- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Assessing Membrane Fluidity and Visualizing Fluid Membrane Domains in Bacteria Using Fluorescent Membrane Dyes

Published: Vol 8, Iss 20, Oct 20, 2018 DOI: 10.21769/BioProtoc.3063 Views: 13867
Reviewed by: David CisnerosRon Saar DoverAgnieszka Zienkiewicz
Original research article
The authors used this protocol in:

Feb 2018
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Membrane fluidity is a key parameter of bacterial membranes that undergoes quick adaptation in response to environmental challenges and has recently emerged as an important factor in the antibacterial mechanism of membrane-targeting antibiotics. The specific level of membrane fluidity is not uniform across the bacterial cell membrane. Rather, specialized microdomains associated with different cellular functions can exhibit fluidity values that significantly deviate from the average. Assessing changes in the overall membrane fluidity and formation of membrane microdomains is therefore pivotal to understand both the functional organization of the bacterial cell membrane as well as antibiotic mechanisms. Here we describe how two fluorescent membrane dyes, laurdan and DiIC12, can be employed to assess membrane fluidity in living bacteria. We focus on Bacillus subtilis, since this organism has been relatively well-studied with respect to membrane domains. However, we also describe how these assays can be adapted for other bacteria such as Staphylococcus aureus and Streptococcus pneumoniae.
Keywords: Membrane fluidityBackground
Bacterial membranes have long been viewed as homogenous lipid bilayers following the classical fluid mosaic membrane model. However, many studies have later shown that membranes are in fact highly organized structures and possess distinct domains that can be characterized by containing specific membrane proteins, the enrichment of certain lipid species, or by having a higher or lower membrane fluidity than the neighboring membrane areas (Lopez and Kolter, 2010; Bach and Bramkamp, 2013; Barák and Muchová, 2013; Strahl et al., 2014; Bramkamp and Lopez, 2015; Schneider et al., 2015; Müller et al., 2016). Two types of membrane domains that are characterized by a specific membrane fluidity are rigid lipid and regions of increased fluidity (RIFs). Both have been well-characterized in the Gram-positive model organism Bacillus subtilis (Bach and Bramkamp, 2013; Strahl et al., 2014). Evidence for specific membrane domains has also been found in the pathogenic bacteria Staphylococcus aureus (Garcia-Fernandez et al., 2017 and Weihs et al., 2018) and Streptococcus pneumoniae (Rosch and Caparon, 2005; Vega et al., 2013). Membrane fluidity appears to be a key factor that distinguishes these specific domains from the rest of the membrane. This is achieved by enrichment of fluidizing lipid species, i.e., branched-chain, unsaturated, and short-chain fatty acid-containing lipids. Bacteria can adapt their membrane fluidity by changing the ratios of branched/non-branched, unsaturated/saturated, and short-chain/long-chain fatty acids. In B. subtilis, lipid desaturation (rapid adaptation) and adjusting the ratio of iso and anteiso branched chain fatty acids (long-term adaptation) are the main mechanisms to adapt membrane fluidity in response to environmental challenges (Beranova et al., 2008; Kingston et al., 2011).
Recently, it has emerged that membrane fluidity and membrane domains of specific fluidity play a key role in the mechanism of action of membrane-active antibiotics (Epand and Epand, 2009; Müller et al., 2016; Saeloh et al., 2018). Therefore, it is crucial to be able to assess changes in both overall membrane fluidity and membrane domains when studying the in vivo activity of these compounds. We have recently established protocols for measuring membrane fluidity using two different fluorescent membrane probes, laurdan (Figure 1A) and DiIC12 (Figure 1B) (Strahl et al., 2014; Müller et al., 2016; Saeloh et al., 2018). Laurdan is a fluorescence probe that intercalates into the membrane bilayer and displays an emission wavelength shift depending on the amount of water molecules in the membrane (Parasassi and Gratton, 1995; Sanchez et al., 2007).
Laurdan generalized polarization (GP) can therefore be used as a reporter for head group density and fatty acyl spreading (Figure 1C). Laurdan fluorescence can be measured in a fluorescence plate reader and allows both end-point and kinetic measurements. It works well in 96-well plate format and allows relatively high throughput screening. Laurdan fluorescence can also be visualized under the microscope and used for single-cell analysis. It can further be employed for measuring the fluidity of liposomes, which can be important to distinguish direct and indirect antibiotic effects.
DiIC12 displays affinity for membrane areas of increased fluidity due to its short hydrocarbon tail (Baumgart et al., 2007; Zhao et al., 2013). Since fluid membranes are typically thinner (Reddy et al., 2012; Karabadzhak et al., 2018), it can be used as a reporter for membrane thickness and fluidity (Figure 1D). DiIC12 has been of key importance in the discovery of RIFs, fluid membrane microdomains that harbor the cell wall synthetic machinery in B. subtilis and E. coli (Strahl et al., 2014; Müller et al., 2016; Oswald et al., 2016). These domains are easily disturbed by membrane-active antibiotics and appear to play a key role in the mechanism of action of the last resort antibiotic daptomycin and the new antibiotic candidate rhodomyrtone (Müller et al., 2016; Saeloh et al., 2018). DiIC12 is ideally suited to visualize fluid membrane domains, whether natural or antibiotic-induced, in living cells. It can, however, also be used to stain liposomes. Together, laurdan and DiIC12 constitute a very useful assay combination to study both overall membrane fluidity and membrane domain formation in bacteria. We here describe the detailed protocols for measuring overall fluidity in batch culture and in liposomes as well as the distribution of membrane domains of different fluidity in single cells.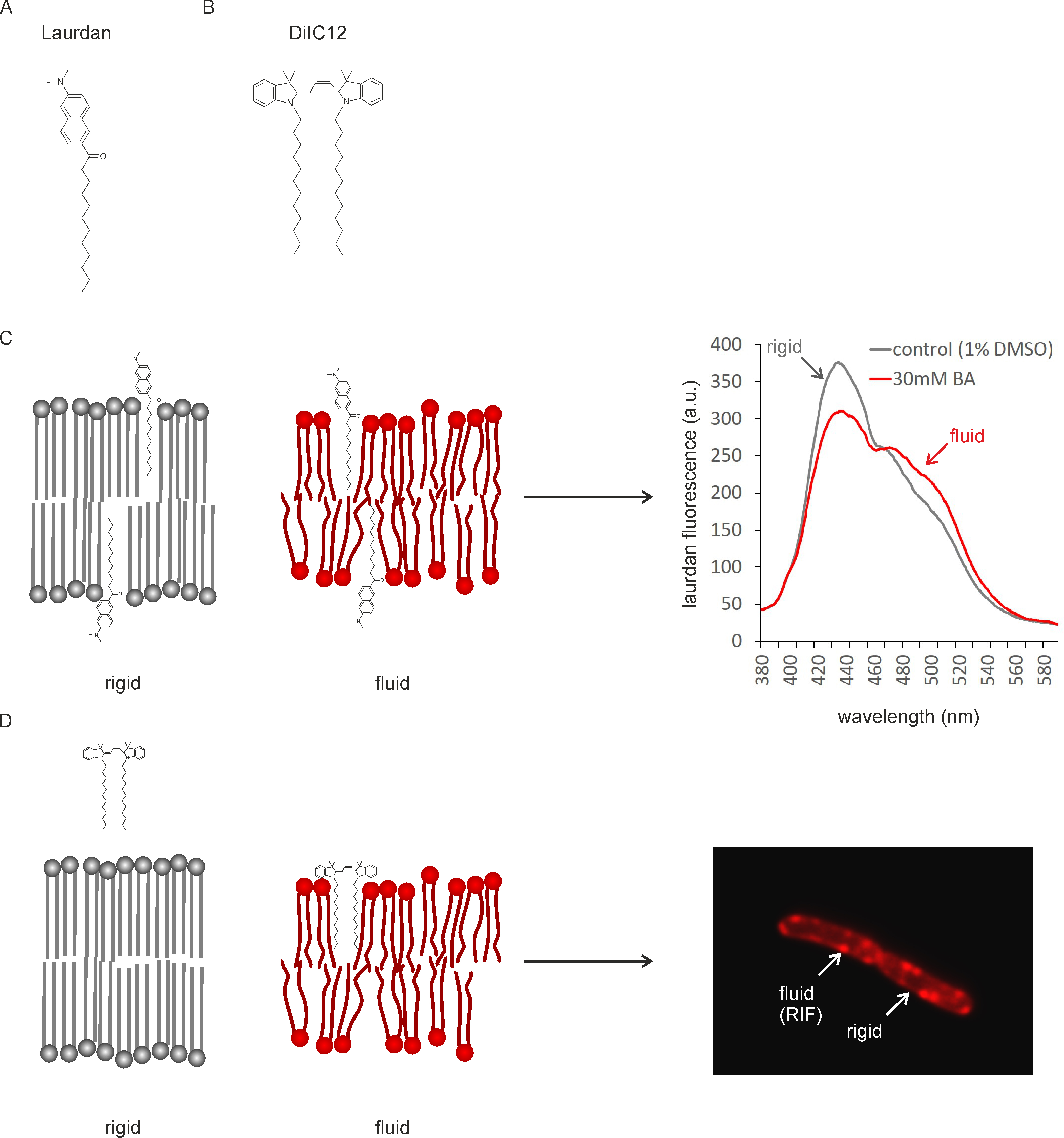
Figure 1. Mechanism of fluidity measurements with Laurdan and DiIC12. A. Structure of Laurdan; B. Structure of DiIC12; C. Laurdan inserts into membrane bilayers of different fluidity. Head group spreading and fatty acyl chain mobility determine the amount of water molecules around the laurdan molecule, which in turn causes a peak shift of the fluorescence probe. D. DiIC12 displays affinity for fluid membranes due to its short hydrocarbon tail, which is better accommodated in flexible, thin lipid bilayers. As a result, the dye accumulates in fluid membrane regions (RIFs). Note that due to better readability, dyes and lipids are not depicted to scale (dyes would be smaller).
