- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Kinetic Characterization of the Shigella Type Three Secretion System ATPase Spa47 Using α-32P ATP
Published: Vol 8, Iss 21, Nov 5, 2018 DOI: 10.21769/BioProtoc.3074 Views: 5596
Reviewed by: Andrea PuharLionel SchiavolinKanika Gera
Original research article
The authors used this protocol in:

Apr 2018
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
ATPases represent a diverse class of enzymes that utilize ATP hydrolysis to support critical biological functions such as driving ion pumps, providing mechanical work, unfolding/folding proteins, and supporting otherwise thermodynamically unfavorable chemical reactions. We have recently shown that the Shigella protein Spa47 is an ATPase that supports protein secretion through its specialized type three secretion apparatus (T3SA), supporting infection of human host cells. Characterizing ATPases, such as Spa47, requires a means to accurately determine enzyme activity (ATP hydrolysis) as a function of time, reaction conditions, and potential cofactors, regulators, inhibitors, etc. Here, we describe a detailed protocol for characterizing the enzyme kinetics of Spa47 using a direct α-32P ATPase assay.
Keywords: ATPaseBackground
The study of enzyme kinetics arguably gave rise to the field of biochemistry and remains an important and diverse area of research today. While many important factors are considered when characterizing enzyme kinetics, they ultimately all rely on a robust and accurate means of determining the rate of enzyme-catalyzed product formation under a series of carefully controlled conditions. One important and highly diverse class of enzymes includes those that couple the hydrolysis of ATP to downstream processes such as driving ion pumps, providing mechanical work, unfolding/folding proteins, and supporting otherwise thermodynamically unfavorable chemical reactions (Stock et al., 2000; Sauer et al., 2011; Miller et al., 2016). Despite what processes these enzymes are ultimately responsible for, they are all collectively referred to as ATPases for the critical role ATP hydrolysis plays in their overall function.
The biological importance and diversity of ATPases have driven the development of several methods to quantify ATPase activity. Examples include a wide range of enzyme-coupled assays that rely on the generation of either ADP or inorganic phosphate to support a subsequent enzyme-catalyzed reaction or series of reactions to ultimately generate a product that provides a detectable change in signal (often photon absorbance) (Sehgal et al., 2016; Hoskins et al., 2018). This provides a means for real-time monitoring of ATP hydrolysis though conditions for each enzyme-catalyzed reaction in the scheme must be carefully considered and can limit the utility of this method. Some colorimetric and fluorescence assays have more recently been developed to monitor ATP depletion or product generation without the need for coupling to additional enzyme-catalyzed reactions, but they remain reliant on interaction/reaction with reporter proteins/molecules and can be highly sensitive to reaction buffer composition and contamination (Soyenkoff, 1947; Baykov et al., 1988; Li et al., 2005). Perhaps one of the most robust means for monitoring ATP hydrolysis relies on a radioactive ATP substrate such as γ-32P ATP or α-32P ATP. The radioactive 32P ATP substrate is hydrolyzed by the enzyme, producing either ADP and 32P (inorganic phosphate) or α-32P ADP and inorganic phosphate from γ-32P ATP and α-32P ATP, respectively (King, 1995). The radioactive products and remaining substrate can then be separated by thin layer chromatography, detected by exposure to either film or a storage phosphor screen and the concentration of product formed quantified as a function of time the reaction was allowed to proceed during the assay. Though this method does not readily support real-time monitoring of product formation, it carries many inherent advantages including high signal-to-noise, capability with essentially any buffer conditions required to support enzyme stability, and no downstream coupling reactions required for product detection/quantitation.
Here, we provide a detailed α-32P ATP hydrolysis protocol for kinetic characterization of the Shigella type three secretion system ATPase, Spa47 that has provided insight into working with this specific protein (Burgess et al., 2016a and 2016b; Case and Dickenson, 2018). This protocol is intended, however, to serve as a guide for the characterization of any enzymes that hydrolyze ATP as the basis of the protocol is unchanged by the enzyme being studied.
