- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Electroporation of Labeled Antibodies to Visualize Endogenous Proteins and Posttranslational Modifications in Living Metazoan Cell Types

Published: Vol 8, Iss 21, Nov 5, 2018 DOI: 10.21769/BioProtoc.3069 Views: 6433
Reviewed by: David CisnerosYi CuiTrinadh Venkata Satish Tammana
Original research article
The authors used this protocol in:

Apr 2018
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
The spatiotemporal localization of different intracellular factors in real-time and their detection in live cells are important parameters to understand dynamic protein-based processes. Therefore, there is a demand to perform live-cell imaging and to measure endogenous protein dynamics in single cells. However, fluorescent labeling of endogenous protein in living cells without overexpression of fusion proteins or genetic tagging has not been routinely possible. Here we describe a versatile antibody-based imaging approach (VANIMA) to be able to precisely locate and track endogenous proteins in living cells. The labeling is achieved by the efficient and harmless delivery of fluorescent dye-conjugated antibodies or antibody fragments (Fabs) into living cells and the specific binding of these antibodies to the target protein inside of the cell. Our protocol describes step by step the procedure from testing of the suitability of the desired antibody, over the digestion of the antibody to Fabs until the labeling and the delivery by electroporation of the antibody or Fab into the cells. VANIMA can be adapted to any monoclonal antibody, self-produced or commercial, and many different metazoan cell lines. Additionally, our method is simple to implement and can be used not only to visualize and track endogenous factors, but also to specifically label posttranslational modifications, which cannot be achieved by any other labeling technique so far.
Keywords: AntibodiesBackground
The fluorescent labeling of proteins to follow in real time their spatiotemporal localization in living cells was mainly achieved until now by using transgenic or overexpression-based approaches. However, the labeling of specific endogenous proteins or even posttranslational modifications in living cells is not yet routinely possible. Imaging of cellular structures and processes is typically performed by either immunofluorescence (IF) labeling on fixed cells or by exogenously overexpressing fluorescent fusion proteins in living cells. Although these well-established techniques showed to be very powerful to locate or follow proteins inside the cells, they inherit also some important drawbacks. In IF, the cells need to be chemically fixed and permeabilized to be able to incubate them with specific primary and secondary antibodies. Despite many variables and potential artifacts (Schnell et al., 2012; Teves et al., 2016) like fixation-related protein denaturation or permeabilization efficiency, IF is still often used to visualize target proteins in fixed cells or tissues. Otherwise, imaging of proteins in living cells is mainly achieved through the exogenously expression of fluorescent fusion proteins (Ellenberg et al., 1999; Betzig et al., 2006; Schneider and Hackenberger, 2017) or by knock-in of a fluorescent tag into the endogenous locus using the CRISPR/Cas9 technology (Ratz et al., 2015). Although fluorescent fusion proteins have been proven to be very powerful, they often do not behave as their endogenous counterparts due to their increased levels when exogenously overexpressed (Burgess et al., 2012). On the other hand, endogenous fusion proteins containing knocked-in tags are difficult to obtain as knock-in efficiencies are often very low. Consequently, there is a need for new and easy to implement imaging approaches to visualize endogenous target proteins in single living cells. Previous studies and methods, like FabLEM or the expression of mintbodies, showed that intracellular labeling of proteins with fluorescently labeled antibody fragments can give new insights into the dynamics of histone modifications (Hayashi-Takanaka et al., 2009; Hayashi-Takanaka et al., 2011; Sato et al., 2013). However, these techniques suffer from lower delivery efficiencies into living cells, or potential poor solubility of the intracellular expressed mintbodies. Recently, another method achieved fluorescent labeling of endogenous proteins by using a bacterial toxin called streptolysin O, which creates pores in the membrane of cells and allows for the delivery of fluorescent probes into living cells (Teng et al., 2016). However, this method requires additional steps to be able to reseal the membrane after treatment which can be quite harmful for the cells and can decrease cell viability. In contrast, our versatile antibody-based imaging approach (VANIMA) uses fluorescent dye-conjugated antibodies or Fabs, which are delivered into the cells by electroporation (Freund et al., 2013; Brees and Fransen, 2014). The antibody labeling reaction is highly efficient and can result in up to 5-7 fluorescent dyes per molecule of antibody depending on the antibody and the labeling kit used. The transduction of the antibodies has a very high delivery efficiency and viability of the cells is above 90% in human cancer cell lines such as U2OS. Afterwards, the transduced antibodies will bind to the endogenous target protein inside the cell and for nuclear targets they will be transported with the target protein into the nucleus (piggyback mechanism). Otherwise, for faster delivery into the nucleus of the cells, the antibodies can be digested to produce Fabs which can freely diffuse into the nucleus to find and bind their target. Thus, even proteins with posttranslational modifications in the nucleus can be visualized specifically using fluorescently-labeled Fabs against the target. Considering that there are several thousands of commercially-available antibodies that specifically recognize intracellular target proteins with high affinity, VANIMA can be used to uncover the dynamical behavior of a plethora of targets in living cells (Conic et al., 2018). Besides nuclear targets, the antibodies could also be used to label and image cytoplasmic structures/proteins. However, it is important to note that only proteins that are either directly accessible for the antibodies/Fabs or that can be reached through the piggyback mechanism can be labeled using this technique. We were already able to label α-tubulin in the cytoplasm but other accessible targets like the mitochondrial membrane or cytoplasmic vesicles could also be tested for labeling with VANIMA. However, specific labelling of cytoplasmic targets would only be possible if the target molecules are highly expressed. If their abundance in the cell is below the one of the introduced antibodies, a large fraction of the antibodies does not bind and will generate background staining. Additionally, the method is easy to implement in any laboratory and can also be used to perform multicolor imaging with different targets just by labeling two different antibodies with different dyes or by combining it with an already established endogenous knock-in clone. Finally, VANIMA can also be used with identified inhibiting antibodies to disrupt protein functions inside living cells.