Materials and Reagents
- 0.1-10 µl pipette tips (Gilson, catalog number: F161630)
- 0.1-2 µl pipette tips (Eppendorf, catalog number: 3120000011)
- 0.5-10 µl pipette tips (Eppendorf, catalog number: 3120000020)
- 0.2 µm Filtropur filters S (SARSTEDT, catalog number: 83.1826.001)
- 10 ml combitips for multistep pipette (Eppendorf, catalog number: 0030069.269)
- 100-1,000 µl pipette tips (Greiner, catalog number: 686290)
- 2 ml micro tubes (Greiner, catalog number: 623201)
- 2-20 µl pipette tips (Eppendorf, catalog number: 3120000038)
- 2-200 µl pipette tips (Greiner, catalog number: 739290)
- 5 ml combitips (Eppendorf, catalog number: 0030069.250)
- 50 ml Falcon tubes (SARSTEDT, catalog number: 62.547.254)
- 96-well plates, black flat-bottom (Screening Devices b.v., catalog number: 324002)
- Glass coverslips (Carl Roth GmbH, catalog number: NK75.1)
- Glass slides (Fisher-Emergo B.V., catalog number: 361000)
- Multi-spot glass slides (Hendley-Essex, catalog number: SM011, white)
- TetraSpeck microspheres, 0.2 µm, fluorescent blue/green/orange/dark red (Invitrogen via Thermo Fisher, catalog number: T7280)
- Black clear bottom microtiter plates (Greiner, catalog number: 675096)
- B. subtilis 168 (DSMZ, catalog number: 402)
- Agarose (Sphaero Q, catalog number: S103b)
- Ammonium iron (II) sulfate (Fe(II)NH4 citrate) (Sigma-Aldrich, catalog number: F5879)
- Ammonium sulfate ((NH4)2SO4) (Sigma-Aldrich, catalog number: A6387)
- Benzyl alcohol (Sigma-Aldrich, catalog number: 305197)
- Calcium chloride dihydrate (CaCl2•2H2O) (Merck, catalog number: 102382)
- Casein hydrolysate (casamino acids) (Duchefa, catalog number: C1301.0250)
- DiIC12 (1,1'-didodecyl-3,3,3',3'-tetramethylindocarbocyanine perchlorate) (Anaspec, catalog number: AS-84902)
- Dimethylformamide (DMF) (Sigma-Aldrich, catalog number: 227056)
- Dimethylsulfoxide (DMSO) (Sigma-Aldrich, catalog number: D8418)
- Disodiumhydrogenphosphate (Na2HPO4) (VWR, catalog number: 26932.290)
- E. coli polar lipid extract (Avanti Polar Lipids, catalog number: 100600P)
- Glucose (Duchefa Biochemie, catalog number: G0802)
- L-glutamic acid potassium salt (Sigma-Aldrich, catalog number: G1501)
- Gramicidin S (Sigma-Aldrich, catalog number: G0900)
Note: This product has been discontinued. The batch used in this study was kindly supplied by Marina Rautenbach, Stellenbosch University. - Hydrochloric acid (Merck, catalog number: 1003171000)
- Iron sulfate heptahydrate (FeSO4•7H2O) (Sigma-Aldrich, catalog number: 215422)
- Laurdan (6-Dodecanoyl-N,N-dimethyl-2-naphthylamine) (Sigma-Aldrich, catalog number: 40227)
- Magnesium sulfate heptahydrate (MgSO4•7H2O) (Roth, catalog number: T888.1)
- Manganese sulfate tetrahydrate (MnSO4•4H2O) (Fischer Scientific, catalog number: M/2250/53)
- MP196 (synthesized by solid-phase peptide synthesis according to Albada et al., 2012 and Sanchez et al., 2007); the batch used in this study was kindly supplied by Nils Metzler-Nolte, Ruhr University Bochum
- Potassium chloride (KCl) (VWR, catalog number: 26764.298)
- Potassiumdihydrogenphosphate (KH2PO4) (Merck, catalog number: 104873)
- Rhodomyrtone (purified form Rhodomyrtus tomentosa leaves according to Limsuwan et al. (Zhao et al., 2013); the batch used in this study was kindly supplied by Supayang Voravuthikunchai, Prince of Songkla University)
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: 1.06404.1000)
- Todd-Hewitt broth (Sigma-Aldrich, catalog number: T1438-500G)
- Tris (Merck, catalog number: 1083821000)
- Trisodium citrate dihydrate (Na3citrate•2H2O) (Merck, catalog number: 106448)
- Tryptone (Duchefa Biochemie, catalog number: T1332,1000MG)
- L-tryptophane (Sigma Aldrich, catalog number: T0254)
- Yeast extract (Duchefa Biochemie, catalog number: Y133)
- K2HPO4
- Agarose solution (see Recipes)
- BMM basic medium (see Recipes)
- BMM supplements (see Recipes)
- DiIC12 washing medium (see Recipes)
- Laurdan buffer (see Recipes)
- Lysogeny broth (LB) medium (see Recipes)
- SMM basic medium (see Recipes)
- SMM supplements (see Recipes)
Equipment
- 100-1,000 µl pipette (Eppendorf, catalog number: 3120.000062)
- 20-200 µl pipette (Eppendorf, catalog number: 3120000054)
- HeraCell 150 stove (Kendro, catalog number: 50075549B)
- Cy3 filter cube (Nikon, catalog number: MXU96213)
- DAPI filter cube (Nikon, catalog number: MBE41300)
- Laurdan custom filter cube composed of:
- C-FL filter block, frame only (Nikon, catalog number: MXA22030)
- FF01-360/23-25 excitation filter (Nikon, catalog number: MXR00637)
Important note: The same filter as in the DAPI filter cube. - Di02-R405-25x36 dichroic mirror (Nikon, catalog number: MXR00604)
Important note: The same dichroic mirror as in the DAPI filter cube. - FF01-535/5--25 emission filter (Nikon, catalog number: MXX99999)
- Multi step pipette (Eppendorf, catalog number: 022260201)
- Nikon Eclipse Ti inverted epi-fluorescence (wide-field) microscope (Nikon, catalog number: MEA53100) equipped with:
- CFI Plan Apochromat DM 100x NA 1.45 oil objective (Nikon, catalog number: MRD31905)
- Intensilight HG 130 W light source (MBF72665)
- C11440-22CU Hamamatsu ORCA Flash USB 3.0 camera (Nikon, catalog number: MHC11441)
- NIS elements AR software (Nikon, catalog number: MQS31000)
- Plate reader (BioTek Synergy Mx, catalog number: SMATBCL)
- Temperature-controlled microtube centrifuge (Eppendorf, model: 5424R, catalog number: 5404000010)
- Thermomixer (Eppendorf, catalog number: 5350000013)
- Water bath (GFL, catalog number: 5905985)
- Incubator Heraeus HERAcell (Kendro Laboratory Products, catalog number: 50042307)
- Autoclave (Sanyo, catalog number MLS-3780)
- Monochromators
- Spectrofluorometer Quantamaster 2000-4 (Photon Technology International)
Software
- ImageJ (https://imagej.nih.gov/ij/download.html)