Materials and Reagents
- Nitrile gloves
- Low retention pipette tips (VWR, catalog numbers: 89187-848, 89079-476 and 89079-486)
- PEI Cellulose F TLC plates (Millipore, catalog number: 105579)
- 0.65 ml Microcentrifuge Tubes (VWR, catalog number: 87003-290)
- Plastic wrap
- Soft Pencil
- Permanent marker
- Sterile 0.22 μm PVDF syringe Filter (Sartorius, catalog number: 17761-Q)
- Container for collecting 32P-contaminated pipette tips and tubes (VWR, catalog number: 13915-114)
- Bovine Serum Albumin (BSA) (Thermo Fisher Scientific, catalog number: 23209)
- Dithiothreitol (DTT) (Gold Bio, catalog number: DTT100)
- 12% SDS-PAGE gel(s) (Bio-Rad, catalog number: 4561046)
- BLUEstain Protein Ladder (Gold Bio, catalog number: P007-500)
- Adenosine triphosphate (ATP) (Sigma-Aldrich, catalog number: A2383)
- Ethylenediaminetetraacetic acid (EDTA) Disodium, dehydrate (Gold Bio, catalog number: E-210-1)
- Tris-HCl (VWR Life Science, catalog number: 77-86-1)
- NaCl (Sigma-Aldrich, catalog number: 7647-14-5)
- SDS (Ameresco, catalog number: M107-500G)
- NaN3 (Sigma-Aldrich, catalog number: S2002)
- Glycine (VWR Life Sciences, catalog number: 56-40-6)
- Acetic acid (Pharmco AAPER, catalog number: 281000ACSCSLTGL)
- Methanol (Fischer Chemical, catalog number: 67-56-1)
- Coomassie blue R250 (Gold Bio, catalog number: C-461-100)
- Magnesium chloride (MgCl2) Anhydrous (Ameresco, catalog number: J364-500G)
- α-32P ATP (PerkinElmer, catalog number: BLU003H250UC)
- Potassium monophosphate (K2HPO4) (VWR Life Science, catalog number: 0781-1KG)
- Bromophenol blue
- Glycerol
- Milli-Q water
- NaOH
- 1 M MgCl2
- 3x SDS sample buffer (see Recipes)
- SDS running buffer (see Recipes)
- Total protein SDS-PAGE gel stain (see Recipes)
- SDS-PAGE gel de-stain (see Recipes)
- Spa47 buffer (see Recipes)
- ATP reaction Mix (see Recipes)
- Potassium monophosphate (K2HPO4) buffer (see Recipes)
- 0.5 M EDTA, pH 7.9 (see Recipes)
Note: Many of these reagents can be hazardous. Please familiarize yourself with the potential hazards of each chemical prior to performing this protocol.
Equipment
- Ruler
- Timer
- Microcentrifuge Tube rack(s)
- Thermal cycler
- Rocking platform
- -20 °C freezer
- Electrophoresis system and modules for SDS-PAGE (Bio-Rad, catalog number: 125BR)
- Electrophoresis power supply (Bio-Rad, model: PowerPac 300)
- Adjustable pipettes (Eppendorf, catalog numbers: 3120000011, 3120000020, 3120000046, 3120000038 and 3120000062)
- Gel imager (Bio-Rad Chemi Doc MP imaging system)
- Spectrophotometer (Thermo Spectronic, model: Genesys 10 UV)
- Space designated (approved) for radioactive work
- Radiation shield (Nalgene, catalog number: 6700-1812)
- Geiger counter (Technical Associates, model: TBM-15)
- Vortexer mini (VWR, catalog number: 58816-121)
- TLC developing tank (Sigma-Aldrich, catalog number: Z126195-1EA)
- Storage phosphor screen (Kodak, model number: SO230)
- STORM 860 scanner (Molecular Dynamics)
Software
- Image Lab Software 5.2.1 (Bio-Rad Laboratories)
- Image Quant 5.1 (Molecular Dynamics)
- Microsoft Excel (Microsoft)
- Sigma Plot 12 (Systat Software)
- ImageJ
- STORM scanner control software
Procedure
Day 1
- Spa47 can be purified using previously published methods (Burgess et al., 2016b). Determine the concentration of Spa47 via in gel densitometry or other appropriate methods for proteins with low A280 extinction coefficients. (Spa47 does not contain any tryptophan residues and the A280 extinction coefficient is extremely low, but protein concentration determination by A280 can be used here for most other proteins)
Note: Following purification, Spa47 is in “Spa47 buffer” (Recipe 5).- Prepare BSA standards (0.025, 0.05, 0.1, 0.25, 0.5 mg/ml) by diluting a certified 2.0 mg/ml standard in Spa47 buffer. Prepare each BSA standard and the Spa47 solution of unknown concentration in 0.65 ml microcentrifuge tubes by adding 3x SDS sample buffer, and DTT (20 μl protein sample, 10 μl 3x SDS sample buffer, 1 μl 1.5 M DTT). Heat all samples to 99 °C for 10 min using a thermal cycler.
