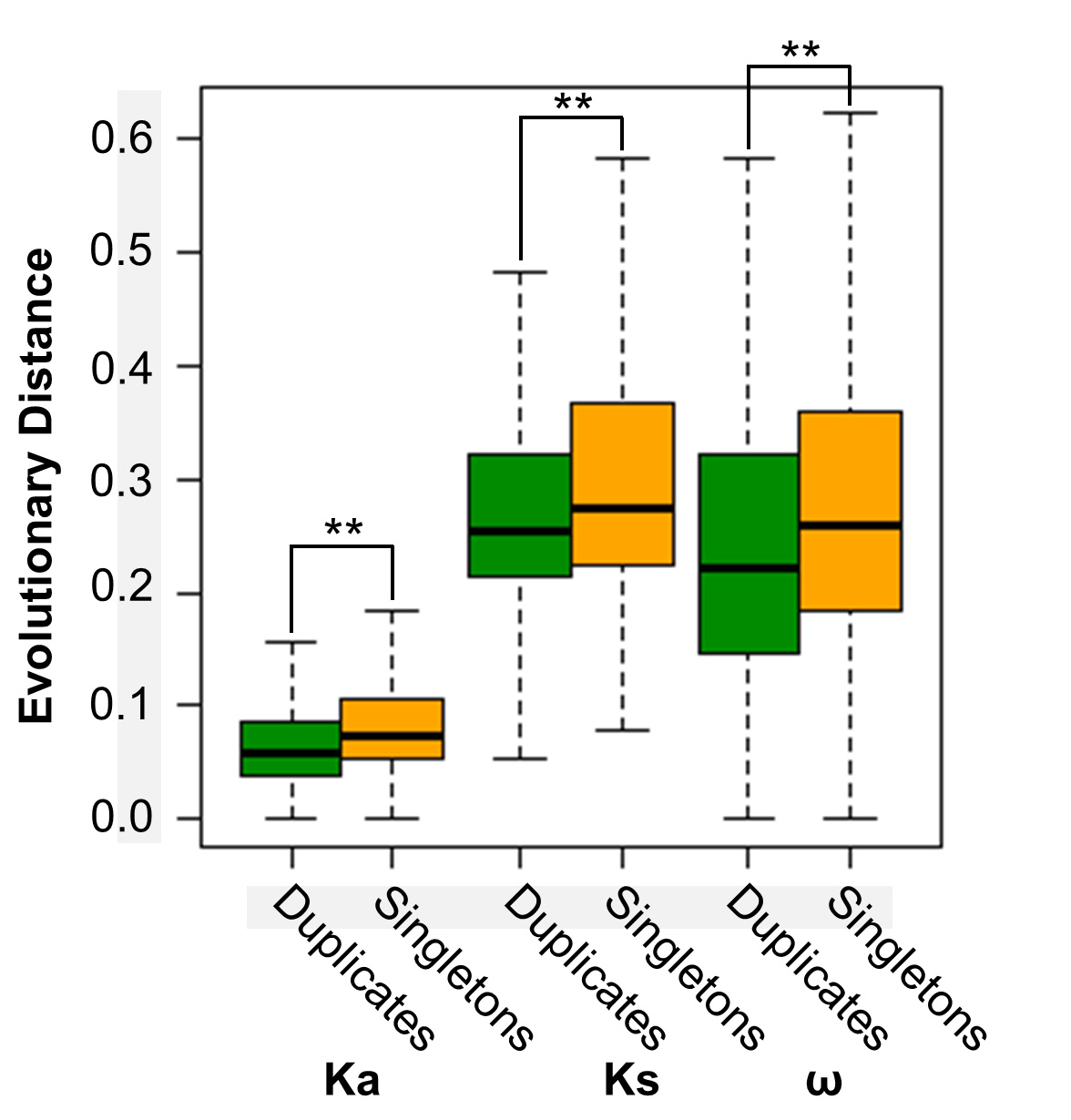
Figure 3. Comparison of the evolutionary distance between duplicates and singletons in soybean. Ka and Ks were calculated by pairwise comparison between soybean and common bean. The statistical analysis was conducted by Student’s t-test. **, P < 0.0001. Ka, non-synonymous substitution; Ks, synonymous substitution; ω, Ka/Ks. The bottom and top boundaries of the box are the first and third quartiles, and the bold lines within individual boxes are the medians, which are referred to as the second quartiles. The ends of the whiskers (the dotted lines) represent the minimum values and maximum values of the data.
Notes
- Some designated singleton genes, the homologs of which were not found in the syntenic region, may belong to duplicated pairs because of potential gene transposition. The homologs of the singletons may be transposed or translocated from the original syntenic regions to elsewhere in the genome, rather than being deleted.
- Genes involved in tandem duplication always have an ambiguous retention status, and thus were separately analyzed.
Acknowledgments
This protocol was adapted from Zhao et al. (2017). This work was supported by soybean check-off funds from the United Soybean Board and Indiana Soybean Alliance and National Science Foundation Grant DBI-0822258 to J.M., by National Science Foundation Grant DBI-1237931 to D.L. and Purdue Startup Funds to D.L, and by Miami University Startup Funds to M.Z.
Competing interests
The authors declare that there are no conflicts of interest or competing interests.
References
- Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D. J. (1997). Gapped BLAST and PSIBLAST: a new generation of protein database search programs. Nucleic Acids Res 25(17): 3389-3402.
- Blanc, G. and Wolfe, K. H. (2004). Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell 16(7): 1667-1678.