- Fluorescence spectrophotometer software (PTI acquisition software FeliX32 version 1.2 Build 56)
- Microplate reader software Gen5 version 2.00 (Biotek)
Procedure
- Laurdan
According to most manufacturers, undissolved laurdan is stable for years if kept at -20 °C and protected from light. However, our experience is that its spectral properties might change much earlier depending on the individual batch. Since this significantly affects the reproducibility of results, we recommend checking the emission wavelength spectrum of the dye when excited at 350 nm. To this end, an emission wavelength scan of laurdan-stained liposomes (see protocol below) should be performed. A typical laurdan spectrum should show a clear peak at 440 nm and a second peak or shoulder at 480-500 nm (Figure 2) Deteriorated laurdan batches display two peaks at ~420 and ~460 nm with a clear drop at 440 nm (Figure 2), which drastically affects the GP measurements. Due to this problem, we recommend the following handling of the laurdan dye. A 10 mM Laurdan stock solution is prepared in DMF. Stock solutions can usually be kept at -20 °C for up to 3 months. In case of visible precipitation or formation of a micelle film on the surface, a new stock needs to be prepared. Every new 10 mM stock should be checked in an emission scan prior to first use. A 1 mM working solution is prepared from this stock by dilution in DMF. This ensures a final DMF concentration of 1%, which helps keeping laurdan soluble in the medium or buffer and increases reproducibility of the results. Aliquoted working solutions are stable for 4-6 weeks at -20 °C. Multiple freeze-thaw cycles should be avoided and laurdan solutions must be protected from light at all times. For optimal reproducibility, all experiments of one series should be performed with the same stock.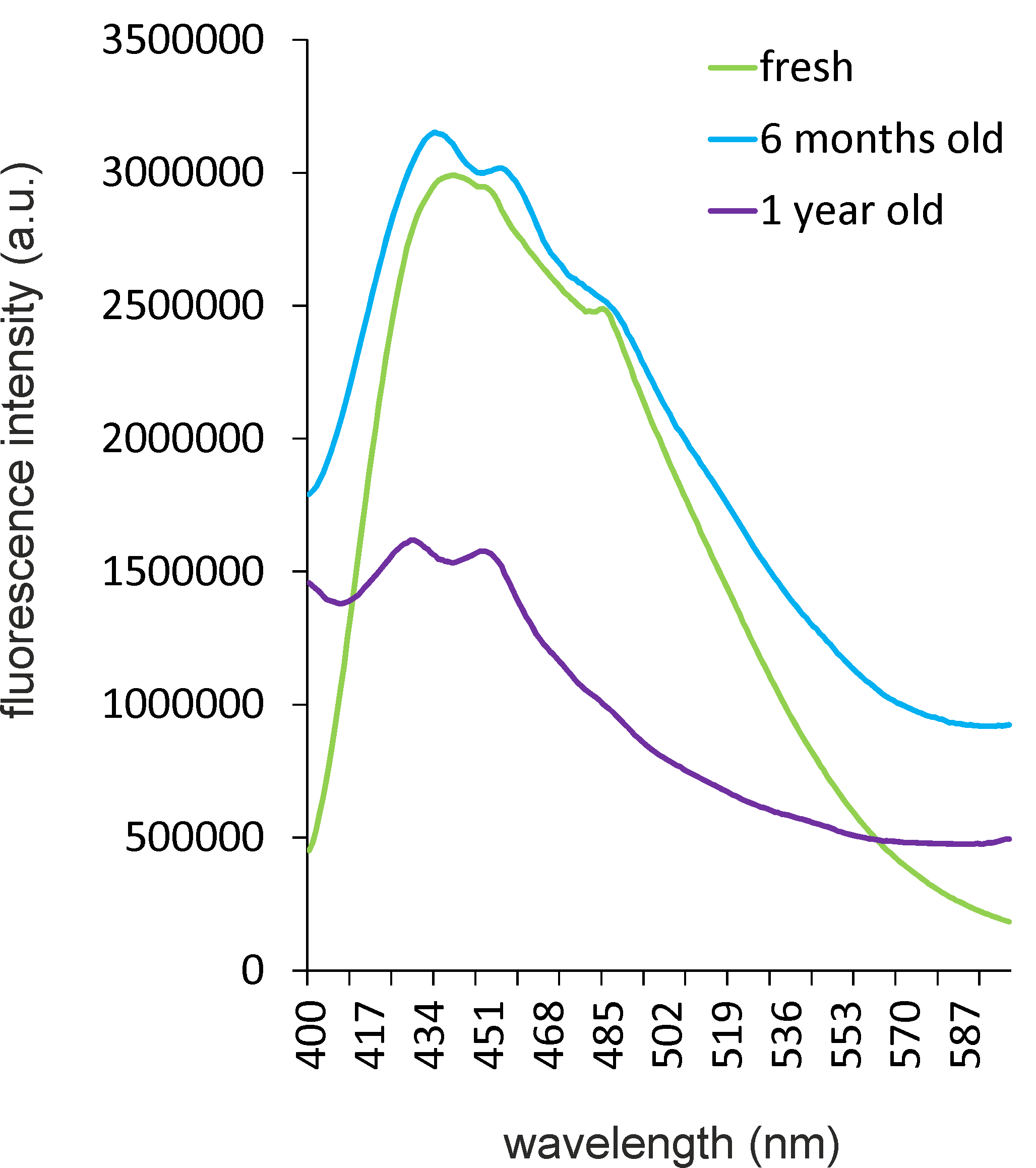
Figure 2. Deterioration of laurdan over time. Emission wavelength scans of laurdan incorporated into 0.2 µm liposomes made from E. coli polar lipid extract were recorded with an excitation wavelength of 350 nm. Liposomes were stained as described in Step A1b. Measurements were taken in 5 mM Tris-HCl, pH 7.4 at constant 30 °C. Note the change of the 440 nm peak into two peaks at 420 and 460 nm. Laurdan was kept undissolved at -20 °C and was protected from light constantly.- Laurdan Spectroscopy
Spectroscopic membrane fluidity measurements with laurdan are straight-forward and easy to implement. Optimal results are obtained with a plate reader equipped with monochromators, but appropriate filter sets covering 350 nm excitation and 440 and 500 nm emission wavelengths are also suitable. The protocol can also be adapted for the use in fluorimeters, yet they do not offer the high throughput of the 96-well plate format (Note 1). A constant temperature is pivotal for membrane fluidity measurements and the plate reader must be equipped with a precision temperature control. Ideally, sample preparation is carried out in a climate room to ensure stable temperature. If this is not possible, samples must be handled quickly and all solutions and plastic ware must be pre-warmed. Pipetting steps are carried out in a thermomixer. If samples must be transported to another location, they must be kept warm at all times. Multistep or multichannel pipettes are recommended when preparing several samples to save sample handling time. In case of B. subtilis, which is extremely sensitive to oxygen depletion, cells also must be kept shaking at all times. In order to prevent membrane damage by shearing forces, pipetting of cells should be kept to a minimum and cell pellets should never be resuspended by pipetting up and down.- Laurdan Spectroscopy of bacteria (Note 2)
- Set up overnight cultures in lysogeny broth (LB) or other medium of choice and grow overnight at the desired temperature (typically 30 or 37 °C; we used 30 °C for all experiments in this paper) (Notes 3 and 4).
- Dilute overnight cultures 1:100 in fresh LB or another medium of choice (Notes 3 and 4).
- Grow at the desired temperature until the desired cell density (standard: 30 or 37 °C, log phase) (Note 5).
- Add 10 µM laurdan from 1 mM working solution and incubate for 10 min.
- Harvest cells by centrifugation using pre-warmed (30 °C) 2 ml microtubes in a pre-warmed microtube centrifuge (up to 16,000 x g, 30 s, 30 °C). (Note 6)
- Wash 4x in pre-warmed (30 °C) laurdan buffer (Note 7). Keep tubes warm using a thermomixer while pipetting. Resuspend cells by vortexing shortly rather than pipetting up and down to avoid shearing forces that could damage the membrane.
For end point measurements (Figure 3A)- Resuspend cells to a final OD600 of 0.4.
- Add 200 µl (Note 8) of cell suspension per well to a pre-warmed (30 °C) black flat and clear bottom microtiter plate using a fluorescence plate reader. Depending on the number of samples, this step should be facilitated by using a multistep pipette (Note 9).
- Measure fluorescence in a suitable plate reader (excitation: 350 nm, emission: 460 and 500 nm).
For end point measurements with antibiotics (Note 10) (Figures 3B and 3D)- Resuspend cells to a final OD600 of 0.4
- Add 220 µl of cells to 2 ml pre-warmed (30 °C) micro tubes and add appropriate antibiotic concentrations.
- Incubate in a thermomixer at constant temperature and shaking for the desired time. Typically, 5-10 min of treatment are sufficient for most antibiotics. Treatment with 30-50 mM of the membrane fluidizer benzyl alcohol serves as positive control. As additional controls measure (i) the supernatants of the washing steps (to determine the efficiency of washing) and (ii) laurdan buffer (to subtract the background). The fluorescence intensity of stained cells should be at least 10 times higher than the fluorescence intensity of the laurdan buffer background control.
