- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Qualitative in vivo Bioluminescence Imaging

Published: Vol 8, Iss 18, Sep 20, 2018 DOI: 10.21769/BioProtoc.3020 Views: 10963
Reviewed by: Nicoletta CordaniLaura J. LambertAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Bioluminescence imaging (BLI) technology is an advanced method of carrying out molecular imaging on live laboratory animals in vivo. This powerful technique is widely-used in studying a variety of biological processes, and it has been an ideal tool in exploring tumor growth and metastatic spread in real-time. This technique ensures the optimal use of laboratory animal resources, particularly the ethical principle of reduction in animal use, given its non-invasive nature, ensuring that ongoing biological processes can be studied over time in the same animal, without the need to euthanize groups of mice at specific time points. In this protocol, the luciferase imaging technique was developed to study the effect of co-inoculating pericytes (contractile, αSMA+ mesenchymal stem cell-like cells, located abluminally in microvessels) on the growth and metastatic spread of ovarian cancers using an aggressive ovarian cancer cell line–OVCAR-5–as an example.
Keywords: BioluminescenceBackground
The principle of bioluminescence imaging (BLI) is based on the light-emitting properties of a relatively simple biochemical process, i.e., luciferase-mediated oxidation of the molecular substrate luciferin to produce light. In cancer research, BLI is a popular tool (Contag et al., 2000) used to study the metastatic spread of luciferase-transduced cancer cells in live animals in vivo. Most animal tissues have little to no baseline bioluminescent properties, which ensures that there is a very high signal to noise ratio in BLI experiments. Nevertheless, it is always pragmatic to ensure that BLI experiments are conducted with non-substrate injected rodents as a negative control group. A few crucial factors are key to ensuring best practice in performing BLI experiments on rodents for the detection of luciferase-tagged cancer cells. Firstly, BLI is a powerful tool that allows easy detection of luciferase expressing-cells by imaging emitted bioluminescence under anesthesia (Figure 1). However, this technique is essentially qualitative and attempts to quantitate BLI signal can be misleading given that the strength of signal is dependent on several factors including duration of exposure, anesthetic technique, time elapsed after injecting luciferin, etc. Moreover, published evidence indicates that light emission (i.e., quantity of photons) is not directly related to luciferase activity (Rice et al., 2001). Secondly, the location or depth of the tissue of interest, particularly its distance from the skin’s surface, and the size of the metastatic cell mass are important considerations in planning a BLI experiment (Weissleder, 2001). This is because photon loss occurs as the signal travels through the tissue mass–consequently, luciferase-tagged cells/tissues that are closer to the skin’s surface tend to appear brighter as do larger metastases. Micrometastases can be detected but may require sacrificing the animal and imaging the organs directly after skin removal in place of intact animals. Notwithstanding these and other challenges, BLI is an efficient and powerful, non-invasive technique for studying biological processes in vivo.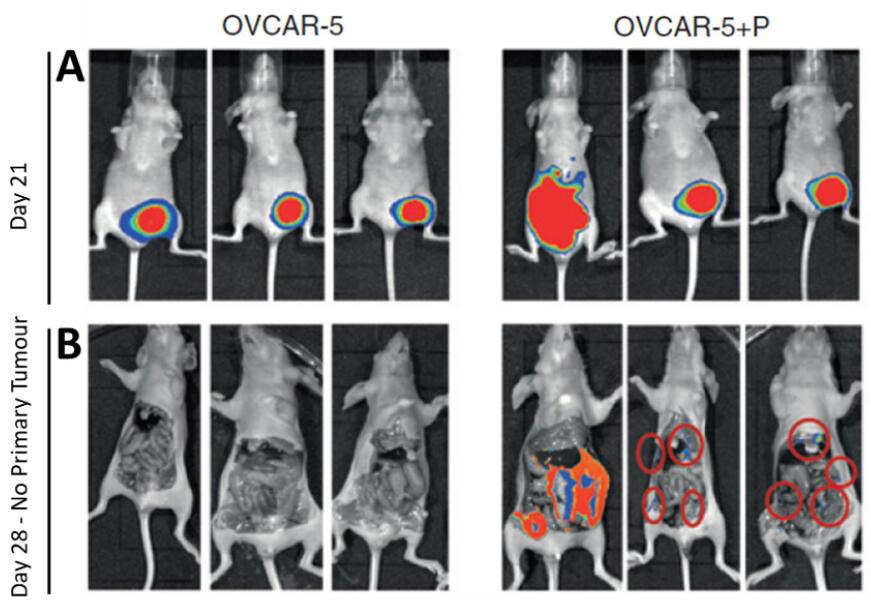
Figure 1. Imaging primary subcutaneous OVCAR-5 tumors and metastases. A. GFP-luciferase labeled OVCAR-5 cells, xenografted alone (OVCAR-5) or co-injected with pericytes (OVCAR-5+P) generated tumors that were imaged at regular intervals. The increased BLI signal observed in images of pericyte co-injected xenografts show that pericytes promote OVCAR-5 tumor growth rate and induce metastases compared to the control group. B. At day 28 the BLI signal from the primary tumor is saturated, and the mice were sacrificed and the primary tumors (along with the abdominal skin excised to permit clearer imaging of the metastatic nodules–metastatic nodules marked with red rings).