Materials and Reagents
- 15 ml conical Falcon tubes (Corning, Falcon, catalog number: 352095)
- 1.5 ml Eppendorf tubes (Sigma-Aldrich, Eppendorf, catalog number: Z66505-100EA)
- 0.5 ml Eppendorf tubes (Sigma-Aldrich, Eppendorf, catalog number: Z666491-100EA)
- Falcon 12-well clear flat bottom cell culture plate (Corning, catalog number: 351143)
- µ-slide 8-well glass bottom: No. 1.5H (170 µm +/- 5 µm) (Ibidi, catalog number: 80827)
- 18 mm high precision cover glasses (Marienfeld, catalog number: 117580)
- Microscope slides ground edges plain (VWR, catalog number: 631-1552)
- Poly-Prep chromatography column (Bio-Rad, catalog number: 731-1550)
- DiaEasy dialyzer (3 ml) MWCO 6-8 kDa (Biovision, catalog number: K1013-25)
- DiaEasy dialyzer (800 µl) MWCO 6-8 kDa (Biovision, catalog number: K1019-25)
- Amicon Ultra-4 centrifugal filter units 10 kDa (Merck-Millipore, catalog number: UFC801024)
- Amicon Ultra-0.5 centrifugal filter units 10 kDa (Merck-Millipore, catalog number: UFC501096)
- CountessTM cell counting chamber slides (Thermo Fisher, catalog number: C10312)
- Sterile individually packaged 5 ml pipettes (Sigma-Aldrich, catalog number: SIAL1487)
- Sterile individually packaged 10 ml pipettes (Sigma-Aldrich, catalog number: SIAL1488)
- U2OS osteosarcoma cells [American Type Culture Collection (ATCC, catalog number: HTB-96)]
- Neon transfection 10 µl kit (including the Neon 10 µl tips) (Invitrogen, catalog number: MPK1096)
- AlexaFluor-488 antibody labeling kit (Invitrogen, catalog number: A20181)
- Dulbecco's Modified Eagle Medium (DMEM) (Thermo Scientific, Gibco, catalog number: 10567-014)
- Heat-inactivated fetal calf serum (FCS) (Gibco, catalog number: 15750-037)
- Gentamicin (Gibco, catalog number: 15750-037)
- 16% Paraformaldehyde (16% PFA) (Electron Microscopy Sciences, catalog number: 50-980-487)
- Phosphate Buffered Saline (PBS) (GE Healthcare, catalog number: SH30013.03)
- Triton X-100 (Sigma-Aldrich, catalog number: X100-100ML)
- Vectashield antifade mounting medium with DAPI (Vector-Laboratories, catalog number: H-1200-10)
- Alexa Fluor 488 goat-anti-mouse IgG (life technologies, catalog number: A11001)
- Protein G Sepharose FastFlow (GE Healthcare, catalog number: GE17-0618-01)
- Protein A Sepharose FastFlow (GE Healthcare, catalog number: GE17-5280-01)
- Papain-coated magnetic beads (Spherotech, catalog number: PAPM-40-2)
- Tris-HCl (Sigma-Aldrich, catalog number: 10812846001)
- Glycine (Sigma-Aldrich, catalog number: G8898)
- Sodium bicarbonate (Sigma-Aldrich, catalog number: S5761-1KG)
- Tris(2-carboxyethyl) phosphine hydrochloride (TCEP) (Sigma-Aldrich, catalog number: C4706)
- Sodium dodecyl sulfate (SDS) (Euromedex, catalog number: EU0660)
- Acrylamide/Bis-acrylamide 40% solution (Euromedex, catalog number: EU0077-B)
- Tetramethylethylendiamin (TEMED) (Serva, catalog number: 35930.01)
- Ammonium persulfate (APS) (Sigma-Aldrich, catalog number: A3678)
- Trypsin (Sigma-Aldrich, catalog number: T4799)
- U2OS growth medium (see Recipes)
- 4% Paraformaldehyde (4% PFA) (see Recipes)
- 10x Phosphate Buffered Saline (10x PBS) (see Recipes)
- Triton X-100 solutions (see Recipes)
- 10% Triton X-100
- 0.1% Triton X-100
- 0.02% Triton X-100
- 1 M Tris-HCl pH 8.2 (see Recipes)
- 0.1 M glycine-HCl pH 2.7 (see Recipes)
- Sodium bicarbonate buffers (see Recipes)
- 1 M sodium bicarbonate pH 8.2
- 0.1 M sodium bicarbonate
- 2.5% Trypsin (see Recipes)
Equipment
- Pipetman P2 pipette (Gilson, catalog number: F144801)
- Pipetman P20 pipette (Gilson, catalog number: F123600)
- Pipetman P200 pipette (Gilson, catalog number: F123601)
- Pipetman P1000 pipette (Gilson, catalog number: F123602)
- Jewelers forceps, Dumont No. 5 (Sigma-Aldrich, Dumont, catalog number: 6521)
- Pipette boy (Corning, Falcon, catalog number: 357469)
- Water bath (Julabo, model: ED (v.2))
- Magnetic tube rack (Diagenode, catalog number: B04000001)
- Fume hood Hera Safe KS (Thermo Scientific, catalog number: 51023175)
- Cell culture incubator with CO2 supply (Sanyo, catalog number: MCO-19AIC)
- SP8UV confocal microscope (Leica)
- Eppendorf centrifuge 5804 R (Eppendorf, model: 5804 R, catalog number: 805000620)
- Beckman Coulter Allegra centrifuge (Beckman, catalog number: 21R)
- NanoDrop 2000 spectrophotometer (Thermo Scientific, model: NanoDropTM 2000, catalog number: ND2000)
- Countess Cell Counter (Thermo-Fisher, catalog number: AMQAX1000)
- Neon Transfection System (Invitrogen, catalog number: MPK5000S)
Software
- Fiji/Image J (https://fiji.sc/)
Procedure
Notes:
- This protocol was optimized for U2OS cells but can be adapted to any adherent metazoan cell line.
- Use 500 μl of buffer for every wash-step if 12-well plates are used, otherwise the volume needs to be adjusted.
- Validation of antibody for VANIMA by immunofluorescence (see Note 1)
- Seed around 105 cells of U2OS cells in a 12-well plate containing growth medium and a glass coverslip.
- Let cells re-attach to the coverslip surface overnight.
- Pre-warm 4% PFA, diluted in PBS to 37 °C in a water bath.
- Remove growth medium from the cells and rinse them twice with PBS.
- Fix the cells by adding the pre-warmed 4% PFA for 5 min at RT.
- Rinse the cells two times with PBS.
- Permeabilize the cells by incubating them in PBS containing 0.1% Triton X-100 for 20 min at RT.
- Rinse the cells twice with PBS.
- Incubate the cells with different dilutions of your antibody in PBS supplemented with 10% FCS for 1 h at RT.
- Wash the cells twice with PBS supplemented with 0.02% Triton X-100 for 5 min at RT.
- Wash the cells once with PBS for 5 min at RT.
- Incubate the cells with the corresponding fluorescently-labeled secondary antibody in the supplier recommended dilution (like Alexa Fluor 488 goat-anti-mouse IgG in a dilution of 1/3,000) in 10% FCS in PBS for 1 h at RT.
- Repeat the wash-steps as mentioned in Steps A10 and A11.
- Mount the coverslip on a microscope slide using 6-10 μl of Vectashield mounting medium containing DAPI.
- Observe your cells under the microscope to define if the antibody shows the expected staining of the target (see Figure 1 and Note 2).