- Add 200 µl of the incubated cell suspension per well to a pre-warmed (30 °C) black flat and clear bottom microtiter plate (Note 10).
- Measure fluorescence in a fluorescence plate reader (excitation: 350 nm, emission: 460 and 500 nm).
For kinetic measurements with antibiotics (Note 9) (Figures 3C and 4)- Resuspend cells to a final OD600 of 0.8.
- Add 100 µl of stained cell suspension to 100 µl of pre-warmed (30 °C) laurdan washing buffer (control samples) in a pre-warmed (30 °C) black flat and clear bottom microtiter plate (Notes 6 and 8).
- Measure fluorescence in a fluorescence plate reader in 2 min intervals over 6-10 min to record the pre-treatment baseline (Note 11).
- Add 100 µl of stained cell suspension to 100 µl of pre-warmed (30 °C) laurdan washing buffer containing twice the desired antibiotic concentration in a pre-warmed (30 °C) black flat and clear bottom microtiter plate. If necessary, use a multistep pipette to reduce sample handling time.
- Immediately measure fluorescence in 2 min intervals over 30 min (Notes 12 and 13).
- If done correctly, laurdan GP measurements of untreated cells are very reproducible, even with different batches of growth medium (Figure 4). Antibiotic-treated samples might show a bit more variation but are usually very well reproducible as well (Figure 4).
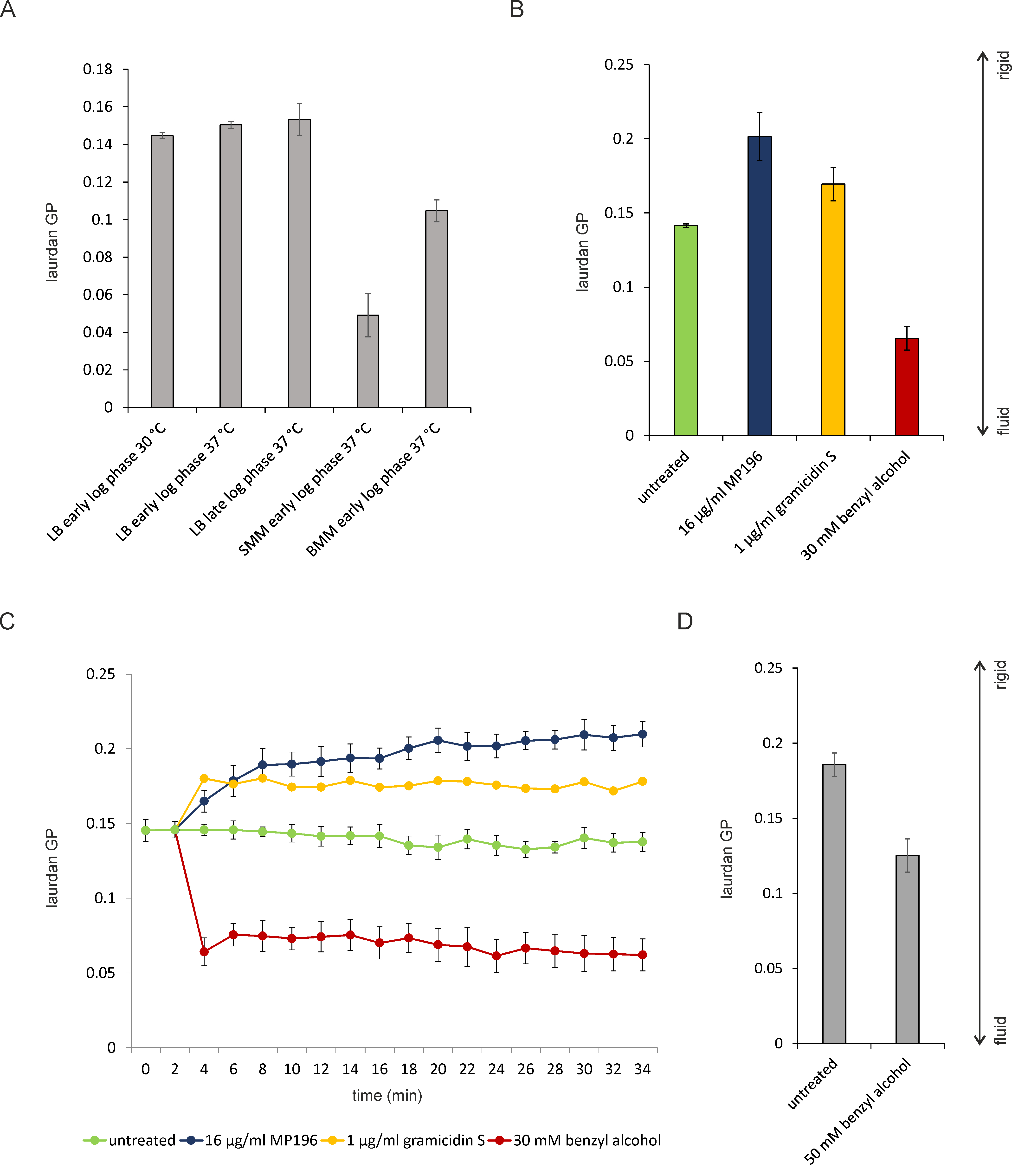
Figure 3. Spectroscopic Laurdan measurements in living bacteria. A. One point measurements of B. subtilis 168 (Anagnostopoulos and Spizizen, 1961) grown under different conditions. SMM (Spizizen minimal medium) and BMM (Belitzky minimal medium) are minimal media commonly used for B. subtilis (Anagnostopoulos and Spizizen, 1961; Stülke et al., 1993; Wenzel et al., 2011). Note that different media have a far more drastic effect on membrane fluidity than moderate differences in temperature and growth phase. Early log phase refers to an OD600 of 0.3. Late log phase refers to an OD600 of 0.8. B. One point Laurdan measurements of B. subtilis 168 treated with antibiotics for 10 min (growth temperature 30 °C). Minimal inhibitory concentrations were used. C. Kinetic Laurdan measurements of B. subtilis 168 treated with the same antibiotics under the same conditions as in (B). Note the gradual effect of the antimicrobial hexapeptide MP196 compared to the immediate effect of the cyclic β-sheet peptide gramicidin S and the membrane fluidizer benzyl alcohol. D. One point Laurdan measurements of S. aureus NCTC 8325 (Bæk et al., 2013). S. aureus was grown in LB at 37 °C. Measurements were taken at an OD600 of 0.3. Note the different membrane fluidity that S. aureus and B. subtilis display under the same growth conditions.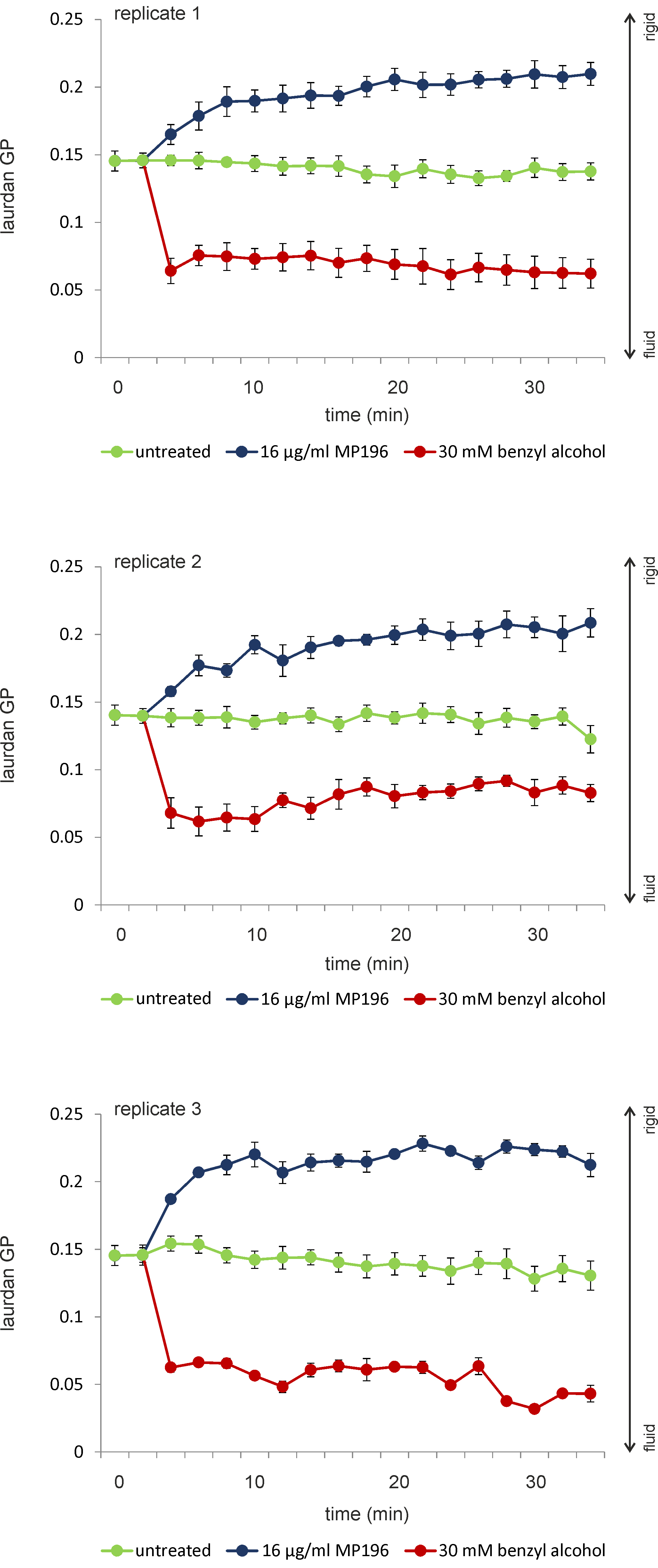
Figure 4. Reproducibility of laurdan measurements using B. subtilis. B. subtilis 168 was grown in LB at 30 °C under continuous shaking. Three independent biological replicates from three different days are shown. Laurdan stocks, buffers, and media were separately prepared for each replicate. Error bars show the standard deviation of 5 technical replicates. Untreated samples are highly reproducible, while antibiotic-treated samples show a slight variation. - Laurdan spectroscopy of liposomes
- Prepare liposomes from the desired lipids with your method and buffer of choice. Here we used 10 mg/ml 0.4 µm liposomes prepared from E. coli polar lipid extract in 50 mM Tris-HCl, pH 7.4, using the detergent-dialysis method described in Strahl and Hamoen (2010).
- Dilute liposome stock with double-distilled water to a final concentration of 1 mg/ml liposomes and 5 mM Tris-HCl, pH 7.4.
- Add 10 µM laurdan from 1 mM working stock solution in DMF and incubate at 30 °C for 30 min. Keep the temperature stable at all times (here 30 °C).
For one point measurements with antibiotics (Figure 5A)- Add 20 µl of stained liposomes to 180 µl 5 mM Tris-HCl, pH 7.4, with and without antibiotic compound in a black flat and clear bottom microtiter plate (pre-warmed to 30 °C) and incubate for the desired time period (2-10 min are typically sufficient) (Note 13).
- Measure samples in a microtiter plate reader.
For kinetic measurements with antibiotics (Figure 5B)- Add 20 µl of stained liposomes to 180 µl pre-warmed (30 °C) 5 mM Tris-HCl, pH 7.4 (control samples) in a black flat and clear bottom microtiter plate (Note 13).
- Measure fluorescence in 2 min intervals over 6-10 min to record the pre-treatment baseline.
- Add 20 µl of stained liposomes to 180 µl pre-warmed (30 °C) 5 mM Tris-HCl, pH 7.4 containing antibiotic compound (final concentrations calculated for 200 µl final volume) in a black flat and clear bottom microtiter plate.
- Measure samples for 30 min in 2 min intervals (Note 14).
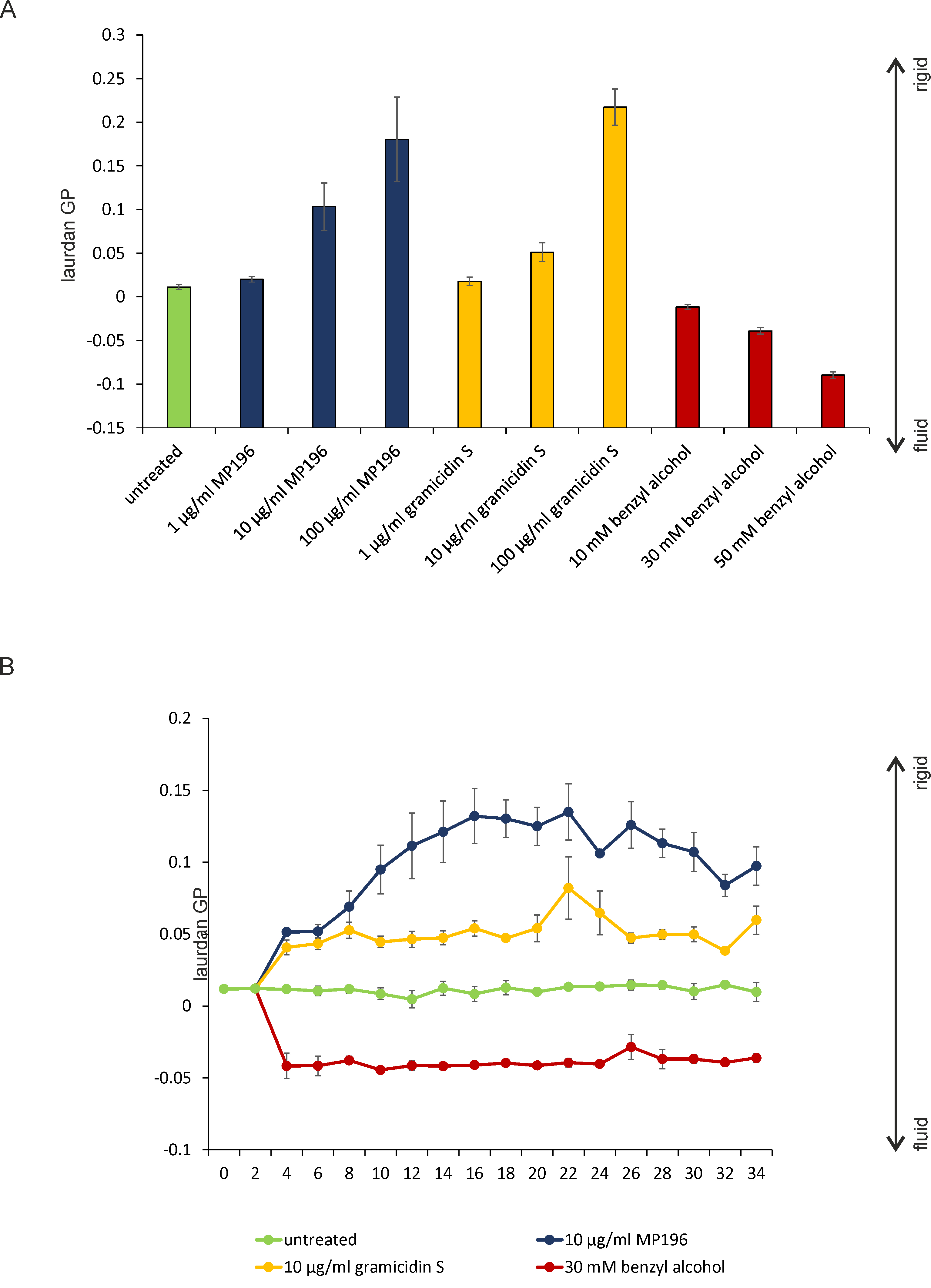
Figure 5. Laurdan measurements of liposomes composed of E. coli polar lipid extracts. A. One point Laurdan measurements with different concentrations of antibiotic. Liposomes were kept at constant 30 °C. Note the concentration-dependent rigidification/fluidization effect. Since lipid to compound ratios in vivo cannot be exactly determined due to unknown biological factors such as binding to cell wall components, they cannot be compared to in vitro lipid to compound ratios using artificial liposomes. We therefore recommend testing a range of concentrations to control, e.g., total membrane lysis at too high concentrations. B. Kinetic Laurdan measurements under the same conditions used in (A). Note the gradual effect of the antimicrobial hexapeptide MP196 compared to the immediate effect of the cyclic β-sheet peptide gramicidin S and the membrane fluidizer benzyl alcohol, which corresponds very well to the in vivo measurements (Figure 3C).