Materials and Reagents
- Materials
- Pipette tips (Interpath Services, catalog numbers: 39770 , 39730 )
- Hamilton® syringe (Hamilton, catalog number: 80366 )
- Tissue culture plasticware (6-well plates BD; 2, 5 and 10 ml pipettes SARSTEDT)
- Steritop-GP polyethersulfone with low binding PES membrane (0.22 μm pore size) (Merck, Millipore, catalog number: SCGPS05RE )
- Pasteur pipette (Biologix, catalog number: 30-0138A1 )
- Biological materials
- Lentiviral vector pFUGW-Pol2-ffLuc2-eGFP (Addgene, catalog number: 71394 )
- OVCAR-5 cell line was obtained from NCI, and authenticated using short tandem repeat markers to confirm cell identity against the Genome Project Database (Wellcome Trust Sanger Institute)
- HIV-1 packaging vector pCMV-deltaR8.2 (Addgene, catalog number: 8455 ), a kind gift from Dr. Cameron Johnstone, Anderson Lab, Peter MacCallum Cancer Centre, Melbourne
- Packaging cell line HEK293T (ATCC, catalog number: CRL-3216 ), a kind gift from Dr. Cameron Johnstone, Anderson Lab, Peter MacCallum Cancer Centre, Melbourne
- Mice: 6-8 weeks old female athymic nude Balb/c mice were obtained from Walter Eliza Hall Institute, housed in a pathogen-free 12 h light–dark environment, fed ad libitum were used for tumorigenicity assays. This age range of mice is optimal for good tumor take rates which decline if older mice (e.g., 10 weeks old) are used.
- Reagents
- Bovine serum albumin (Sigma-Aldrich, catalog number: A9418-500G )
- DAPI:4’, 6’-Diamidino-2-Phenylindole Dihydrochloride (Sigma-Aldrich, catalog number: D9542-5MG )
- DMEM (Thermo Fisher Scientific, GibcoTM, catalog number: 11965092 )
- Diflucan (Fluconozole) (Sigma-Aldrich, catalog number: F8929 )
- D-Luciferin: Sodium salt 4,5-Dihydro-2-(6-hydroxy-2-benzothiazolyl)-4-thiazolecarboxylic acid sodium salt (Gold Biotechnology, catalog number: LUCNA-1G )
- FuGENE 6 (Roche Diagnostics, Mannheim, Germany)
- Endothelial basal media (EBMTM-2) (Lonza, catalog number: CC-3156 )
- Endothelial growth media (EGMTM-2) SinglequotsTM (Lonza, catalog number: CC-4147 )
- Fetal Calf Serum (FCS) (Thermo Fisher Scientific, GibcoTM, catalog number: 10099141 )
- Forthane/Isoflurane (Sigma-Aldrich, catalog number: 792632 )
- HEPES pH 7.4 (Sigma-Aldrich, catalog number: H0887-100ML )
- MatrigelTM (standard) (BD, catalog number: 356234 )
- PBS without Ca2+ and Mg2+ (GE Healthcare, Hyclone, catalog number: SH30256.FS )
- Penicillin-Streptomycin (Thermo Fisher Scientific, GibcoTM, catalog number: 15140122 )
- Polybrene (Sigma-Aldrich, catalog number: 107689 )
- Potassium chloride (Sigma-Aldrich, catalog number: P5405 )
- Potassium dihydrogen phosphate (Sigma-Aldrich, catalog number: P5655 )
- RMPI-1640 (Thermo Fisher Scientific, InvitrogenTM, catalog number: 11875 )
- Sodium bicarbonate (Sigma-Aldrich, catalog number: S5761 )
- Sodium chloride (Astral Scientific, catalog number: AMX190 )
- Sodium hydrogen phosphate (Sigma-Aldrich, catalog number: S5136 )
- Trypsin 0.05% (Thermo Fisher Scientific, GibcoTM, catalog number: 25300120 )
- Trypan blue (Thermo Fisher Scientific, GibcoTM, catalog number: 15250061 )