- Load 1 μl of protein ladder and 10 μl of each BSA standard and unknown concentration protein sample in the wells of a 12% SDS-PAGE gel. Run the separation at 180 V until the blue dye front from the SDS sample buffer reaches the bottom of the gel plate (about 60 min).
- Remove the gel from the gel plates and place it in the total protein SDS-PAGE gel stain solution for 30 min on a rocking platform. Move the gel to SDS-PAGE gel de-stain solution for 1-2 h on a rocking platform or until the gel is evenly and completely de-stained. This may require the de-stain solution to be changed once or twice during the de-staining procedure.
- Image the stained gel using a Bio-Rad Chemi Doc MP imaging system set to epi-illumination. Use the rectangle selection tool found in the volume tools menu in the associated Image lab software to draw boxes around the protein band in each of the BSA standards and Spa47 samples. Include an additional box in a region of the image that does not contain any proteins and set it to represent the global background signal in the gel. Open the analysis table to show the “adjusted volumes” (background subtracted intensities) of the BSA standards. Plot the adjusted volumes versus the known concentrations of the BSA standards and fit the data points to a linear standard curve. The linear standard curve should result in an R2 ≥ 0.98 and the equation of the fit is then used to determine the concentration of Spa47 (see Figure 1). Alternatively, protein densitometry analyses can be performed with open source software such as ImageJ.
Note: In our hands, a typical monomeric Spa47 concentration is 9 μM and a typical oligomeric Spa47 concentration is 3 μM. Highest levels of Spa47 ATPase reproducibility result from ≥ 95% pure recombinant protein.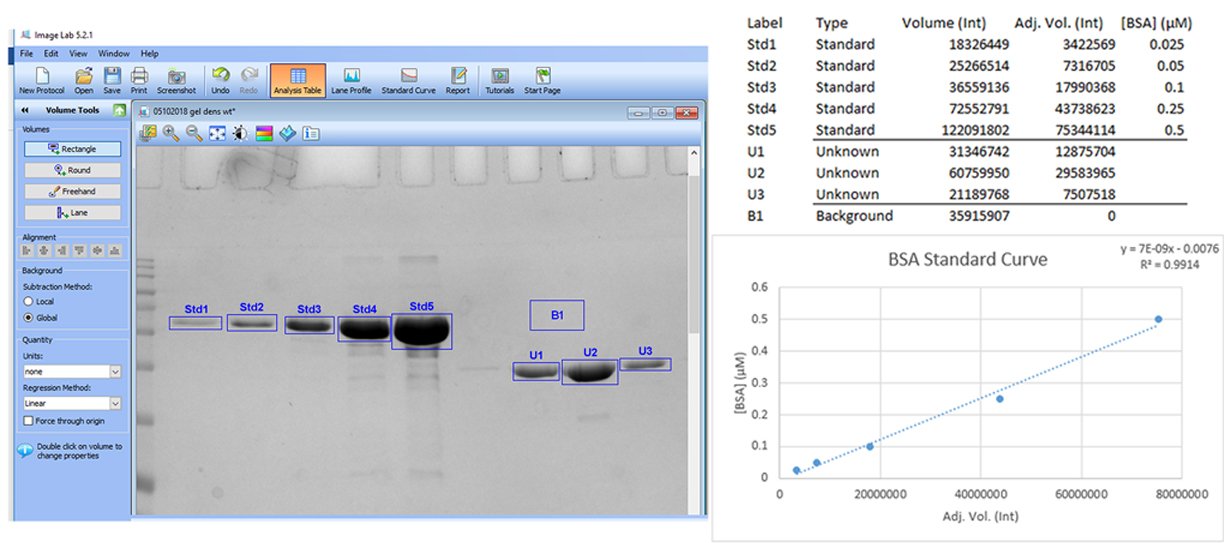
Figure 1. Using Image Lab and in gel densitometry of BSA standards to determine the concentration of Spa47
- Prepare stock solution of ATP
- Dissolve ATP in Milli-Q water, aiming for a concentration of 100 mM (55.114 mg ATP/ml).