- Laurdan Spectroscopy of bacteria (Note 2)
- Laurdan microscopy
Laurdan microcopy requires a fluorescence light microscope with a high quality 100x oil immersion objective with correction for chromatic aberration (see Step A2a), a temperature chamber, and a specific set of filters. 350 nm excitation and 440 nm emission are usually covered by standard DAPI filter sets. For the second emission wavelength a special filter set with the exact same excitation filter and dichroic mirror as the DAPI set, but an emission filter that covers 500-540 nm, is necessary (see filters described in the Equipment section). Since temperature-dependent changes in membrane fluidity happen rapidly, a temperature-controlled chamber is of key importance for reliable and reproducible results.- Microscope calibration
For laurdan microscopy, a separate picture is taken for each emission wavelength. For correct GP calculation a perfect overlay of these pictures is essential. Therefore, it is necessary to determine wavelength-dependent differences in magnification and z-offset prior to laurdan microscopy experiments. These aberrations depend on the objective type and can be greatly reduced using a high-quality objective with a high degree of chromatic aberration correction, such as the Nikon APO or Zeiss APOCHROMAT series. In our laurdan setup using a Nikon CFI Plan Apochromat objective (see Equipment section for details on the objective and filters), chromatic aberration is neglectable. Calibration only needs to be done once the objective or filters are changed.- Prepare a microscope slide covered with a 1.2% agarose film (Note 15) and spot an appropriate dilution of multicolor fluorescent beads (e.g., TetraSpeck).
- Select a field of view with beads located close the edges of the field.
- Perform a z-stack (200 nm step size, 5 µm depth) with both wavelengths.
- Select the exact focal plane for each individual wavelength by examining the captured z-stacks. Here the optimal focus is determined by measuring the point spread function (PSF) of a chosen fluorescent bead. The focal plane with the optimal focus is characterized by the narrowest PSF. This step can be carried out in ImageJ by determining the fluorescence intensity with line scan profile measured across the bead. If the optimal focal plane differs between the wavelengths used, correct this error by introducing a corresponding wavelength-dependent z-offset in the image acquisition routine of your microscope software.
- Select two beads at opposite edges of the field of view and measure the distance between the centers of the fluorescent beads for each wavelength. Depending on the objective, the distance (measured in pixels or in µm) will be slightly larger for the 440 nm emission wavelength picture. Use this value to later adjust the size of the 520 nm emission wavelength pictures during data analysis in ImageJ.
- Laurdan microscopy (Note 2) (Figure 6)
- Set up an overnight culture in lysogeny broth (LB) or other medium of choice and grow overnight at the desired temperature (typically 30 or 37 °C) (Notes 3 and 4).
- Dilute overnight cultures 1:100 in fresh LB or other medium of choice (Notes 3 and 4).
- Grow at the desired temperature until the desired cell density (standard: 30 or 37 °C, log phase) (Note 5).
- Add 10 µM laurdan from 1 mM working solution and incubate for 10 min (Note 16).
- Harvest cells by centrifugation using pre-warmed (30 °C) 2 ml microtubes in a pre-warmed microtube centrifuge (up to 16,000 x g, 30 s, 30 °C).
- Wash 4x in pre-warmed (30 °C) laurdan buffer (Note 6). Keep tubes warm using a thermomixer while pipetting. Resuspend cells by vortexing rather than pipetting up and down to avoid shearing forces that could damage the membrane (Note 17).
- Resuspend in laurdan buffer to an OD600 of 0.4.
- Prepare 50-200 µl aliquots in pre-warmed (30 °C) 2 ml microtubes and add antibiotics (or apply other condition of interest). Incubation times of 5-10 min are typically sufficient (Note 18).
- Withdraw 0.5 µl and spot on a glass slide covered with a thin layer of 1.2% agarose (Note 15). Wait until the droplet has just dried up (~60 s) and immediately add a coverslip. Prevent the formation of air bubbles between coverslip and agarose.
- Search for a good field of view with a sufficient number of cells using phase contrast or bright field. Perfect focus is crucial for laurdan microscopy.
- Take a picture in the laurdan custom channel followed by a second picture in the DAPI channel (typically 1 s exposure time each) (Notes 19, 20 and 21).

Figure 6. Laurdan microscopy of B. subtilis 168. Separate pictures were taken with 440 nm (A) and 520 nm (B) emission wavelength using two filter cubes with exactly the same excitation filter (360 nm) and dichroic mirror (405 nm). After correction of wavelength-dependent size aberration and z-drift, a GP map can be generated using the ImageJ Calculate GP plugin (C). Since GP values will be calculated for any pixel, it is important to apply an individual background correction for each picture. However, nonsense GP values will still appear in the cytoplasmic space of the cells and for the untrained eye it can be difficult to distinguish the cell membrane from the rest of the cells. It is therefore helpful to create an overlay of the laurdan GP picture (grey) with one of the fluorescence channel pictures (red) to determine the exact position of the cytoplasmic membrane in the GP picture (D). This also greatly facilitates measuring GP values in an area of interest (e.g., membrane patch or rest membrane) using ImageJ. This figure was adapted from Saeloh et al. (2018).
- Microscope calibration
- Laurdan Spectroscopy
- DiIC12
DiIC12 microscopy can in principle be carried out with any good fluorescence microscope. It is advised to use a Cy3 filter (here 535 nm excitation and 590 nm emission) for an optimal signal. While detection of DiIC12 with standard RFP filters (around 570 nm excitation and 645 nm emission) is possible, the much lower signal often results in less clear RIFs. While the staining procedure is simple, observation of clear RIFs in B. subtilis critically depends on the growth phase, and stressed cells either display smooth DiIC12 stains or large patches. Therefore, stable culturing conditions are essential, especially with respect to temperature and oxygen supply.
Fluid membrane domain staining with DiIC12 can be easily adapted for other Gram-positive bacteria, e.g., S. aureus and S. pneumoniae (Figure 7). However, RIFs in strict sense are defined fluid foci as observed in B. subtilis and Escherichia coli and seem to be a phenomenon of MreB-dependent longitudinal cell elongation (Strahl et al., 2014; Oswald et al., 2016), and do not occur in bacteria that grow differently (Saeloh et al., 2018) (Figure 7). DiIC12 can be used in Gram-negative bacteria but may result in less intense membrane staining. A successful method for E. coli has been established by Oswald et al. (2016).- DiIC12 microscopy (Figures 7 and 8)
- Set up overnight cultures in lysogeny broth (LB) or other medium of choice and grow overnight at the desired temperature (typically 30 or 37 °C) (Notes 3 and 4).
- Dilute overnight cultures 1:100 in fresh LB or other medium of choice (Notes 3, 4 and 22).
- Add 1 µg/ml DiIC12 from a 100 µg/ml stock in DMSO (1% final DMSO concentration) (Notes 23 and 24).
- Grow at the desired temperature until the desired cell density (Notes 25 and 26).
- Harvest cells by centrifugation using pre-warmed (30 °C) 2 ml microtubes in a pre-warmed microtube centrifuge (up to 16,000 x g, 30 s, 30 °C).
- Wash 4x in pre-warmed (30 °C) LB supplemented with 1% DMSO (and other supplements like inducers or co-factors, where appropriate) (Notes 25 and 28). Keep tubes warm (30 °C) using a thermomixer while pipetting. Resuspend cells by vortexing rather than pipetting up and down to avoid shearing forces that could damage the membrane (Note 17).
- Resuspend in the same medium to an OD600 of 0.4.
- For antibiotic treatment: Prepare 50-200 µl aliquots in pre-warmed (30 °C) 2 ml microtubes and add antibiotics (or apply other condition of interest). Incubation times of 5-10 min are typically sufficient (Note 18).
- Withdraw 0.5 µl and spot sample on a glass slide covered with a thin layer of 1.2% agarose (Note 15). Wait until the droplet has just dried and add a coverslip without air bubbles.
- Search for a good field of view with a sufficient number of cells using phase contrast or bright field.
- Take a picture in the Cy3 channel. If the microscope is not equipped with a Cy3 filter cube, DiIC12 can also be visualized with a standard RFP filter, yet this will yield a lower fluorescence signal (Notes 27 and 28).