- Phosphate buffered Saline (PBS) (see Recipes)
- RPMI-1640 media (see Recipes)
Equipment
- Pipettes (Corning, catalog numbers: 4487 , 4488 , 4489 )
- Hemocytometer (ProSciTech, catalog number: SVZ2NI0U )
- Cell culture incubator (NuAire, model: NU-5510/E )
- Centrifuge (Beckman Coulter, model: Allegra X-12 )
- Electronic calipers (Fisher Scientific, catalog number: 14-648-17 )
- SW28 Rotor (Beckman Coulter, catalog number: 342207 )
- Ultracentrifuge (Beckman Coulter, model: OptimaTM XE-100 )
- Fluorescent Microscope (Nikon Instruments, model: Nikon A1+ Confocal Microscope )
- Xenogen Realtime Imaging System (IVIS Lumina II)
- Becton Dickinson Biosciences FACS DivaTM cell sorter
Software
- Prism 6 (GraphPad Inc.)
- Photoshop CS 6.0 (Adobe Inc.)
- Metamorph (Molecular devices)
- ImageJ (NIH software)
Procedure
In the protocol below we describe luciferase imaging to measure the metastatic spread of OVCAR-5 cells after the establishment of primary tumors following subcutaneous injection in immunocompromised mice. However, other cell types are equally amenable to this approach, and we have used this protocol to track OVCAR-8 cells for metastatic spread and monitor the persistence and survival of pericytes in vivo, when co-injected with OVCAR cells (Sinha et al., 2016).
- Lentiviral vector
All work with lentiviruses was performed with due regard to biosafety concerns in line with guidelines set down by the Office of the Gene Technology Regulator of the Government of Australia and with Institutional approval (#09/2006). The replication defective 3rd generation lentiviral vector pFUGW-Pol2-ffLuc2-eGFP (Addgene plasmid #71394) expressing green fluorescent protein (GFP) from jellyfish Aequorea victoria and the firefly luciferase gene (Day et al., 2009) is used to transduce OVCAR-5 cells in this protocol, by co-transfection of the HIV-1 packaging vector and VSV-G envelope glycoprotein (Sinha et al., 2016). - Lentiviral production
The packaging cell line HEK293T is used to produce lentivirus for transduction of OVCAR-5 cells (Sinha et al., 2016). Briefly, HEK293T cells are plated at 2 x 105 cells per 60 mm plate in 3 ml DMEM containing 10% FCS, and allowed to adhere and grow overnight at 37 °C in a humidified incubator containing 5% (v/v) CO2. HEK293T cells are then transfected using FuGENE 6 (Roche Diagnostics, Mannheim, Germany) at a FuGENE (μl): pFUGW-Pol2-ffLuc2-eGFP plasmid DNA (μg) ratio of 3:1 as follows:- Add an appropriate volume of FuGENE 6 to serum free media and incubate for 5 min at room temperature.
- Determine the concentration of plasmid DNA using 260 nm absorption; then add it to the FuGENE/media mix at the appropriate concentration, and incubate for 15 min at room temperature.
- Add the FuGENE/DNA/media mixture dropwise to the plated cells at 100 μl per 60 mm plate. Incubate the plate for 24-48 h at 37 °C.
- Collect viral supernatant from HEK293 cells, place it in a sterile tube and concentrate by spinning at 1,000 x g at 25 °C for 90 min in an ultracentrifuge using an SW28 rotor; aspirating the media and resuspending the viral pellet in 1 ml of RPMI-1640 media.