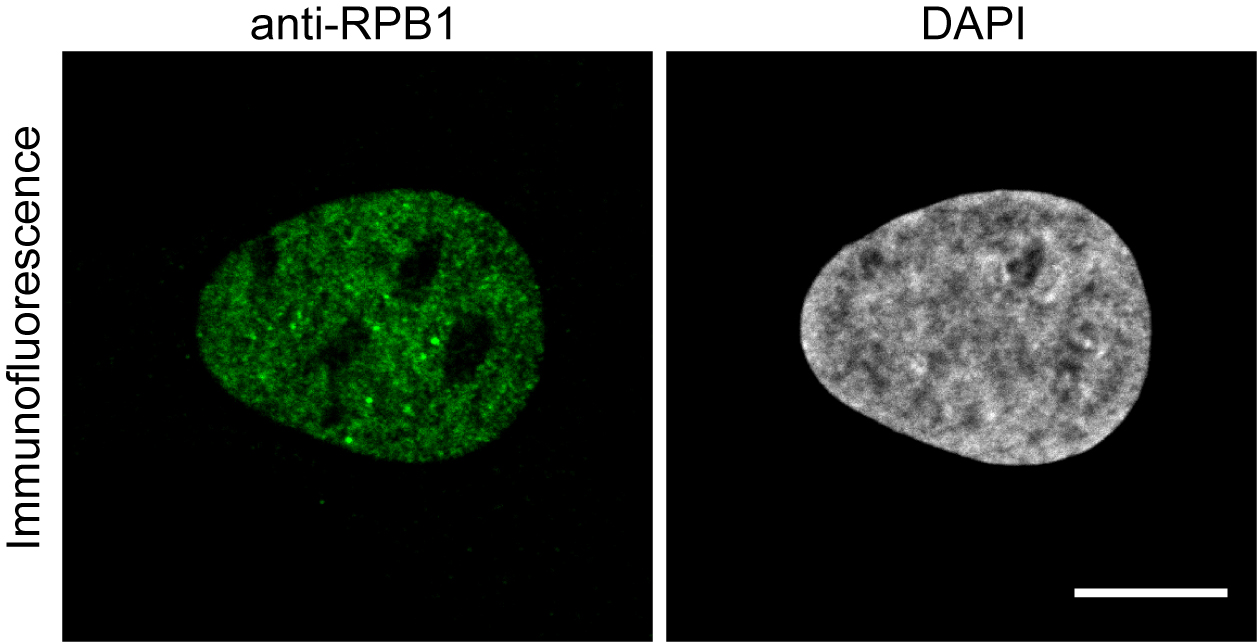
Figure 1. Immunofluorescence image using the anti-RPB1 antibody. U2OS cells were fixed and treated with anti-RPB1 antibody and an Alexa488 labeled anti-mouse secondary antibody to image RPB1. Scale bar = 10 μm.
Note: Labeling of antibodies or Fabs can only be performed with purified proteins. The purification of antibodies is described in the following section B. However, procedure B can be skipped if the antibody is already pure, and/or if a commercially available antibody is in PBS (see Notes of Procedure D).
- Purification of mouse monoclonal antibodies for electroporation (see Figure 2A)
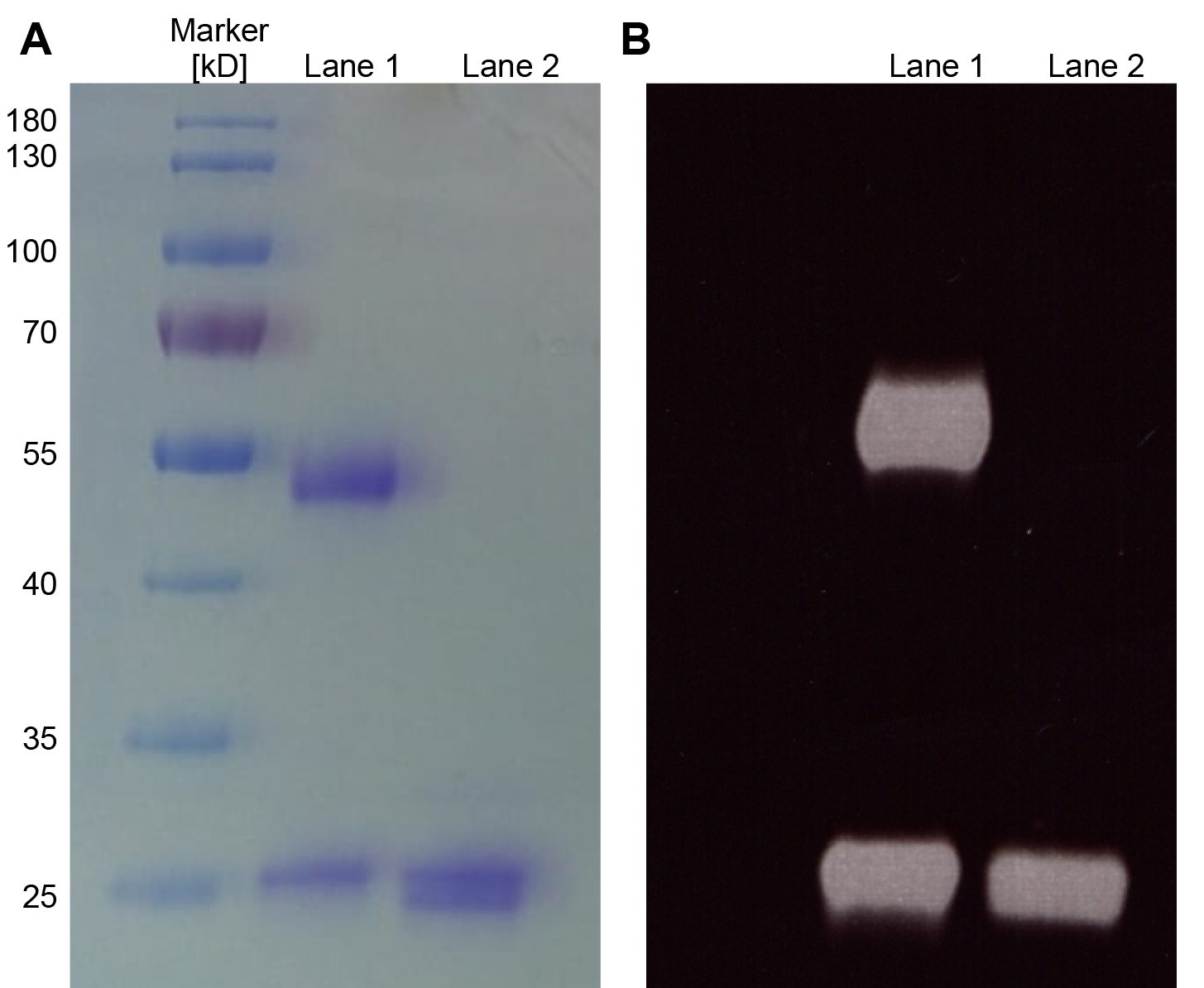
Figure 2. Analysis of antibodies and Fabs by SDS-PAGE. A. Purified aliquots of antibodies (Lane 1) or Fabs (Lane 2) were chemically-labeled with Alexa488 and subsequently analyzed by a 12% SDS-PAGE and Coomassie staining. Five microgram of the antibody and Fab were loaded on the SDS-PAGE. For the antibody (Lane 1), the heavy (50 kDa) and light chain (25 kDa) are visible and for the Fab (Lane 2) a double band is detected which correspond to the Fd chain and the light chain of the Fab. B. Corresponds to the same gel shown in A analyzed under UV illumination (wavelength range from 10 nm to 400 nm) before staining to excite the Alexa488 fluorophore and visualize the fluorescent dyes on the antibodies/Fabs.- Transfer 1 ml of Protein G Sepharose FastFlow beads into a 15 ml tube.