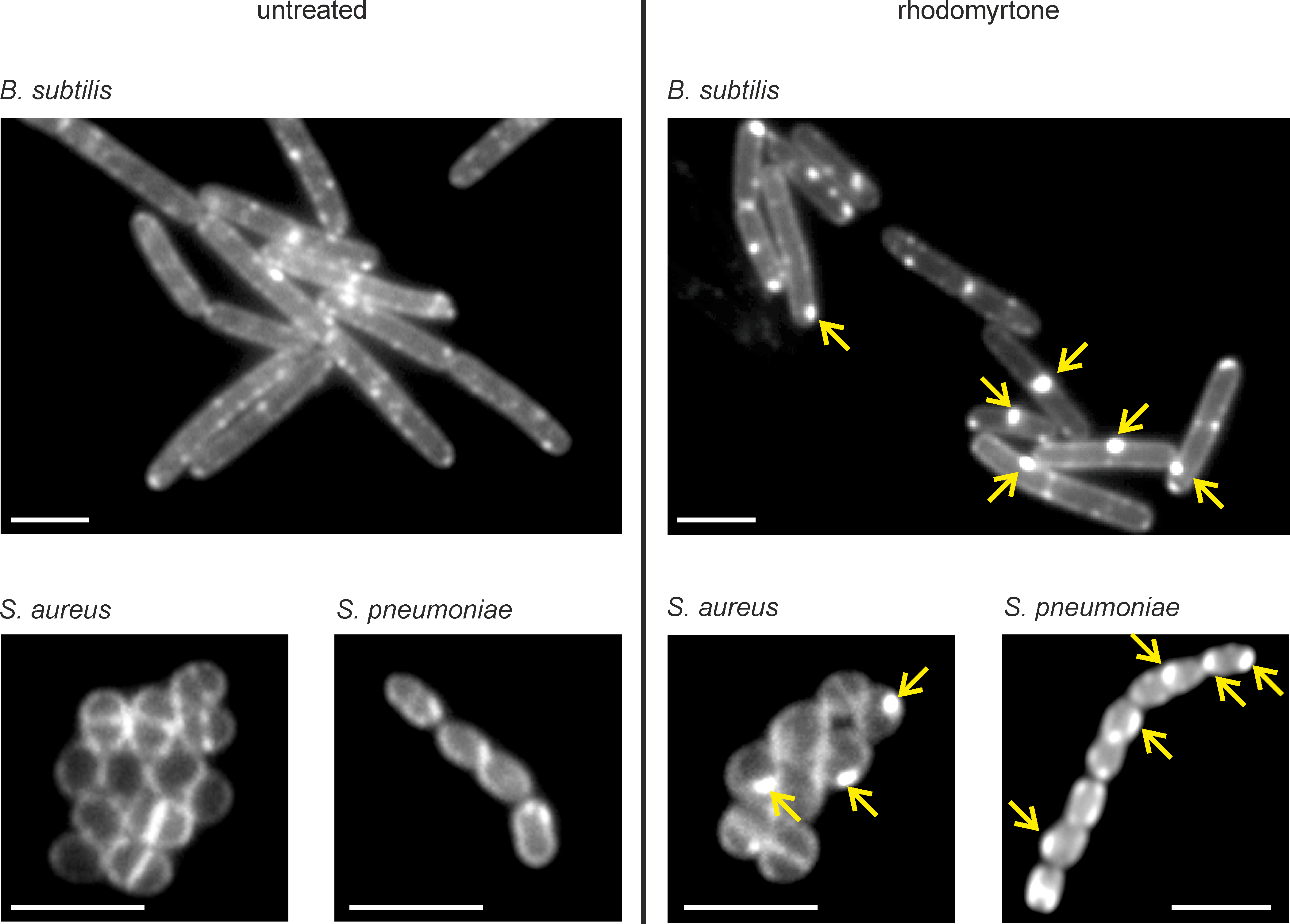
Figure 7. DiIC12 stain of logarithmically growing B. subtilis 168, S. pneumoniae D39 (Lanie et al., 2007), and S. aureus NCTC 8325. RIFs in B. subtilis are organized by the actin homologue MreB. Interestingly, S. pneumoniae and S. aureus, both of which do not have MreB homologues, do not display clearly distinct RIFs. However, both bacteria do not show completely smooth fluorescent membranes, and the slight irregularity of the membrane stain is indicative of fluid microdomains. Treatment with rhodomyrtone, which causes fluid membrane domains (Saeloh et al., 2018), results in a clear accumulation of the DiIC12 dye in fluid membrane foci in all bacteria (yellow arrows). Cells were grown at 37 °C in LB (B. subtilis, S. aureus) or Todd-Hewitt broth supplemented with 0.5 % yeast extract (S. pneumoniae). Scale bars = 2 µm.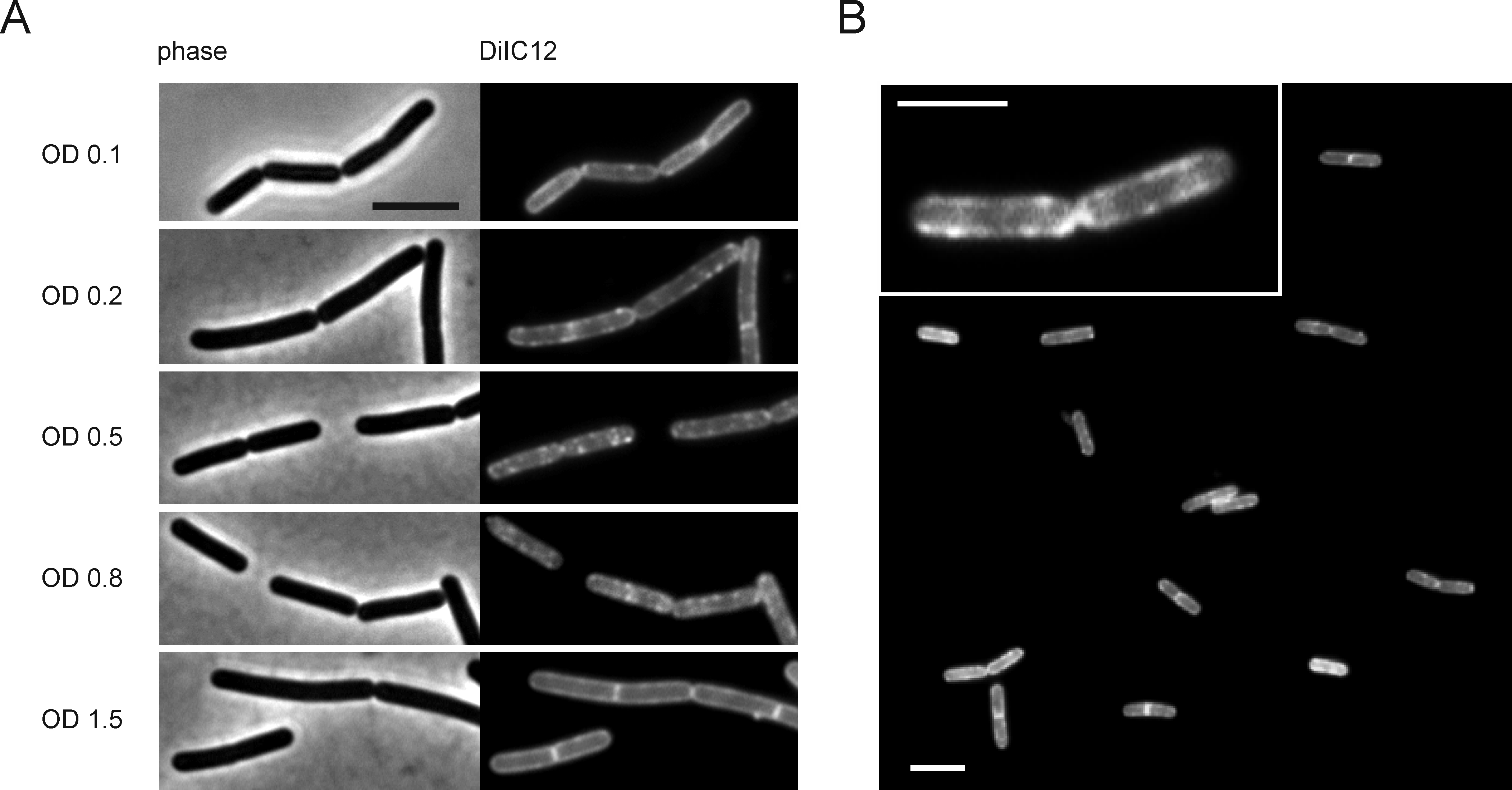
Figure 8. RIFs become clearly visible during logarithmic growth phase in B. subtilis. A. Development of RIFs over time. B. subtilis 168 was grown in LB at 37 °C under continuous shaking. Samples were withdrawn for microscopy at different growth phases. B. RIFs also occur in minimal medium. B. subtilis 168 was grown in BMM at 37 °C under continuous shaking. Scale bars = 2 µm.
- DiIC12 microscopy (Figures 7 and 8)
Data analysis
- Laurdan Spectroscopy
- Calculate Laurdan GP according to the formula: GP = (I460 - I500)/(I460 + I500).
- For kinetic measurements plot GP against time.
Table 1. Exemplary laurdan experiment. The experiment was conducted at 30 °C as described under Step A1a ‘End point measurements with antibiotics’. Background-subtracted values were used for GP calculation.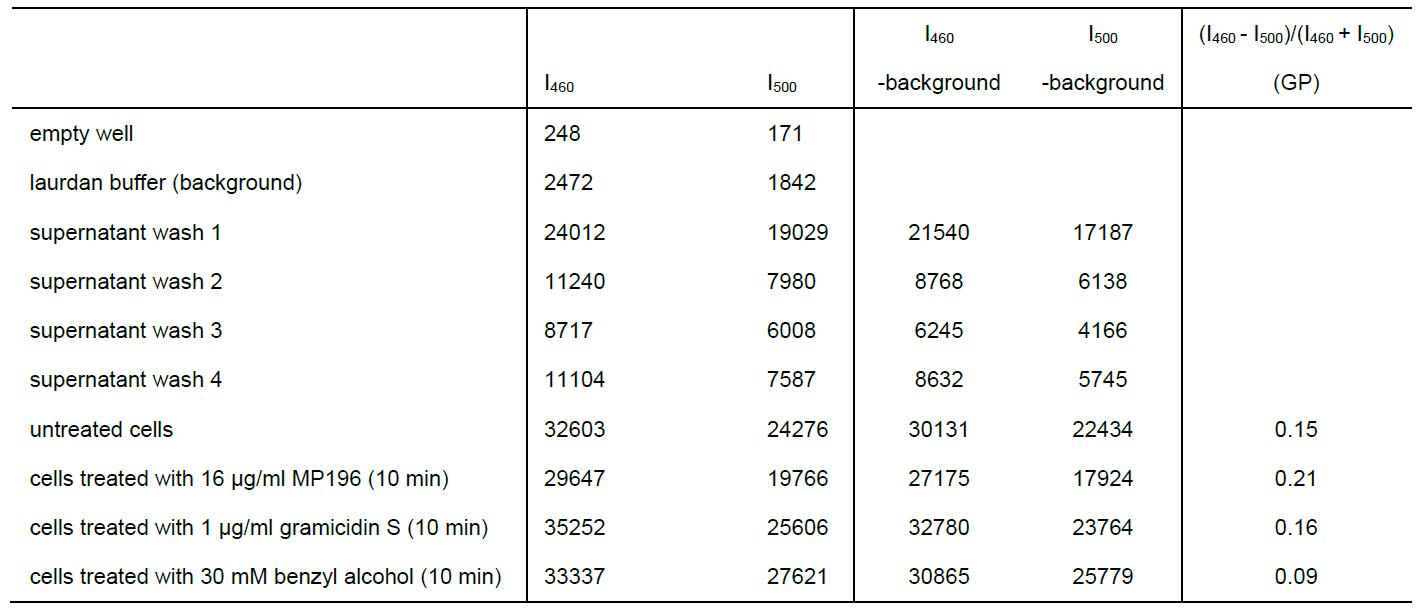
- Laurdan Microscopy
- Rename the DAPI channel picture as ‘A’ and the Laurdan custom channel picture as ‘B’ (see Supplementary File 1 for example pictures).