- Lentiviral transduction
- One day prior to infection, plate OVCAR-5 cells into 6-well plates at 1 x 105 cells per well in RPMI-1640 (Sinha et al., 2016).
- For each well of OVCAR-5 cells to be transduced, add 2.5 μl of 10 mg/ml polybrene to 2 ml of media, and add to 500 μl of viral supernatant. This is the infection cocktail.
- Overlay OVCAR-5 cells with infection cocktail for 12 h at 37 °C with 5% CO2, aspirate virus-containing medium and replace with fresh medium.
- Assess transduction efficiency of OVCAR-5 cells using fluorescence microscopy, as judged by the number of GFP positive cells.
- Propagate transduced cells and expand in culture for up to two passages monitoring GFP transduction efficiency by fluorescence microscopy, prior to sorting for enrichment for GFP-positive cells. To sort, trypsinize cells and resuspend in blocking buffer (2% [v/v] FCS and 2% BSA in PBS without Ca2+ and Mg2+) containing DAPI at a final concentration of 2 μg/ml. Viable (DAPI negative) GFP+ cells are sorted and collected on a BD FACS DivaTM sorter and expanded prior to use in luciferase imaging experiments. Lentiviral transduction efficiencies are extremely high and transduction efficiencies of 85-90% were routinely achieved in our laboratory even with primary cells such as pericytes.
- GFP-luciferase+ OVCAR-5 cell culture and preparation for injection into mice
- Culture GFP-luciferase+ OVCAR-5 cells to 80-90% confluency.
- Aspirate culture media using sterile Pasteur pipette.
- Wash cell monolayer with a small volume of pre-warmed PBS without Ca2+ and Mg2+.
- Add an appropriate volume of 0.05% trypsin at 37 °C and incubate for 1-2 min.
- Tap plate gently to dislodge adherent cells.
- Quench trypsin activity with an equal volume of media containing FCS.
- Transfer cell suspension to centrifuge tube and spin at 400 x g for 5 min at 4 °C.
- Discard supernatant.
- Perform cell count using trypan blue stain and hemocytometer.
- Resuspend 8 x 105-8 x 106 cells in 100 μl of a PBS/MatrigelTM (5 μg/ml) mixture per mouse on ice.
- Animal preparation
- Inject immunocompromised mice with 100 μl cell suspension subcutaneously under the front flank using a Hamilton® syringe with a 26 G needle. (See video clip of procedure at https://youtube.com/watch?v=nrOTzLiWC8U)
- Keep mice under daily observation after injection to ensure full recovery, looking for signs of stress.
- Monitor tumor volume by taking metric measurements using electronic calipers.
Note: OVCAR-5 tumors were monitored every alternate day. Modify protocol depending on the tumor growth rate of cell lines in use.
- Luciferase Imaging
- Inject mice subcutaneously on the non-tumor bearing flank with 150 μg per gram of body weight in 100 μl sterile PBS of D-luciferin, the substrate for the luciferase enzyme.
Note: Acquire imaging within 10-12 min of injecting with D-Luciferin. - Allow mice free movement for 6-8 min.
- Anesthetize mice with 2.5% Forthane (commercial isoflurane anesthetic) in oxygen.
Note: Take care not to anesthetize for longer than 3 min. - Place anesthetized mice to inhalation cones in induction chamber of Xenogen Real-Time Imaging System on the heated platform.
- Close chamber door securely.
- Acquire images at the same exposure length and time interval for all experimental groups within 10-12 min of luciferin injection prior to signal decay.
- Inject mice subcutaneously on the non-tumor bearing flank with 150 μg per gram of body weight in 100 μl sterile PBS of D-luciferin, the substrate for the luciferase enzyme.
- End point
- Repeat above luciferase imaging (Steps F1-F6) every 7 days to continue to track metastatic spread.
- At the experimental end point, the luminescent signal from large primary tumors is saturated and can block the signal from the smaller metastatic nodules. In order to overcome this, inject mice with D-luciferin and allow free movement for 6-8 min, euthanize the animals and surgically excise the primary tumors. Open the peritoneal cavity surgically, remove skin tissue over the anatomical sites to be imaged and proceed with BLI, adjusting exposure length to ensure signals from smaller metastatic nodules are captured.