- Centrifuge the bead solution at 277 x g for 3 min at 4 °C to pellet the beads.
- Remove the storage solution and add 5 ml of PBS. Resuspend the beads and centrifuge them again as mentioned before.
- Repeat Steps B2 and B3 for 4 times to equilibrate the beads in PBS and to remove all the storage solution.
- Remove all PBS from the beads and add 2 ml of antibody solution with a concentration of around 1 mg/ml to the beads.
- Incubate the beads for 2 h at 4 °C under constant shaking.
- Centrifuge the beads for 5 min at 277 x g at 4 °C.
- Remove the supernatant and keep it on ice. This is the flow through (FT) which shouldn’t contain any antibodies anymore.
- Add 2 ml of PBS to the beads, resuspend them and transfer them to a Poly-Prep chromatography column.
- Add a total of 20 ml of PBS to wash the beads and to remove all unspecific bound proteins.
- Prepare ten 1.5 ml Eppendorf tubes with 70 μl of 1 M Tris-HCl pH 8.2 for fractionation and neutralization.
- After all the PBS passed through the column, start the elution of the antibody from the beads by adding stepwise 10 ml of 0.1 M glycine-HCl pH 2.7 in 1 ml steps and collect the fractions in the prepared Eppendorf tubes containing the neutralization buffer.
- Analyze an aliquot of every elution fraction by SDS-PAGE using a 12% SDS-acrylamide gel. Fifteen microliter of the following samples can be loaded:
- The input antibody solution
- The flow-through (FT)
- All ten fractions collected
- Perform Coomassie staining after the electrophoresis and pool all the fractions containing the purified antibodies.
- Dialyze the pooled fractions against a total of 4 L of PBS in two steps using DiaEasy dialyzer tubes, the first step overnight and the second for 4 h with 2 L of PBS each at 4 °C.
- Measure the concentration of the dialyzed antibody by 280 nm absorption and an extinction coefficient of 1.37 using a NanoDrop spectrophotometer. Concentrate the purified antibodies using the 4 ml Amicon filter units with a cutoff of 10 kDa by centrifugation at 4,000 x g until the concentration is 1 mg/ml or higher.
Note: This protocol is optimized for an antibody input of 200 μg which corresponds to antibody samples that are commercially available.
- Digestion of monoclonal antibodies to Fab fragments (see Note 3 and Figure 2A)
- Prepare 200 μg of monoclonal antibodies in PBS (1 mg/ml) (see Procedure B).
- Add 1.2 μl of 0.17 M TCEP (1 mM final concentration) in 200 μl of antibody solution.
- Transfer 100 μl of magnetic Papain coated bead solution per digestion into 0.5 ml Eppendorf tubes.
- Use an Eppendorf magnetic stand to fix the magnetic beads and remove the storage solution.
- Wash the beads 3 x each with 300 μl of PBS by resuspending them in the buffer and afterwards removing the washing buffer again with the help of the magnet.
- Remove all PBS and add the antibody solution with TCEP to the beads.
- Incubate for 3 h at 37 °C under shaking.
- Remove and collect the supernatant (S1) from the beads using the magnet (this includes the Fab’s).
- Wash the beads again 3 x with PBS and store them at 4 °C in PBS to be able to reuse them.
- For the purification of the Fab fragments, transfer 100 μl of Protein A Sepharose beads into a 0.5 ml Eppendorf tube.
- Equilibrate the beads by washing them 4 x each with 300 μl of PBS. Centrifuge them for each wash-step for 3 min at 277 x g.
- Remove all PBS and add supernatant (S1) from the digestion to the beads.
- Incubate for 30 min at 4 °C under shaking.
- Centrifuge for 5 min at 277 x g and collect the supernatant (S2).
- Wash beads with 300 μl of PBS and centrifuge again.
- Collect wash step and pool with supernatant (S2).
- Concentrate the fraction S2 using an Amicon filter unit with a cutoff of 10 kDa (0.5 ml or 4 ml tubes) to about 100 μl volume (5 min at 14,000 x g). This is now fraction S3.
- Determine the concentration of the Fab by measuring the absorption at 280 nm and an extinction coefficient of 1.37 using the NanoDrop spectrophotometer.
- Perform an SDS-PAGE using a 4%-15% acrylamide gradient gel and 1 µl of each of the following samples that need to be boiled for 5 min before loading:
Input antibody solution
Supernatant S1
Supernatant S2
Supernatant S3
Filtrate from the Amicon concentration step - The Fab fragments are now purified and ready for labeling.
- Fluorescent labeling of monoclonal antibodies or Fab fragments for VANIMA (see Note 4)
Note: In our experience, all fluorophores from the Alexa and Atto families are working very well. Other dyes and kits would need to be tested.- Prepare 1 L of 0.1 M sodium bicarbonate pH 8.2 (see Recipes).
- Dialyze 100 μg of antibody/Fab solution in a volume of 100 μl (1 mg/ml) against 1 L of 0.1 M sodium bicarbonate for 4 h at
4 °C using DiaEasy dialyzing tubes.
Note: The dialysis against the labeling buffer (0.1 M sodium bicarbonate) is an important step as it will raise the pH of the antibody solution over a pH of 8 which will increase labeling efficiency. The dialysis also showed to give better labeling efficiencies (3-4 dyes per molecule more) than the pH raising step described in the Invitrogen labeling protocol. - Mix the antibody/Fab solution with fluorescent dyes as written in the manufacturer’s protocol and incubate at RT for 1 h in the dark. Mix the solution every 15 min by inverting the tube. The labeling kits used to label the antibodies/Fabs are the Alexa Fluor Monoclonal Antibody labeling kits from Invitrogen (see Materials and Reagents).
- Remove non-bound dyes by purifying the labeling mix using the gel filtration columns supplied in the labeling kit.