- Open pictures in ImageJ. Make sure picture ‘A’ is opened first, followed by picture ‘B’.
- Correct the image size using the correction value determined from microscope calibration (see Step A2a Microscope calibration).
- Execute the ‘Align’ plugin to perfectly align the cell(s) of interest (Supplementary File 2).
- Determine background values (average intensity) in both pictures by using the rectangle tool and executing ‘measure’.
- Execute the ‘Calculate GP’ plugin (Supplementary File 3). This will cause several pictures to appear. ‘A’ and ‘B’ are the background-subtracted images, ‘A+B’ and ‘A-B’ are the images used for calculating the GP, and ‘ratio’ is the final image that visualizes the GP values.
- Set significance to 10% of the highest pixel intensity in the field of view for A and B, respectively. This value represents the minimal signal intensity required to be included in the GP calculation. Use the measured background values (average intensity) for background subtraction for A and B. It must be noted that these values typically provide a good starting point in our hands, but since the optimal values depend on the individual sample type, specific research question, and given microscope setup, it might be needed to modify these values in order to get an optimal image. Try different values until the resulting picture clearly shows the cells without extracellular background. Intracellular background will always remain but should be excluded from the analysis. Make sure not to subtract too much background since that could result in cutting off the membrane signal. To control for this, an overlay of the resulting GP picture with the fluorescence picture ‘A’ can help finding the exact position of the cell membrane in the GP picture (see Figure 6D). Note that these modifications only change the visualization of the data but not the GP values themselves.
- Set Lookup Table (LUT) to ‘Rainbow RGB’.
- Edit LUT -> Open -> GP map.lut (Supplementary File 4).
- The picture now shows the GP values when going over the picture with the cursor.
- Save this picture and use it for all quantification purposes, such as line scans etc. When opened in other image software, it will appear in grayscale.
- For display purposes, export the picture as RGB TIFF. This picture should be used for display purposes only, since RGB color format eliminates the GP information.
Notes
- Spectroscopic laurdan measurements can be performed in 96-well format and are in principle suitable for high throughput approaches. However, constant temperature and oxygen supply during sample handling and measurements must be ensured. In case of B. subtilis and other species with a similar sensitivity to oxygen depletion, cells cannot be stopped from shaking for longer than maximum 2 min. This must be kept in mind when designing the experiment (e.g., with respect to measurement intervals and shaking times in the plate reader).
- These assays have been established and optimized for B. subtilis but can be easily adapted for other Gram-positive organisms (see S. aureus, Figure 3D). For Gram-negative bacteria the outer membrane should be permeabilized, e.g., by addition of polymixin B nonapeptide.
- Incubation in a water bath provides better temperature stability than incubation in dry shakers or temperature chambers.
- Glassware is prone to release remains of disinfectant of soap from cleansing solutions, which affect membrane fluidity. Using disposable plastic ware (such as Falcon tubes) ensures that no chemical contaminations affect the experiments. For B. subtilis, which is very sensitive to oxygen depletion, growing 2 ml of culture in a 50 ml falcon tube in a water bath has been proven optimal for fluidity experiments.
- For antibiotic studies an OD600 of 0.3-0.5 has been proven optimal. Since cultures are diluted later, always grow double the desired volume. Do not grow to double the desired OD, since the growth phase affects membrane fluidity.
- While the effects of centrifugation on membrane fluidity have not yet been studied directly, it is possible that centrifugal force influences the cell membrane. Therefore, we do not recommend higher centrifugal force than necessary to produce a solid cell pellet and suggest to keep centrifugation times at a minimum.
- The yellow color and weak autofluorescence of LB and comparable media interferes with laurdan fluorescence. Hence, laurdan buffer is used for washing and further incubation. When using clear media, such as Spizizen or Belitzky Minimal Medium, laurdan buffer might be replaced with the respective clear medium supplemented with 1% DMF.
- Sample volume might be adjusted depending on the shaking speed of the plate reader. Fast shaking as required for B. subtilis might lead to cross-contamination by splashing when using high sample volumes.
- Experiments should be performed in technical triplicates.
- If constant temperature and aeration is ensured, e.g., by using a thermomixer with microtiter plate holder, antibiotic treatment can more simply be performed in the microtiter plate directly. To this end, simply use the procedure described for ‘kinetic measurements with antibiotics’ and perform an end point instead of kinetic measurement.
- Kinetic measurements require the presence of glucose in the laurdan buffer in order to keep cells energized and membranes polarized during the 30 min measurement. Longer measurements are not recommended. However, it is possible to take separate samples at any time point, stain, wash, and make a one-point measurement. Using a clear culture medium prevents this problem. However, be aware that different growth media drastically change membrane fluidity (Figure 3A).
- An OD600 of 0.4 at the time of the measurement is required to keep the measurements stable over time. Lower ODs result in unstable (continuously decreasing or ‘jumping’) GP values in untreated control cells.
- It depends on the lipid mixture (since laurdan inserts better into more fluid membranes), liposome preparation method, plate reader, and quality of the stain, how much the sample has to be diluted. Therefore, it is advisable to run a test sample prior to running the actual experiment.
- While not essential for in vitro experiments, liposome samples are also kept shaking continuously during sample handling and measurements to keep the conditions as similar as possible to the in vivo experiments.
- A thin and perfectly flat agarose film is pivotal for laurdan microscopy. Detailed instructions on how to prepare suitable slides can be found in (Te Winkel et al., 2016). The same protocol should be followed to prepare slides for DiIC12 microscopy.
- While laurdan does not exhibit relevant spectral overlap with other membrane dyes like Nile red or FM5-95, we have observed drastic effects of such dyes on laurdan GP in co-staining experiments. We therefore strictly advise to run laurdan-only controls, if co-staining with other membrane dyes is desired.
- Note that physical and chemical stress factors such as shearing forces, temperature shock or residual chemicals in the tubes might all affect membrane fluidity and results in patchy membrane stains that compromise both laurdan and DiIC12 microscopy experiments.
- Depolarizing agents such as the proton ionophore carbonyl cyanide m-chlorophenyl hydrazine (CCCP) are known to cause fluid membrane patches in B. subtilis (Strahl et al., 2014) and can be used as positive control. To date, the only compound that has been published to cause fluid membrane patches in laurdan microscopy experiments in other bacteria is rhodomyrtone, which can at least be used as positive control for B. subtilis, S. aureus, and Streptococcus pneumoniae (Saeloh et al., 2018).
- Oxygen depletion, e.g., by keeping the cells under the coverslip for too long, leads to membrane depolarization, which also leads to fluid membrane patches (Strahl and Hamoen, 2010; Strahl et al., 2014). It is advised to finish microscopy within 10-15 min after mounting the cells to avoid such artifacts. Always image the untreated control first. Then after finishing with the last sample, examine it again for any signs of aberrant membrane stains due to oxygen depletion. Exposure times may vary depending on the light source and filters, but need to be the same for both channels.
- After 2x 1 s of fluorescence light exposure the laurdan dye is moderately affected by photobleaching. Since a good signal-to-noise ratio is important for good GP calculation, the same area should only be exposed once per channel. We typically reach a signal-to-noise ratio of at least 5:1 with untreated control cells in both channels.
- Overnight cultures should not be overly stationary or even in death phase, since this results in longer lag phase and might affect RIFs.
- Do not add DiIC12 to glassware, since it largely irreversibly stains glass surfaces.
- A final concentration of 1% DMSO helps keeping DiIC12 soluble and should be constantly maintained. However, high concentrations of DMSO might affect the cell membrane and cause a bacterial stress response. Higher DMSO concentrations are therefore not recommended.
- RIFs are only clearly visible in logarithmic growth phase (in B. subtilis OD600 of 0.2-0.8). In our hands, lag phase and stationary phase cells always showed a smooth membrane stain (Figure 8A). Cells depleted of oxygen or exposed to temperature shifts might also show smooth or aberrant DiIC12 patterns with large membrane patches.
- DiIC12 is not toxic to B. subtilis and is also suitable for time-lapse experiments. However, since RIFs are growth phase dependent in this organism, they might be less pronounced in cells growing under a gene frame due to the reduced growth rate (compare also cells grown in minimal medium, Figure 8B). A microfluidic setup has been shown to be well-suited for DiIC12 time-lapse microscopy (Saeloh et al., 2018).
- DiIC12 tends to form large precipitates resulting in a strong background. Cells need to be washed thoroughly to reduce this background to a minimum. When following a time course, it might be necessary to wash the samples again immediately prior to microscopy.
- A good stain with a proper Cy3 filter cube will only require 50-300 ms exposure time. Since DiIC12 is also detected in the mCherry channel, it cannot be used together with red-fluorescing stains or RFP-fusions.
- Bleed-through to the GFP channel is low but existing. This should be kept in mind when combining DiIC12 with GFP fusions with a weak fluorescence signal, and bleed-through controls should be included. For yet unknown reasons, DiIC12 might also influence signal intensity of GFP fusions (Strahl et al., 2014), which might require some adjustment of inducer and dye concentration.
Recipes
Note: All solutions are prepared with double-distilled water.