Note: All experiments were performed in compliance with Institutional ethics committee guidelines which stipulate that animals should not be subjected to undue distress–experimental animals were monitored daily for signs of distress and were sacrificed when tumors reached a volume of 1,500 mm3 (where tumor volume = [width]2 x length x 0.5) even if this occurs before the planned end point of the experiment. No experiments were taken beyond Day 35 after injection.
Data analysis
- Metastatic spread detected by BLI is a qualitative assay permitting the investigator to determine whether particular experimental conditions (in this case co-injection with pericytes) leads to metastasis or not. It is also possible to compare the time of onset of metastases under specific experimental conditions–in this case establishing that co-injection of OVCAR-5 cells with pericytes, leads to earlier onset of metastases than OVCAR-5 cells alone, by imaging at different time points. However, to establish that OVCAR-5 cells consistently yield metastatic nodules when co-injected with pericytes at specific time points, 10 mice per experimental group were imaged at several time points and each experiment replicated twice. BLI can also be used to track the persistence of non-tumor cells–as shown in Figure 2, by transducing pericytes with the GFP-luciferase lentivirus and co-injecting with untagged OVCAR-5 cells.
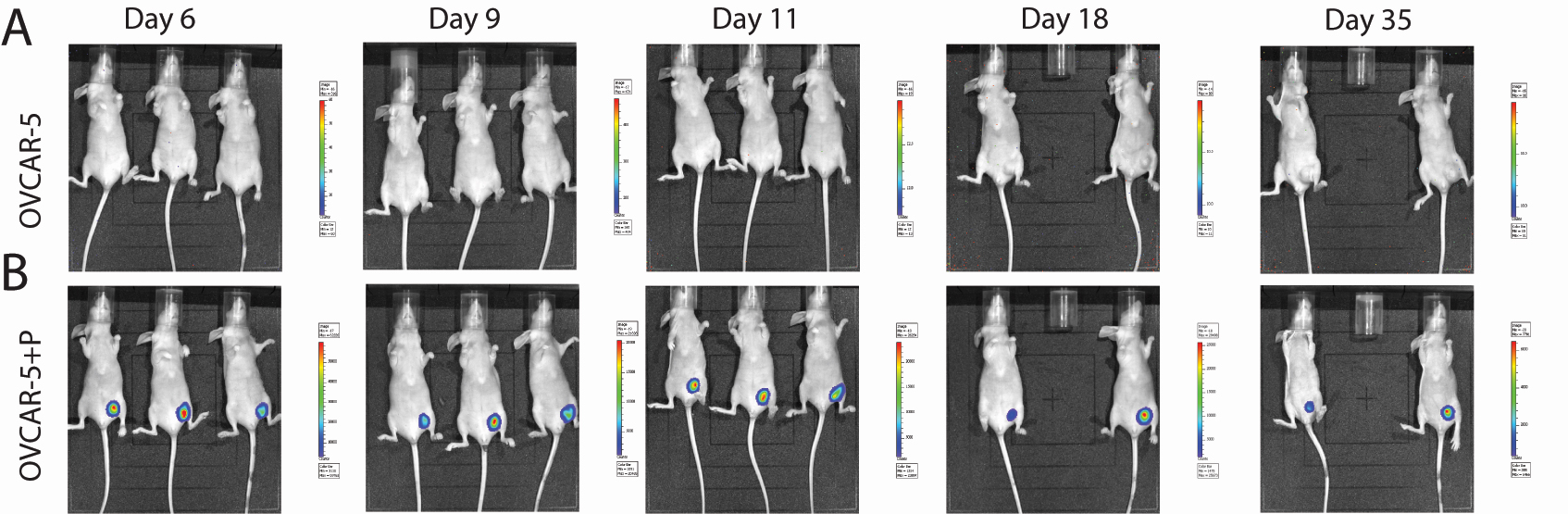
Figure 2. Tracking pericytes in OVCAR-5 xenografts in vivo (Sinha et al., 2016). BLI images of xenografts generated by unlabeled OVCAR-5 cells alone (A); or OVCAR-5 cells co-injected with GFP-luciferase labeled pericytes (B). Imaging was conducted in 3 mice per group per time point, in 2 replicate experiments. Unlabeled OVCAR-5 tumors acted as a negative control, giving no signal despite luciferin injection (A). Sequential BLI of OVCAR-5 tumors co-injected with GFP-luciferase labeled pericytes displaying a positive BLI signal from Day 6 to Day 35, suggesting the persistence of pericytes in xenografts (B). - BLI can be used to quantitate the number of metastatic nodules obtained with OVCAR-5 cells alone versus OVCAR-5 cells co-injected with pericytes but is best determined at the endpoint of the experiment after sacrificing the animals and enumerating the number of nodules detected by BLI from 10 mice per group from two independent experiments. Data are then expressed as mean number ± SEM of metastatic nodules per mouse and statistical analysis for differences between experimental groups performed using one-way ANOVA (Figure 3). In addition, metastases must be verified independently to confirm their OVCAR-5 cell origin by an independent means–thus each metastatic nodule is harvested, processed for histological analysis via paraffin sections that are immunostained for GFP expression using an antibody to GFP.