- Filter the freshly made stock ATP solution with a sterile 0.22 μm PVDF syringe filter and make 3 separate 1 to 10,000 dilutions of the stock ATP. Measure the absorbance of each solution at 259 nm using a Genesys 10 UV spectrophotometer.
- Use the extinction coefficient of ATP (15 x 103 M-1 cm-1) to determine the concentration of each diluted ATP solution. Determine the average concentration of the three and record this as the concentration of the stock ATP solution (ensure that the three measurements are in good agreement with one another). The ATP stocks can be stored for long periods if stored at -20 °C and aliquoted to avoid freeze/thaw cycles.
- Prepare TLC plates
Using a ruler measure and cut a 20 x 20 cm Millipore TLC plate in half. Measure 1 cm up the 10 cm side and lightly draw a pencil line across the plate. Place tick marks at 1 cm intervals across the line.
Note: Must use soft pencil and draw lightly to avoid scraping stationary phase off the TLC plate. Do not use plate if the stationary phase is damaged. - Set up 0.65 ml Micro-centrifuge tubes as shown in Figure 2
- Pipette 7.5 μl of 0.5 M EDTA into each of the “time point tubes” for the reaction as well as the “background quencher” tube. Label these tubes with the appropriate time point to be collected and protein condition/ATP concentration.
- Add the appropriate amount of Spa47 and Spa47 buffer to the reaction tube to reach a final concentration of 0.5 μM Spa47 in the final 75 μl volume of the reaction (this volume will allow for up to nine time points).
Note: It is important to note that because you will add 7.5 μl of ATP Mix to each tube to initiate the reaction and we want the final Spa47 concentration to be 0.5 μM in the reaction, the tubes should contain 67.5 μl of 0.555 μM Spa47 prior to addition of ATP. - Make a background sample by adding 67.5 μl of Spa47 buffer to a tube and making a corresponding background quencher tube with 7.5 μl of 0.5 M EDTA.
Note: If you are performing analyses at multiple ATP concentrations, you must make background tubes for each ATP concentration used in the experiment.

Figure 2. Tube setup for α-32P ATP ATPase activity assay - Make ATP reaction Mix (see Recipes) behind an appropriate radiation shield in an area designated for handling radioactive material.
Note: If you are performing kinetic analyses at multiple ATP concentrations, you will need to perform serial dilutions to generate ATP reaction Mix solutions that will result in the desired final ATP concentrations. Also note that the mix contains radioactive α-32P ATP and should be handled carefully. - ATPase activity Assay
- Starting with a setup as illustrated in Figure 2, work behind an appropriate radiation shield and open all tubes. Start a timer and at the same time add 7.5 μl ATP reaction Mix to the reaction tube to initiate the reaction. Quickly close the reaction tube and briefly vortex. Re-open tube.
- Remove 7.5 μl of the enzyme reaction from the reaction tube and quench it at the pre-determined time point by adding it to one of the time point tubes containing 7.5 μl 0.5 M EDTA. Repeat this step at appropriate time points until all desired time points have been collected and all desired reaction conditions completed.
Note: Common time points for monomeric Spa47 assayed at 0.5 µM include 1, 5, 10, 15 and 30 min, while common time points for oligomeric Spa47 at 0.5 µM include 1, 2.5, 5, 10 and 15 min. - Add 7.5 μl reaction mix (contains no enzyme) to the background tube, close and vortex this tube. Remove 7.5 μl from the background tube and quench it in the background quencher tube containing 7.5 μl 0.5 M EDTA.
Note: This step can be done during the assay or directly after.
- Separate reactions on TLC plates
- Pipet 1 μl of the quenched reaction from each time point onto the TLC plate (pipette the solution directly onto the intersection of the horizontal line and vertical “tick mark” you drew on the TLC plate earlier (Step 3) (see Figure 3). Spot the background in triplicate.

Figure 3. Separation of α-32P ATP and α-32P ADP. A. Layout for spotting quenched Spa47 reaction samples on a TLC plate (dashed circles showing where to spot 1 µl sample). B. An example of spotted TLC plate in buffer chamber is included as well. - Let the spots on the TLC plate dry and then place the plate into buffer chamber containing enough potassium monophosphate buffer (see Recipes) to cover the bottom of the chamber, but not so much that it makes contact with the location of the spotted protein samples.