Note: Washing of the gel filtration column with PBS prior to loading of the labeling reaction is recommended as it will remove any traces of NaN3 that is present in the storage buffer of the gel filtration beads. - Concentrate the labeled antibody/Fab by using 0.5 ml Amicon filter units with a cut-off of 10 kDa. Centrifuge for 10 min at 14,000 x g to concentrate the solution to a volume of approximately 50 μl.
- Measure the concentration of the labeled antibody using a NanoDrop spectrophotometer and the Protein and Labels mode.
- Labeling efficiency can be visualized by SDS-PAGE and UV illumination (see Figure 2B) and quantified by measuring the absorption at 280 nm and at the dye specific wavelength. The dye/antibody labeling ratio can then be calculated using the formula mentioned in the protocol of Invitrogen.
Note: To label antibodies or Fabs, the labeling kits are using N-hydroxysuccinimide ester fluorophores that react with the amine group at the tip of the side chain of lysines. This works fine with antibodies that do not harbor lysine residues in their binding site (paratope). If the binding capacity of the labeled antibodies or Fab (that can be easily tested by IF) is affected by this technique, we propose to set up a site-directed labeling. This labeling procedure consists in the preparation of (Fab’)2 fragments, which can be specifically labeled at the typical cysteine residues in the C-terminal of the Fab’ (hinge region) with maleimide-activated fluorophores upon mild reduction. Whilst this method preserves theoretically the antibody-binding site from any deleterious chemical alteration, it allows the addition of a maximum of two to three fluorophore molecules only per antibody or Fab.
- Electroporation procedure for monoclonal antibodies or Fab fragments (see also Video 1)
Notes:- The Neon transfection system and the corresponding Neon transduction kits are used for antibody transduction.
- The purified antibody, or Fab fragments, need to be diluted in sterile PBS with no other ingredients prior to electroporation as preservatives like sodium azide or traces of BSA will lower the viability of your cells after electroporation significantly.
- Furthermore, the working solution of labeled antibodies or Fab fragments should have a concentration of 1-2 mg/ml. The concentration of the antibody/Fab solution should be as high as possible to decrease the volume needed for electroporation since the volume needed for one electroporation should not exceed 5 µl to avoid that the cells are getting too diluted.
- This protocol describes electroporation using the 10 μl Neon tips, but 100 μl Neon tips can be used as well. In this case the number of cells and antibodies/Fabs need to be adjusted accordingly.
- All buffer and solutions need to be filtered and sterile for electroporation to avoid contamination of the antibodies and cells.
- Pre-warm 2.5% trypsin and the growth culture medium without antibiotics to 37 °C prior to the experiment.
 Video 1. Electroporation procedure
Video 1. Electroporation procedure- Transfer the volume corresponding to 1-5 μg of antibody/Fab solution (between 1-2 mg/ml) into a 1.5 ml Eppendorf tube and keep them in the dark on ice. For example, 2 µl of antibody solution with a concentration of 1 mg/ml if you want to electroporate 2 µg of antibodies.
- Add antibiotics free growth medium into the cell culture plate (12-well plate with glass coverslip) or live-imaging chamber slides (μ-slides 8-well glass bottom from Ibidi) and store them in the incubator at 37 °C.
- Wash the U2OS cells twice with PBS.
- Detach the cells from the surface by incubating them with 2.5% trypsin for 4 min and dilute them in antibiotic-free growth medium.
- Count the cells and transfer the volume of cell suspension needed for a final cell number of 8 x 105 cells to a 15-ml tube and pellet the cells by centrifuging them at 200 x g for 5 min. Every electroporation uses 105 cells which means that with this pellet one can perform 8 transductions in total.
Note: A total of 8 electroporation experiments can be performed with a pellet of 8 x 105 cells (105 cells per transduction). Even if a lower number of electroporations are planned it is still better to keep a higher cell number as the lower number will result in very small pellets and inaccuracies which can lower cell viability drastically. - Remove the growth medium and resuspend the cell pellet in 4 ml of PBS and centrifuge them again.
- Remove the PBS and resuspend the pellet in 80 μl of resuspension buffer (R-buffer).
Note: As soon as the cells are resuspended in R-buffer, it is important to proceed quickly to the electroporation. Cell viability could decrease significantly if the cells remain longer than 15 min in R-buffer. The resuspension buffer (R-buffer) is part of the electroporation kit from Invitrogen and therefore its composition is confidential. However, we also tested 250 mM sucrose buffer in 1x PBS (Brees and Fransen, 2014) or just simple 1x PBS and they also worked for the electroporation of antibodies. - Mix 10 μl of the resuspended cells with the antibody/Fab solution and pipette this mix with the Neon pipette using the 10 μl Neon tips. Be careful that no bubble can be seen in the Neon tip as this will induce an electric short-circuit which will kill the cells during the electroporation.
- Put the Neon pipette into the pipette station and electroporate the cells using the following parameters:
Voltage: 1550 V
Number of pulses: 3
Time width of pulse: 10 ms - Pipette the cells after the electric pulse directly into the cell culture plate prepared in Step E2.
- Repeat Steps E8-E10 until all transduction have been performed.
- Incubate the cells in the incubator at 37 °C with 5% CO2.
- For Fab transductions, the cells can be observed or fixed as early as 6 h post electroporation (see Figure 3C).
- For full-length antibody transductions, the incubation time can vary depending on the neo-synthesis rate of the target protein in the cell and its localization (see Note 3 and Figures 3A, 3B and 3D).
- The same protocol as in Procedure A can be followed if the cells need to be fixed starting with Step A3 but except that for the electroporation of labeled antibodies/Fabs Steps A6-A12 can be skipped.
- For live-cell imaging, the samples can be observed under the microscope after the incubation time (see Figure 3).