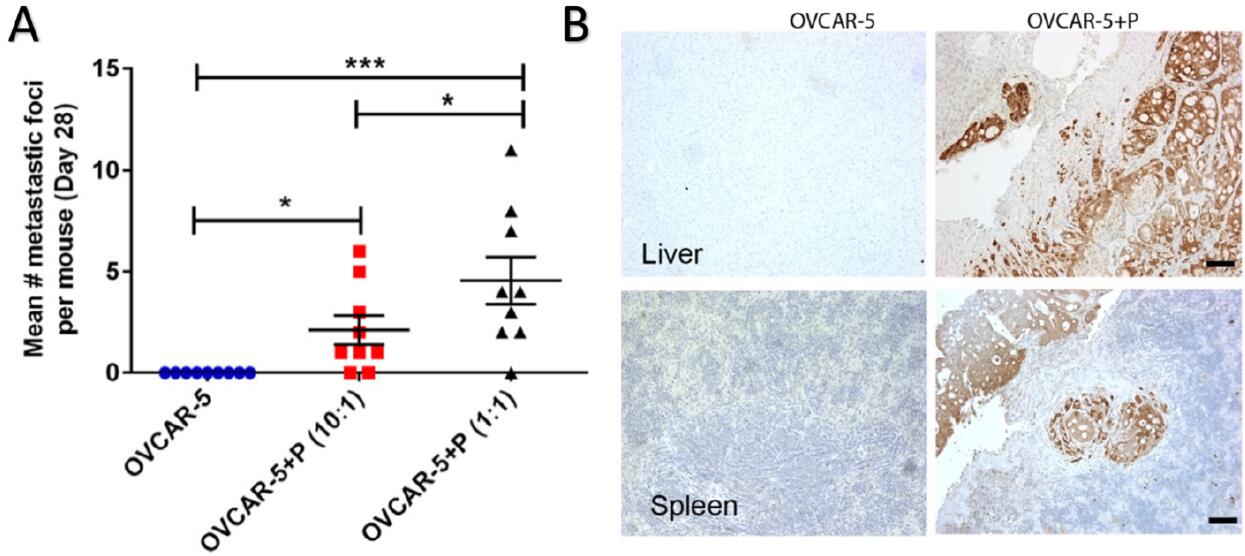
Figure 3. Quantification and immunohistochemical verification of metastatic nodules (Sinha et al., 2016). A. The number of metastatic nodules obtained under varying experimental conditions was counted at the experimental endpoint, revealing that OVCAR-5 cells do not yield metastases at Day 28 whereas co-injection with pericytes does. Increasing the ratio of pericytes: tumor cells leads to an increase in metastatic nodules (compare OVCAR-5: pericyte ratio of 10:1 versus 1:1). B. GFP staining of GFP-luciferase+OVCAR-5 tumors generated from OVCAR-5 cells alone or with pericyte co-injection (OVCAR-5+P) in the liver and spleen verifying that the metastatic nodules detected by BLI are derived from OVCAR-5 cells and not attributed to spurious signal.
Notes
- The absolute amount of BLI signal detected is dependent on the number of GFP-luciferase+ cells being imaged and the duration of exposure after luciferin injection. Thus, imaging smaller tumors at early stages of tumor development or the lower number of pericytes can be enhanced by increasing the duration of exposure after luciferin injection. However, care has to be taken to ensure that the same exposure is used across all experimental groups of animals compared within an experiment.