- Agarose solution
1.2%, freshly prepared - BMM basic medium
50 mM Tris-HCl, pH 7.5
15 mM (NH4)2SO4
8 mM MgSO4•7H2O
27 mM KCl
7 mM Na3citrate•2H2O
Autoclave - BMM supplements
200 mM KH2PO4 (autoclave), add 0.3 ml to 100 ml BMM basic medium
1 M CaCl2•2H2O (autoclave), add 0.2 ml to 100 ml BMM basic medium
0.5 mM FeSO4•7H2O (filtrate), add 0.2 ml to 100 ml BMM basic medium
25 mM MnSO4•4H2O (filtrate), add 0.04 ml to 100 ml BMM basic medium
0.5 M glutamic acid (filtrate), add 0.9 ml to100 ml BMM basic medium
39 mM tryptophane (filtrate), add 2 ml to 100 ml BMM basic medium
20% glucose (filtrate), add 1 ml to 100 ml BMM basic medium - DiIC12 washing medium
LB (autoclaved) (see below)
1% DMSO
Freshly prepared - Laurdan buffer
137 mM NaCl
2.7 mM KCl
10 mM Na2HPO4
1.8 mM KH2PO4
0.2% glucose
1% DMF
Filtrate - Lysogeny broth (LB) medium
10 g/L tryptone
5 g/L yeast extract
10 g/L NaCl
Autoclave - SMM basic medium
13.9 g/L K2HPO4
6 g/L KH2PO4
2 g/L (NH4)2SO4
2 g/L Na3citrate•2H2O
Autoclave - SMM supplements
40% glucose (filtrate), add 1.25 ml to 100 ml SMM basic medium
5 mg/ml tryptophane (filtrate), add 1 ml to 100 ml SMM basic medium
1 M MgSO4•7H2O (filtrate), add 0.6 ml to 100 ml SMM basic medium
20% casamino acids (filtrate), add 0.1 ml to 100 ml SMM basic medium
0.22% Fe(II)NH4 citrate (filtrate), add 0.05 ml to 100 ml SMM basic medium
Acknowledgments
This paper describes in detail the methods used to characterize changes in membrane fluidity in response to antibiotics (Saeloh et al., 2018). MP196 was supplied by Nils Metzler-Nolte, Ruhr University Bochum, gramicidin S by Marina Rautenbach, Stellenbosch University, and rhodomyrtone by Supayang Voravuthikunchai, Prince of Songkla University. The authors wish to thank Annelies Eenhoorn for technical assistance. This work was financially supported by the Netherlands Organization for Scientific Research (NWO, http://nwo.nl/en, STW-Vici 12128 to LWH). MW was supported by a postdoc stipend from the Amsterdam Infection and Immunity Institute.
Author contributions: MW and HS established experimental procedures. NOEV and HS wrote the ImageJ ‘Align’ and ‘Calculate GP’ plugins. LWH supervised experiments. All authors contributed to writing and editing the paper.
Competing interests
The authors declare that they have no conflict of interest or competing interest.
References
- Albada, H. B., Chiriac, A. I., Wenzel, M., Penkova, M., Bandow, J. E., Sahl, H. G. and Metzler-Nolte, N. (2012). Modulating the activity of short arginine-tryptophan containing antibacterial peptides with N-terminal metallocenoyl groups. Beilstein J Org Chem 8: 1753-1764.
- Anagnostopoulos, C. and Spizizen, J. (1961). Requirements for transformation in Bacillus subtilis. J Bacteriol 81(5): 741-746.
- Bach, J. N. and Bramkamp, M. (2013). Flotillins functionally organize the bacterial membrane. Mol Microbiol 88(6): 1205-1217.
- Bæk, K. T., Frees, D., Renzoni, A., Barras, C., Rodriguez, N., Manzano, C. and Kelley, W. L. (2013). Genetic variation in the Staphylococcus aureus 8325 strain lineage revealed by whole-genome sequencing. PLoS One 8(9): e77122.
- Barák, I. and Muchová, K. (2013). The role of lipid domains in bacterial cell processes. Int J Mol Sci 14(2): 4050-4065.
- Baumgart, T., Hunt, G., Farkas, E. R., Webb, W. W. and Feigenson, G. W. (2007). Fluorescence probe partitioning between Lo/Ld phases in lipid membranes. Biochim Biophys Acta 1768(9): 2182-2194.
- Beranova, J., Jemiola-Rzeminska, M., Elhottova, D., Strzalka, K. and Konopasek, I. (2008). Metabolic control of the membrane fluidity in Bacillus subtilis during cold adaptation. Biochim Biophys Acta 1778(2): 445-453.
- Bramkamp, M. and Lopez, D. (2015). Exploring the existence of lipid rafts in bacteria. Microbiol Mol Biol Rev 79(1): 81-100.
- Epand, R. M. and Epand, R. F. (2009). Lipid domains in bacterial membranes and the action of antimicrobial agents. Biochim Biophys Acta 1788(1): 289-294.
- Garcia-Fernandez, E., Koch, G., Wagner, R. M., Fekete, A., Stengel, S. T., Schneider, J., Mielich-Suss, B., Geibel, S., Markert, S. M., Stigloher, C. and Lopez, D. (2017). Membrane microdomain disassembly inhibits MRSA antibiotic resistance. Cell 171(6): 1354-1367 e1320.
- Karabadzhak, A. G., Weerakkody, D., Deacon, J., Andreev, O. A., Reshetnyak, Y. K. and Engelman, D. M. (2018). Bilayer thickness and curvature influence binding and insertion of a pHLIP peptide. Biophys J 114(9): 2107-2115.
- Kingston, A. W., Subramanian, C., Rock, C. O. and Helmann, J. D. (2011). A σW-dependent stress response in Bacillus subtilis that reduces membrane fluidity. Mol Microbiol 81(1): 69-79.
- Lanie, J. A., Ng, W. L., Kazmierczak, K. M., Andrzejewski, T. M., Davidsen, T. M., Wayne, K. J., Tettelin, H., Glass, J. I. and Winkler, M. E. (2007). Genome sequence of Avery's virulent serotype 2 strain D39 of Streptococcus pneumoniae and comparison with that of unencapsulated laboratory strain R6. J Bacteriol 189(1): 38-51.
- Lopez, D. and Kolter, R. (2010). Functional microdomains in bacterial membranes. Genes Dev 24(17): 1893-1902.
- Müller, A., Wenzel, M., Strahl, H., Grein, F., Saaki, T. N., Kohl, B., Siersma, T., Bandow, J. E., Sahl, H. G., Schneider, T. and Hamoen, L. W. (2016). Daptomycin inhibits cell envelope synthesis by interfering with fluid membrane microdomains. Proc Natl Acad Sci U S A. 113(45): E7077-E7086.
- Oswald, F., Varadarajan, A., Lill, H., Peterman, E. J. and Bollen, Y. J. (2016). MreB-dependent organization of the E. coli cytoplasmic membrane controls membrane protein diffusion. Biophys J 110(5): 1139-1149.
- Parasassi, T. and Gratton, E. (1995). Membrane lipid domains and dynamics as detected by Laurdan fluorescence. J Fluoresc 5(1): 59-69.
- Reddy, A. S., Warshaviak, D. T. and Chachisvilis, M. (2012). Effect of membrane tension on the physical properties of DOPC lipid bilayer membrane. Biochim Biophys Acta 1818(9): 2271-2281.
- Rosch, J. W. and Caparon, M. G. (2005). The ExPortal: an organelle dedicated to the biogenesis of secreted proteins in Streptococcus pyogenes. Mol Microbiol 58(4): 959-968.
- Saeloh, D., Tipmanee, V., Jim, K. K., Dekker, M. P., Bitter, W., Voravuthikunchai, S. P., Wenzel, M. and Hamoen, L. W. (2018). The novel antibiotic rhodomyrtone traps membrane proteins in vesicles with increased fluidity. PLoS Pathog 14(2): e1006876.
- Sanchez, S. A., Tricerri, M. A., Gunther, G. and Gratton, E. (2007). Laurdan Generalized Polarization: from cuvette to microscope. In: Méndez-Vilas, A. and Díaz, J. (Eds.). Modern Research and Educational Topics in Microscopy. Formatex, 1007-1014.
- Schneider, J., Klein, T., Mielich-Suss, B., Koch, G., Franke, C., Kuipers, O. P., Kovacs, A. T., Sauer, M. and Lopez, D. (2015). Spatio-temporal remodeling of functional membrane microdomains organizes the signaling networks of a bacterium. PLoS Genet 11(4): e1005140.
- Strahl, H. and Hamoen, L. W. (2010). Membrane potential is important for bacterial cell division. Proc Natl Acad Sci U S A 107(27): 12281-12286.
- Strahl, H., Burmann, F. and Hamoen, L. W. (2014). The actin homologue MreB organizes the bacterial cell membrane. Nat Commun 5: 3442.
- Stülke, J., Hanschke, R. and Hecker, M. (1993). Temporal activation of β-glucanase synthesis in Bacillus subtilis is mediated by the GTP pool. J Gen Microbiol 139(9): 2041-2045.
- Te Winkel, J. D., Gray, D. A., Seistrup, K. H., Hamoen, L. W. and Strahl, H. (2016). Analysis of antimicrobial-triggered membrane depolarization using voltage sensitive dyes. Front Cell Dev Biol 4: 29.
- Vega, L. A., Port, G. C. and Caparon, M. G. (2013). An association between peptidoglycan synthesis and organization of the Streptococcus pyogenes ExPortal. MBio 4(5): e00485-00413.
- Weihs, F., Wacnik, K., Turner, R. D., Culley, S., Henriques, R. and Foster, S. J. (2018). Heterogeneous localisation of membrane proteins in Staphylococcus aureus. Sci Rep 8(1): 3657.
- Wenzel, M., Chiriac, A. I., Otto, A., Zweytick, D., May, C., Schumacher, C., Gust, R., Albada, H. B., Penkova, M., Kramer, U., Erdmann, R., Metzler-Nolte, N., Straus, S. K., Bremer, E., Becher, D., Brotz-Oesterhelt, H., Sahl, H. G. and Bandow, J. E. (2014). Small cationic antimicrobial peptides delocalize peripheral membrane proteins. Proc Natl Acad Sci U S A 111(14): E1409-1418.
- Wenzel, M., Patra, M., Albrecht, D., Chen, D. Y., Nicolaou, K. C., Metzler-Nolte, N. and Bandow, J. E. (2011). Proteomic signature of fatty acid biosynthesis inhibition available for in vivo mechanism-of-action studies. Antimicrob Agents Chemother 55(6): 2590-2596.
- Zhao, J., Wu, J. and Veatch, S. L. (2013). Adhesion stabilizes robust lipid heterogeneity in supercritical membranes at physiological temperature. Biophys J 104(4): 825-834.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Wenzel, M., Vischer, N. O. E., Strahl, H. and Hamoen, L. W. (2018). Assessing Membrane Fluidity and Visualizing Fluid Membrane Domains in Bacteria Using Fluorescent Membrane Dyes. Bio-protocol 8(20): e3063. DOI: 10.21769/BioProtoc.3063.
Category
Microbiology > Microbial cell biology > Cell staining
Microbiology > Antimicrobial assay > Antibacterial assay
Cell Biology > Cell staining > Lipid
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.