- Leave the TLC plate(s) in the chamber until the mobile phase wicks to the top of TLC plate. Remove the TLC plate from the chamber and allow it to dry in a fume hood.
Note: The plate now contains radioactive α-32P ATP and α-32P ADP. - Once dry, wrap the TLC plates in plastic wrap and expose overnight (~8 h) to a storage phosphor screen.
- Pipet 1 μl of the quenched reaction from each time point onto the TLC plate (pipette the solution directly onto the intersection of the horizontal line and vertical “tick mark” you drew on the TLC plate earlier (Step 3) (see Figure 3). Spot the background in triplicate.
- Imaging the storage phosphor screen
Remove the TLC plates and place the storage phosphor screen on the Molecular Dynamics STORM 860 scanner. Open STORM scanner control software and image the screen with the following settings: 635 nm excitation, 390 nm emission, and 200-micron pixel size. - Quantifying ATP and ADP concentrations
- Open the gel STORM image file of the storage phosphor screen using Molecular Dynamics ImageQuant software. Using the selection tool in ImageQuant, draw a box tightly around the largest ATP spot as shown in Figure 4. Copy and paste the same box around every ATP and ADP spot to ensure that each box is exactly the same size.
Note: The image contrast can be increased to more easily visualize faint spots in the image and this will not affect the downstream quantitation. - Under the Analysis menu in ImageQuant, generate a volume report and export the intensities to Microsoft Excel for data analysis.
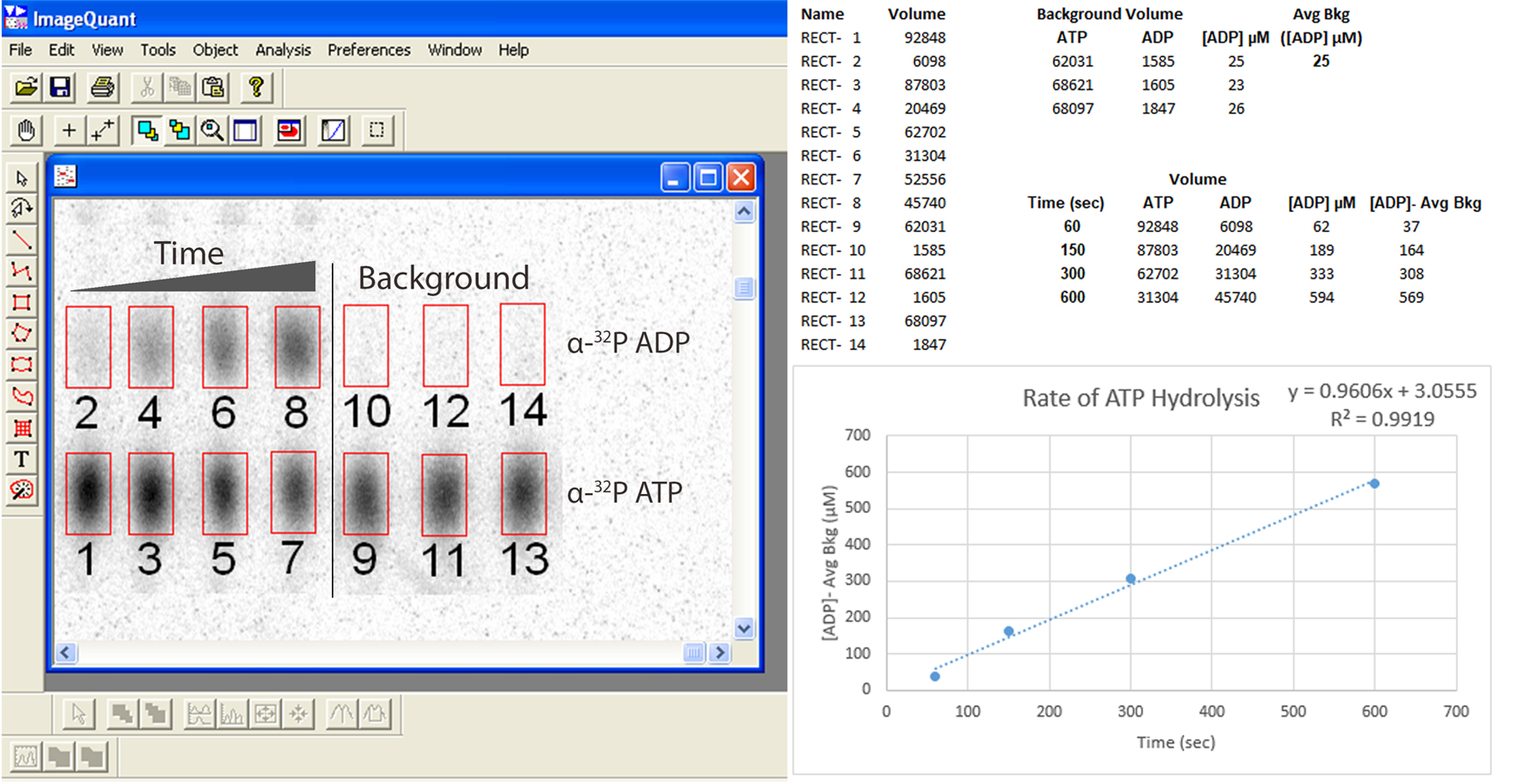
Figure 4. Determining the rate of ATP hydrolysis using ImageQuant analysis of TLC separated α-32P ATP and α-32P ADP
- Open the gel STORM image file of the storage phosphor screen using Molecular Dynamics ImageQuant software. Using the selection tool in ImageQuant, draw a box tightly around the largest ATP spot as shown in Figure 4. Copy and paste the same box around every ATP and ADP spot to ensure that each box is exactly the same size.
Data analysis
A quantitative analysis of Spa47s conversion of ATP to ADP can be performed using the volumes of the α-32P ATP and α-32P ADP spots from the scan of the phosphor storage screen as seen in Figure 3. Using the exported volumes from ImageQuant, divide the volume of ADP for each reaction time point by the volume of ADP + ATP for that time point and multiply by the total concentration of ATP in the original reaction condition (1 mM in this case). This gives you the concentration of ADP in the units you used when multiplying by the total ATP concentration in the previous