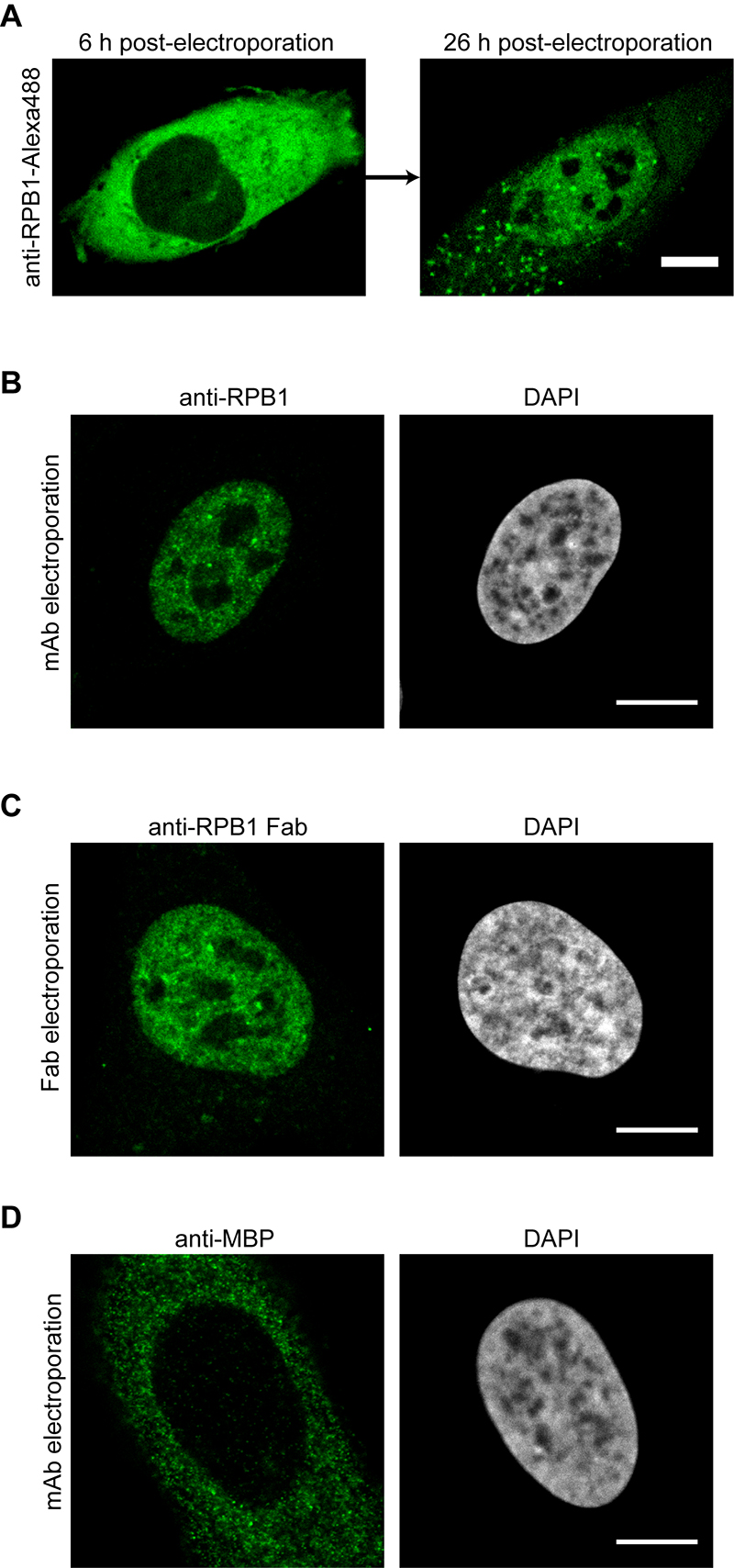
Figure 3. Imaging results after the electroporation of antibodies or Fabs. A. U2OS cells were electroporated with anti-RPB1-Alexa488 labeled antibodies and the same cell was imaged by confocal microscopy 6 h and 26 h post-electroporation. The transport of the labeled antibody from the cytoplasm into the nucleus can be detected. B. Same as in (A), but the cells were fixed 24 h after electroporation and the antibody labeled RPB1 was visualized using confocal microscopy. C. Electroporation as in (A), but this time anti-RPB1-Alexa488 Fab fragments were transduced. The cells were fixed and imaged 6 h post-electroporation by confocal microscopy and specific nuclear staining for RPB1 can be observed. D. U2OS cells were electroporated with an antibody against MBP (maltose binding protein), which is not present in human cells. The cells were fixed 24 h after electroporation and the localization of the antibody was visualized using confocal microscopy. Even after 24 h, the antibody stays in the cytoplasm as it has no target protein for the transport into the nucleus. Scale bars = 10 μm.
Data analysis
- The microscopes used for the analysis were either confocal, spinning disk or 3D-SIM super-resolution microscopes for live-imaging or fixed cell acquisition. For each electroporation experiment, we perform at least three replicates. All samples for one data set were acquired on the same day with the same microscope settings. The use of higher laser power or longer exposure times for these samples is completely normal as labeling of endogenous proteins always results in lower signal intensities. The laser power and exposure times can vary from 5% to 20% or 40 ms to 200 ms respectively depending on the labeling efficiency, target protein as well as the microscope used.
- Microscopic analysis of electroporated cells should be performed within 3 days after transduction. The fluorescent signal intensity will decrease significantly due to degradation of the antibody/Fab or by dilution due to cell division. This loss of signal is a general observation for all antibodies and Fabs and can vary depending on the division rate of the cell line used. The U2OS cells used in this protocol for example, are dividing every 24 h. Thus, almost half of the fluorescent signal is lost after each cell division (see Figure 4).
- Bright spots eventually present in the cytoplasm of cells after electroporation may correspond to antibodies that were structurally altered due to dye conjugation and they thus tend to aggregate in the cytoplasm. A possibility to avoid the formation of these aggregates is to electroporate a lesser amount of antibodies/Fabs or to repeat the labeling experiment using less dye.
- For fixed cell analysis, it is possible that bright spots can be detected on the coverslip outside of the cells. These spots most probably correspond to unbound fluorescent antibodies which were washed out of the cells during the fixation process. Wrong storage of the labeled antibodies can increase the amounts of these artifact spots (see Note below for storage information). However, most of these spots will be removed during the washing steps after fixation using the wash buffer including Triton X-100.
Note: Labeled antibodies and Fabs should be aliquoted after the labeling and can be stored for up to 3 weeks at 4 °C, or for up to 3 months at -80 °C. Antibody solutions should have at least a concentration of 1 mg/ml for freezing. The antibodies should be frozen quickly in liquid nitrogen before moving them to the -80 °C freezer. - Image processing can be performed using classical Fiji/ImageJ software. Different plugins can be used to calculate protein distribution or dynamics using 3D-SIM images. For images of the same data set, the image processing should be performed in the same way.
- It is recommended to perform validation experiments for cell viability and function every time a new antibody/Fab is used for electroporation. Apoptosis and proliferation assays should be performed to test for antibody toxicity. Also, specific validation tests concerning the mechanism involving the target protein inside the cell should be performed to ensure that the antibody is not inhibiting any important function of the protein (e.g., measuring the amount for nascent RNA transcription for a target involved in transcription).