- Du, J., Grant, D., Tian, Z., Nelson, R. T., Zhu, L., Shoemaker, R. C. and Ma, J. (2010). SoyTEdb: a comprehensive database of transposable elements in the soybean genome. BMC Genomics 11: 113.
- Edgar, R. C. (2004). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5: 113.
- Gu, X., Zhang, Z. and Huang, W. (2005). Rapid evolution of expression and regulatory divergences after yeast gene duplication. Proc Natl Acad Sci U S A 102(3): 707-712.
- Ilic, K., SanMiguel, P. J. and Bennetzen, J. L. (2003). A complex history of rearrangement in an orthologous region of the maize, sorghum, and rice genomes. Proc Natl Acad Sci U S A 100(21): 12265-12270.
- Jiao, Y., Wickett, N. J., Ayyampalayam, S., Chanderbali, A. S., Landherr, L., Ralph, P. E., Tomsho, L. P., Hu, Y., Liang, H., Soltis, P. S., Soltis, D. E., Clifton, S. W., Schlarbaum, S. E., Schuster, S. C., Ma, H., Leebens-Mack, J. and dePamphilis, C. W. (2011). Ancestral polyploidy in seed plants and angiosperms. Nature 473(7345): 97-100.
- Kumar, S., Stecher, G., Li, M., Knyaz, C. and Tamura, K. (2018). MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol Biol Evol 35(6): 1547-1549.
- Lavin, M., Herendeen, P. S. and Wojciechowski, M. F. (2005). Evolutionary rates analysis of Leguminosae implicates a rapid diversification of lineages during the tertiary. Syst Biol 54(4): 575-594.
- Makalowski, W. and Boguski, M. S. (1998). Evolutionary parameters of the transcribed mammalian genome: an analysis of 2,820 orthologous rodent and human sequences. Proc Natl Acad Sci U S A 95(16): 9407-9412.
- McClean, P. E., Mamidi, S., McConnell, M., Chikara, S. and Lee, R. (2010). Synteny mapping between common bean and soybean reveals extensive blocks of shared loci. BMC Genomics 11: 184.
- Nekrutenko, A., Makova, K. D. and Li, W. H. (2002). The KA/KS ratio test for assessing the protein-coding potential of genomic regions: an empirical and simulation study. Genome Res 12(1): 198-202.
- Ohno, S. (1970). Evolution by gene duplication. Springer-Verlag, New York, p. 160.
- Otto, S. P. and Whitton, J. (2000). Polyploid incidence and evolution. Annu Rev Genet 34: 401-437.
- Schmutz, J., Cannon, S. B., Schlueter, J., Ma, J., Mitros, T., Nelson, W., Hyten, D. L., Song, Q., Thelen, J. J., Cheng, J., Xu, D., Hellsten, U., May, G. D., Yu, Y., Sakurai, T., Umezawa, T., Bhattacharyya, M. K., Sandhu, D., Valliyodan, B., Lindquist, E., Peto, M., Grant, D., Shu, S., Goodstein, D., Barry, K., Futrell-Griggs, M., Abernathy, B., Du, J., Tian, Z., Zhu, L., Gill, N., Joshi, T., Libault, M., Sethuraman, A., Zhang, X. C., Shinozaki, K., Nguyen, H. T., Wing, R. A., Cregan, P., Specht, J., Grimwood, J., Rokhsar, D., Stacey, G., Shoemaker, R. C. and Jackson, S. A. (2010). Genome sequence of the palaeopolyploid soybean. Nature 463(7278): 178-183.
- Schmutz, J., McClean, P. E., Mamidi, S., Wu, G. A., Cannon, S. B., Grimwood, J., Jenkins, J., Shu, S., Song, Q., Chavarro, C., Torres-Torres, M., Geffroy, V., Moghaddam, S. M., Gao, D., Abernathy, B., Barry, K., Blair, M., Brick, M. A., Chovatia, M., Gepts, P., Goodstein, D. M., Gonzales, M., Hellsten, U., Hyten, D. L., Jia, G., Kelly, J. D., Kudrna, D., Lee, R., Richard, M. M., Miklas, P. N., Osorno, J. M., Rodrigues, J., Thareau, V., Urrea, C. A., Wang, M., Yu, Y., Zhang, M., Wing, R. A., Cregan, P. B., Rokhsar, D. S. and Jackson, S. A. (2014). A reference genome for common bean and genome-wide analysis of dual domestications. Nat Genet 46(7): 707-713.
- Sémon, M. and Wolfe, K. H. (2007). Consequences of genome duplication. Curr Opin Genet Dev 17(6): 505-512.
- Thompson, J. D., Higgins, D. G. and Gibson, T. J. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22(22): 4673-4680.
- Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., Lee, T. H., Jin, H., Marler, B., Guo, H., Kissinger, J. C. and Paterson, A. H. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res 40(7): e49.
- Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24(8): 1586-1591.
- Zhao, M., Zhang, B., Lisch, D. and Ma, J. (2017). Patterns and consequences of subgenome differentiation provide insights into the nature of paleopolyploidy in plants. Plant Cell 29(12): 2974-2994.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Zhao, M., Zhang, B., Ma, J. and Lisch, D. (2018). Genome-wide Estimation of Evolutionary Distance and Phylogenetic Analysis of Homologous Genes. Bio-protocol 8(23): e3097. DOI: 10.21769/BioProtoc.3097.
Category
Plant Science > Plant molecular biology > Genetic analysis
Systems Biology > Genomics > Phylogenetics
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.