step. Do the same for the background spots as shown in Figure 4. Because there is no enzyme in the background reaction conditions, it is likely that there will be very little ADP detected so you may have to use your best judgment when placing the boxes for the corresponding ADP spots on the phosphor storage screen. Now, subtract the average background (spotted and calculated in triplicate) from the concentration of ADP determined at each reaction time point. These values represent the background-subtracted concentrations of ADP formed at each reaction time point. Plotting these concentrations vs. the reaction time will allow you to determine V0 from the slope of a fit linear regression line (Figure 4).
Often times, it is necessary to characterize substrate concentration dependence of an enzyme and this is easily achieved by simply repeating the steps above at multiple starting ATP concentrations in the reaction. The slopes of each of the lines generated from the multiple ATP concentrations can be plotted vs. the corresponding ATP concentration as shown in Figure 5. It is important to note that the substrate concentration will have a significant impact on the initial velocity of each reaction as well as the time required to reach saturation of the reaction. You can see in the graph in Figure 5 that the reactions with the lowest ATP concentrations (25 μM and 50 μM) were allowed to proceed for much shorter periods of time than the other reactions due to loss of linearity at longer time points. The plot of [ATP] versus initial velocity (μmol ADP/s) can then be fit to the Michaelis Menten equation using SigmaPlot (Equation 1).
From the Michaelis Menten fit, the maximum velocity achievable by the enzyme under the tested conditions (Vmax), and the substrate concentration resulting in one-half Vmax (KM) are determined. This allows for calculation of kcat by dividing Vmax by total enzyme concentration and of the specificity constant of the enzyme by dividing kcat by KM. Each reaction condition should then be repeated to allow robust statistical analyses.
Note: The reaction rate for any ATPase can be determined as described above, but that the method described for determination of the kinetic parameters Vmax, KM, kcat, and the specificity constant requires that the enzyme follows Michaelis Menten kinetics.
Figure 5. Example of non-linear kinetic analysis of Spa47 at varying ATP concentrations. A. Rate of ATP hydrolysis at varying ATP concentrations. B. The Michaelis constant (KM) and maximum velocity achievable by the enzyme (Vmax) are illustrated on the Michaelis-Menten plot.