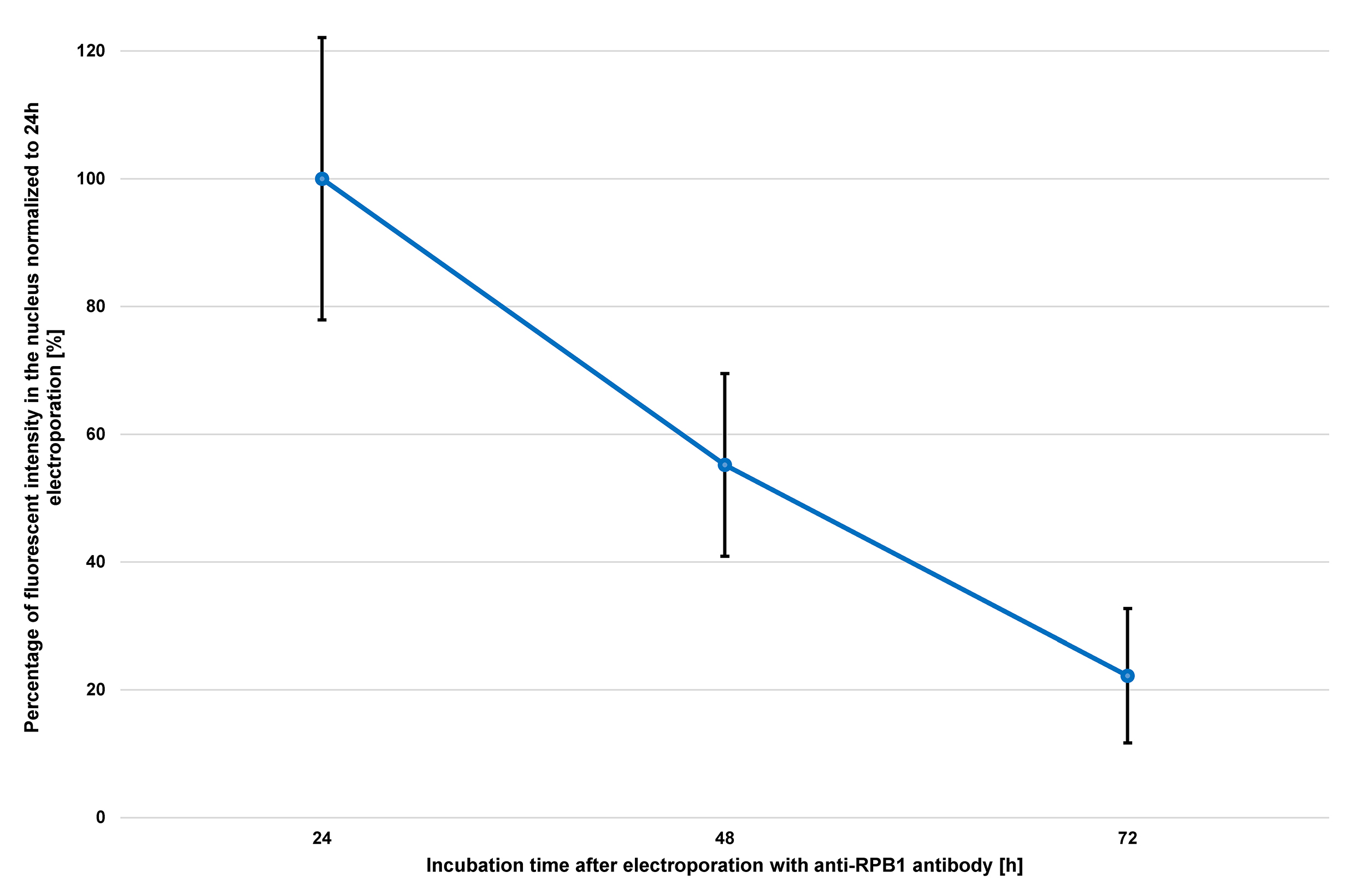
Figure 4. Progression of nuclear fluorescent signal over 72 h after electroporation of anti-RPB1 antibodies. U2OS cells were transduced with 2 µg of anti-RPB1 antibodies and were fixed 24 h, 48 h or 72 h after electroporation and analyzed using confocal microscopy. The fluorescent intensity in the nucleus of the cells (n = 20) for each time point was measured and the mean values were normalized to the sample fixed 24 h after electroporation. Error bars represent the standard deviation obtained with 20 recorded cells for each time point.
Notes
- To verify if the desired antibody is suitable for VANIMA it is recommended to test it first in classical immunofluorescence (IF). Procedure A of this protocol can be skipped if the antibody was already characterized by IF. Depending on the antibody source (hybridoma supernatant, commercial antibody, etc.). It is important to test several dilutions or concentration of the antibody for the first test.
- The expected staining for IF and VANIMA depends on the target antigen that needs to be analyzed. If the antibody recognizes a nuclear target and if its use leads to a strong unspecific signal in the cytoplasm, then this antibody is likely not suitable for VANIMA. Furthermore, the fluorescent signal itself can be also an indication for the suitability of the antibody. Very low fluorescent signals can indicate low binding affinity or accessibility of the antibody to the target protein and should not be used for VANIMA.
- Full-length antibodies are too big to be able to diffuse freely into the nucleus. They need to bind to the newly synthesized target protein in the cytoplasm and get piggybacked with it into the nucleus. Therefore, the time for the transport of the antibody into the nucleus depends highly on the turnover of the target protein and antibodies (see Figure 3D). Note that full-length antibodies against posttranslational modifications in the nucleus will never reach their target and will remain cytoplasmic. In contrast, Fab fragments can diffuse freely into the nucleus and are particularly adapted to target posttranscriptional modifications or proteins with a low cellular turnover.
- To test if the antibody or Fab is functional inside the cell, it is recommended to electroporate them once before they get fluorescently labeled. They can be visualized after fixation with a fluorescently labeled secondary antibody.
- To test a new antibody or Fab it is also important to verify how much antibody is needed to be electroporated to bind and label all of the target protein in the cell. Therefore, for the first electroporation, a titration experiment with different amounts of antibodies or Fabs in a range between 1-5 μg is recommended. Antibody amounts higher than 10 μg should be avoided as these high amounts of protein electroporated will start to get toxic for the cell.