- Detection of metastatic nodules by BLI is limited by their size and depth of location within the body–the deeper their location from the imaged surface, the lower their chance of detection. Low signal from small metastatic nodules can also be obliterated by the strong signal from the primary tumor. Thus, while BLI is a good indicator of the presence or absence of metastasis, it is advisable to use the endpoint of experiments to establish the full extent and number of metastases. At this stage, animals are sacrificed, the primary tumor excised and the skin removed over the region to be imaged for metastases.
- Use of different surgical tools to excise primary tumors from each experimental animal is advisable due to the possibility of transferring luciferase positive cells from one animal to the next, giving rise to false positive BLI readings. This also makes it critical to verify that each “metastatic nodule” detected by BLI is indeed derived from GFP-luciferase labeled tumor cells by harvesting the BLI positive metastasis, processing for histological sectioning and immunohistochemical analyses after staining for GFP as shown in Figure 3B.
Recipes
- Phosphate buffered Saline (PBS)
- Dissolve 1 g KCl; 1 g KH2PO4; 5.75 g Na2HPO4 and 40 g NaCl in approximately 2 L of MillQ H2O
- Make volume up to 5 L with MilliQ H2O and stir well until all ingredients have dissolved
- Measure pH and adjust to pH 7.0-7.5. Ensure osmolarity is 270 ± 13 mM
- Filter sterilize using a 20 L pressure tank and Sartobran filter
- Store at 4 °C
- RPMI-1640 media
- Supplement RPMI-1640 with 25 mM HEPES, 1% penicillin-streptomycin, 1.5% Diflucan and 10% (v/v) heat inactivated FCS
- To heat-inactivate FCS, incubate FCS at 56 °C for 45 min
- Filter sterilize the RPMI-1640 medium using a 20 L pressure tank and Sartobran filter
Acknowledgments
The techniques described in this article were optimized during research work supported by grants from the CASS Foundation, Cancer Council of Victoria #807184 and the NHMRC #1025874 to PK; and an International HDR Ph.D. scholarship from ANU, Canberra to DS. We thank Prof Robin Anderson and Dr. Clare Slaney for valuable discussions and technical advice on BLI. ZP and PK are supported by funds awarded by the Faculty of Health Sciences, Curtin University. This protocol was adapted from a previous study from our laboratory published in Clinical cancer Research (Sinha et al., 2016).
Competing interests
The authors have no conflicts of interest or competing interests to declare.
Ethics
All animal experimentation was conducted with approval from the Peter MacCallum Animal Research Ethics Committee–AEEC (#E394 and #E504) within reference to guidelines of ethical experimentation on animals laid down by the National Health & Medical Research Council of Australia.
References
- Contag, C. H., Jenkins, D., Contag, P. R. and Negrin, R. S. (2000). Use of reporter genes for optical measurements of neoplastic disease in vivo. Neoplasia 2(1-2): 41-52.
- Day, C. P., Carter, J., Bonomi, C., Esposito, D., Crise, B., Ortiz-Conde, B., Hollingshead, M. and Merlino, G. (2009). Lentivirus-mediated bifunctional cell labeling for in vivo melanoma study. Pigment Cell Melanoma Res 22(3): 283-295.
- Rice, B. W., Cable, M. D. and Nelson, M. B. (2001). In vivo imaging of light-emitting probes. J Biomed Opt 6(4): 432-440.
- Sinha, D., Chong, L., George, J., Schluter, H., Monchgesang, S., Mills, S., Li, J., Parish, C., Bowtell, D., Kaur, P. and Australian Ovarian Cancer Study, G. (2016). Pericytes promote malignant ovarian cancer progression in mice and predict poor prognosis in serous ovarian cancer patients. Clin Cancer Res 22(7): 1813-1824.
- Weissleder, R. (2001). A clearer vision for in vivo imaging. Nat Biotechnol 19(4): 316-317.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Sinha, D., Pieterse, Z. and Kaur, P. (2018). Qualitative in vivo Bioluminescence Imaging. Bio-protocol 8(18): e3020. DOI: 10.21769/BioProtoc.3020.
Category
Cancer Biology > Invasion & metastasis > Animal models > Cell invasion
Stem Cell > Adult stem cell > Epithelial stem cell
Cell Biology > Cell engineering > Lentiviral delivery
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.