Recipes
- 3x SDS sample buffer (400 ml)
210 mM Tris-HCl (pH 6.8)
100 mM NaCl
3 M EDTA
200 mM SDS
3 mM NaN3
0.03% bromophenol blue
30% glycerol - SDS running buffer
20 mM Tris-HCl
200 mM glycine
3.5 mM SDS - Total protein SDS-PAGE gel stain
10% acetic acid
40% methanol
2 mM Coomassie blue R250
50% H2O - SDS-PAGE gel de-stain
10% acetic acid
40% methanol
50% H2O - Spa47 buffer
20 mM Tris-HCl
100 mM NaCl
5 mM DTT
Adjust the pH to 7.9 with 6 M HCl - ATP reaction Mix
10 mM ATP
120 mM MgCl2
0.5 μCi (~300 nM) α-32P-ATP - Potassium monophosphate (K2HPO4) buffer
0.6 M K2HPO4
Adjust the pH with HCl to 3.4 - 0.5 M EDTA, pH 7.9
0.5 M EDTA
Add NaOH pellets until EDTA fully dissolves, adjust the pH to 7.9 with 6 M HCl
Acknowledgments
The work presented here was supported in part by a National Institutes of Health Grant 1R15AI124108-01A1 and R. Gaurth Hansen endowment funds to N. E. D.
Competing interests
The authors declare that there are no financial or non-financial competing.
References
- Baykov, A. A., Evtushenko, O. A. and Avaeva, S. M. (1988). A malachite green procedure for orthophosphate determination and its use in alkaline phosphatase-based enzyme immunoassay. Anal Biochem 171(2): 266-270.
- Burgess, J. L., Burgess, R. A., Morales, Y., Bouvang, J. M., Johnson, S. J. and Dickenson, N. E. (2016a). Structural and biochemical characterization of Spa47 provides mechanistic insight into Type III secretion system ATPase activation and Shigella virulence regulation. J Biol Chem 291(50): 25837-25852.
- Burgess, J. L., Jones, H. B., Kumar, P., Toth, R. T. t., Middaugh, C. R., Antony, E., and Dickenson, N. E. (2016b). Spa47 is an oligomerization-activated type three secretion system (T3SS) ATPase from Shigella flexneri. Protein Sci 25 1037-1048.
- Case, H. B. and Dickenson, N. E. (2018). MxiN differentially regulates monomeric and oligomeric species of the Shigella type three secretion system ATPase Spa47. Biochemistry 57(15): 2266-2277.
- Hoskins, J. R., Wickner, S. and Doyle, S. M. (2018). Bacterial Hsp90 ATPase assays. Methods Mol Biol 1709: 199-207.
- King, S. M. (1995). Chapter 21 measurement of ATPase activity using [γ-32P]ATP. In: Dentler, W. and Witman, G. (Eds.). Methods in Cell Biology. Academic Press. pp 141-145.
- Li, C., Numata, M., Takeuchi, M. and Shinkai, S. (2005). A sensitive colorimetric and fluorescent probe based on a polythiophene derivative for the detection of ATP. Angew Chem Int Ed Engl 44(39): 6371-6374.
- Miller, J. M. and Enemark, E. J. (2016). Fundamental characteristics of AAA+ protein family structure and function. Archaea 2016: 9294307.
- Sauer, R. T. and Baker, T. A. (2011). AAA+ proteases: ATP-fueled machines of protein destruction. Annu Rev Biochem 80: 587-612.
- Sehgal, P., Olesen, C. and Moller, J. V. (2016). ATPase activity measurements by an enzyme-coupled spectrophotometric assay. Methods Mol Biol 1377: 105-109.
- Stock, D., Gibbons, C., Arechaga, I., Leslie, A. G. and Walker, J. E. (2000). The rotary mechanism of ATP synthase. Curr Opin Struct Biol 10(6): 672-679.
- Soyenkoff, B. (1947). A micromethod of phosphate determination. J Biol Chem 168(2): 447-457.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Case, H. B. and Dickenson, N. E. (2018). Kinetic Characterization of the Shigella Type Three Secretion System ATPase Spa47 Using α-32P ATP. Bio-protocol 8(21): e3074. DOI: 10.21769/BioProtoc.3074.
Category
Microbiology > Microbial biochemistry > Protein > Activity
Biochemistry > Protein > Activity
Molecular Biology > Protein > Dephosphorylation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.