Recipes
- U2OS growth medium
Supplement 450 ml of DMEM medium with 50 ml of FCS (10% FCS) and 40 μg/ml gentamicin
Store at 4 °C - 4% PFA
Dilute 10 ml of 16% PFA with 30 ml of PBS
Store at -20 °C for a maximum of 1 month - 10x PBS
- Dissolve the whole content of one bottle in 1 L of filtered and sterile dH2O
- Autoclave the solution to get sterile 10x PBS
- Store at RT
- Triton X-100 solutions
- 10% Triton X-100
Dilute 1 ml of Triton X-100 in 9 ml of PBS
Store at RT in the dark for several months - 0.1% Triton X-100
Dilute 0.5 ml of 10% Triton X-100 in 50 ml of PBS
Store at RT in the dark for several months - 0.02% Triton X-100
Dilute 0.1 ml of 10% Triton X-100 in 50 ml of PBS
Store at RT in the dark for several months
- 10% Triton X-100
- 1 M Tris-HCl pH 8.2
- Weigh 15.8 g of Tris-HCl and dissolve it in 100 ml of sterile dH2O
- Adjust the pH to 8.2
- Filter sterilize the solution using a 0.22 μm filter
- Store at RT for several months
- 0.1 M glycine-HCl pH 2.7
- Weigh 3.8 g of glycine and dissolve it in 500 ml of sterile dH2O
- Adjust the pH to 2.7
- Filter sterilize the solution using a 0.22 μm filter
- Store at RT for several months
- Sodium bicarbonate buffer
- 1 M sodium bicarbonate
Dissolve 42 g of sodium bicarbonate in 500 ml of sterile dH2O
Filter the solution through a 0.22 μm filter
Store at 4 °C for a maximum of 2 weeks - 0.1 M sodium bicarbonate
Dilute 100 ml of 1 M sodium bicarbonate with 900 ml of sterile dH2O
Filter the solution through a 0.22 μm filter
Store at 4 °C for a maximum of 2 weeks
- 1 M sodium bicarbonate
- 2.5% Trypsin
- Weigh 1 g of Trypsin and dissolve it in 40 ml of PBS
- Filter the solution through a 0.22 μm filter to sterilize it
- Store at 4 °C for a maximum of one month
Acknowledgments
This work was supported by funds from CNRS, INSERM, University of Strasbourg, Ligue Régionale contre le Cancer (CCIRGE-BFC) (to EW), by the European Research Council (ERC) Advanced grant (ERC-2013-340551, Birtoaction) (to LT) and a grant ANR-10-LABX-0030-INRT, a French State fund managed by the Agence Nationale de la Recherche under the frame program Investissements d’Avenir ANR-10-IDEX-0002-02. The present protocol is based on the following published research papers: Freund et al. (2013); Desplancq et al. (2016) and Conic et al. (2018).
Competing interests
The authors declare no conflict of interest.
References
- Betzig, E., Patterson, G. H., Sougrat, R., Lindwasser, O. W., Olenych, S., Bonifacino, J. S., Davidson, M. W., Lippincott-Schwartz, J. and Hess, H. F. (2006). Imaging intracellular fluorescent proteins at nanometer resolution. Science 313(5793): 1642-1645.
- Brees, C. and Fransen, M. (2014). A cost-effective approach to microporate mammalian cells with the Neon Transfection System. Anal Biochem 466: 49-50.
- Burgess, A., Lorca, T. and Castro, A. (2012). Quantitative live imaging of endogenous DNA replication in mammalian cells. PLoS One 7(9): e45726.
- Conic, S., Desplancq, D., Ferrand, A., Fischer, V., Heyer, V., Reina San Martin, B., Pontabry, J., Oulad-Abdelghani, M., Babu, N. K., Wright, G. D., Molina, N., Weiss, E. and Tora, L. (2018). Imaging of native transcription factors and histone phosphorylation at high resolution in live cells. J Cell Biol 217(4): 1537-1552.
- Desplancq, D., Freund, G., Conic, S., Sibler, A. P., Didier, P., Stoessel, A., Oulad-Abdelghani, M., Vigneron, M., Wagner, J., Mely, Y., Chatton, B., Tora, L. and Weiss, E. (2016). Targeting the replisome with transduced monoclonal antibodies triggers lethal DNA replication stress in cancer cells. Exp Cell Res 342(2): 145-158.
- Ellenberg, J., Lippincott-Schwartz, J. and Presley, J. F. (1999). Dual-colour imaging with GFP variants. Trends Cell Biol 9(2): 52-56.
- Freund, G., Sibler, A. P., Desplancq, D., Oulad-Abdelghani, M., Vigneron, M., Gannon, J., Van Regenmortel, M. H. and Weiss, E. (2013). Targeting endogenous nuclear antigens by electrotransfer of monoclonal antibodies in living cells. MAbs 5(4): 518-522.
- Hayashi-Takanaka, Y., Yamagata, K., Nozaki, N. and Kimura, H. (2009). Visualizing histone modifications in living cells: spatiotemporal dynamics of H3 phosphorylation during interphase. J Cell Biol 187(6): 781-790.
- Hayashi-Takanaka, Y., Yamagata, K., Wakayama, T., Stasevich, T. J., Kainuma, T., Tsurimoto, T., Tachibana, M., Shinkai, Y., Kurumizaka, H., Nozaki, N. and Kimura, H. (2011). Tracking epigenetic histone modifications in single cells using fab-based live endogenous modification labeling. Nucleic Acids Res 39(15): 6475-6488.
- Ratz, M., Testa, I., Hell, S. W. and Jakobs, S. (2015). CRISPR/Cas9-mediated endogenous protein tagging for RESOLFT super-resolution microscopy of living human cells. Sci Rep 5: 9592.
- Sato, Y., Mukai, M., Ueda, J., Muraki, M., Stasevich, T. J., Horikoshi, N., Kujirai, T., Kita, H., Kimura, T., Hira, S., Okada, Y., Hayashi-Takanaka, Y., Obuse, C., Kurumizaka, H., Kawahara, A., Yamagata, K., Nozaki, N. and Kimura, H. (2013). Genetically encoded system to track histone modification in vivo. Sci Rep 3: 2436.
- Schneider, A. F. L. and Hackenberger, C. P. R. (2017). Fluorescent labelling in living cells. Curr Opin Biotechnol 48: 61-68.
- Schnell, U., Dijk, F., Sjollema, K. A. and Giepmans, B. N. (2012). Immunolabeling artifacts and the need for live-cell imaging. Nat Methods 9(2): 152-158.
- Teng, K. W., Ishitsuka, Y., Ren, P., Youn, Y., Deng, X., Ge, P., Lee, S. H., Belmont, A. S. and Selvin, P. R. (2016). Labeling proteins inside living cells using external fluorophores for microscopy. Elife 5: e20378.
- Teves, S. S., An, L., Hansen, A. S., Xie, L., Darzacq, X. and Tjian, R. (2016). A dynamic mode of mitotic bookmarking by transcription factors. Elife 5: e22280.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Conic, S., Desplancq, D., Tora, L. and Weiss, E. (2018). Electroporation of Labeled Antibodies to Visualize Endogenous Proteins and Posttranslational Modifications in Living Metazoan Cell Types. Bio-protocol 8(21): e3069. DOI: 10.21769/BioProtoc.3069.
- Conic, S., Desplancq, D., Ferrand, A., Fischer, V., Heyer, V., Reina San Martin, B., Pontabry, J., Oulad-Abdelghani, M., Babu, N. K., Wright, G. D., Molina, N., Weiss, E. and Tora, L. (2018). Imaging of native transcription factors and histone phosphorylation at high resolution in live cells. J Cell Biol 217(4): 1537-1552.
Category
Cell Biology > Cell-based analysis > Transport
Immunology > Antibody analysis > Antibody detection
Cell Biology > Cell imaging > Live-cell imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.