- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

High-throughput Microscopic Analysis of Salmonella Invasion of Host Cells


Published: Vol 8, Iss 18, Sep 20, 2018 DOI: 10.21769/BioProtoc.3017 Views: 7179
Reviewed by: Emily CopeVishal NehruAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2018
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Salmonella is a Gram-negative bacterium causing a gastro-enteric disease called salmonellosis. During the first phase of infection, Salmonella uses its flagella to swim near the surface of the epithelial cells and to target specific site of infection. In order to study the selection criteria that determine which host cells are targeted by the pathogen, and to analyze the relation between infecting Salmonella (i.e., cooperation or competition), we have established a high-throughput microscopic assay of HeLa cells sequentially infected with fluorescent bacteria. Using an automated pipeline of image analysis, we quantitatively characterized a multitude of parameters of infected and non-infected cells. Based on this, we established a predictive model that allowed us to identify those parameters involved in host cell vulnerability towards infection. We revealed that host cell vulnerability has two origins: a pathogen-induced cellular vulnerability emerging from Salmonella uptake and persisting at later stages of the infection process; and a host cell-inherent vulnerability linked with cell inherent attributes, such as local cell crowding, and cholesterol content. Our method forecasts the probability of Salmonella infection within monolayers of epithelial cells based on morphological or molecular host cell parameters. Here, we provide a detailed description of the workflow including the computer-based analysis pipeline. Our method has the potential to be applied to study other combinations of host-pathogen interactions.
Keywords: Salmonella enterica serovar TyphimuriumBackground
Salmonella enterica serovar Typhimurium infects its host via ingestion of contaminated food or water, causing salmonellosis. Once the bacterium reaches the distal ileum of the gut, they can invade a broad range of host cells, including the intestinal epithelial cells (Watson and Holden, 2010). During the first phase of host cell invasion, Salmonella chooses its targets, using its flagellum to swim and scan the surface of the epithelium (Misselwitz et al., 2012). After several stops on the surface of the cells, the bacterium eventually chooses a site where it docks (Misselwitz et al., 2011; Vonaesch et al., 2013) and triggers its uptake via the Type 3 Secretion System (T3SS) (Haraga et al., 2008; LaRock et al., 2015). The injection of bacterial effectors directly into the cytosol of the targeted cells leads to a strong remodeling of the actin cytoskeleton and the formation of membrane ruffles that engulf the pathogen (Haraga et al., 2008; LaRock et al., 2015). Within the infected cells, Salmonella remains either within a mature endomembrane compartment, the Salmonella-containing vacuole (SCV); or it reaches the cytosol by SCV membrane rupture to replicate at distinct paces within the different intracellular niches (Knodler, 2015; Fredlund et al., 2018).
Eukaryotic cell monocultures display an intrinsic cellular heterogeneity. Indeed, after seeding, cells have different characteristics with regards to their morphology and the local microenvironment, which correlates with differences of the transcriptome, proteome, and lipidome (Snijder et al., 2009; Frechin et al., 2015; Liberali et al., 2015). The selection criteria that determine which host cells are targeted by Salmonella for invasion are poorly understood. Previously, Misselwitz and colleagues proposed that Salmonella preferentially targets the topological obstacles it encountered while swimming near the cell monolayer, such as ruffles or mitotic cells (Misselwitz et al., 2012; Lorkowski et al., 2014). In contrast, Santos and colleagues suggested that cholesterol accumulation during cell metaphase is responsible for mitotic cell targeting (Santos et al., 2013). In a recent study, we investigated at the single-cell level the criteria exploited by Salmonella to select the cell to infect in a naturally heterogeneous monolayer of cells (Voznica et al., 2018).
In order to link the features of each cell with its probability to be infected, we have established a protocol of double infection of HeLa cells with fluorescent bacteria. We imaged the cells with high-throughput microscopy combined with automatic image analysis to obtain the individual features of hundreds of thousands of cells. We observed the distribution of those features in infected and non-infected cells and used a mathematical model to resolve the importance of different cellular parameters in deciphering Salmonella targeting.
This protocol provides a new tool to analyze pathogen targeting of cell features in a non-invasive manner and at the single-cell level. This opens a new path to decipher the cellular and bacterial factors involved in host cell vulnerability to infection.
Materials and Reagents
- Cell culture
- Falcon® 15 ml Polystyrene Centrifuge Tubes, Conical Bottom, with Dome Seal Screw Cap, Sterile (Corning, catalog number: 352095 )
- Counting chamber (KOVA® Glasstic Slide 10 with Grids) (Kova International, catalog number: 87144 )
- 96-well cell culture microplate with clear flat bottom (Greiner Bio One International, catalog number: 655090 )
- 75 cm2 tissue culture flasks with tilting neck and filter caps (TPP Techno Plastic Products, catalog number: 009076 )
- Human epithelial HeLa cells (ATCC, catalog number: CCL-2 )
- Dulbecco’s Modified Eagle’s Medium (DMEM) 1x, High Glucose, GlutaMaxTM (Thermo Fisher Scientific, catalog number: 10566016 ) supplemented with 10% (v/v) heat-inactivated Fetal Bovine Serum (Sigma-Aldrich, catalog number: F7524 )
- DPBS 1x (Thermo Fisher Scientific, catalog number: 14190144 )
- 0.05% Trypsin-EDTA 1x (Thermo Fisher Scientific, catalog number: 25300054 )
- Heat-inactivated Fetal Bovine Serum (Sigma-Aldrich, catalog number: F7524 ) (see Recipes for heat-inactivation)
- Bacteria culture
- Inoculating loops (SARSTEDT, catalog number: 86.1562.010 )
- Falcon® 14 ml round bottom tube with snap cap (Corning, catalog number: 352006 )
- Round Petri plate (Corning, GosselinTM, catalog number: BP93B-102 )
- Bacterial glycerol stock, stored at -80 °C (lab collection)
- SL1344 pM965, expressing GFP under the rpsM promoter. The strain was obtained after transformation of SL1344 with the pM965 plasmid described by Stecher and colleagues (Stecher et al., 2004). See Recipes for transformation.
- SL1344 pGG2, expressing dsRed under the rpsM promoter. The strain was obtained after transformation of SL1344 with the pGG2 plasmid described by Lelouard and colleagues (Lelouard et al., 2010). See Recipes for transformation.
- Electroporation Cuvettes (Bio-Rad Laboratories, catalog number: 1652086 )
- Ampicillin (Sigma-Aldrich, catalog number: A9393 ) (Keep the stock solution of 50 mg/ml at -20 °C)
- Tryptone (BD, catalog number: 211705 )
- Yeast Extract (BD, catalog number: 212750 )
- NaCl (Sigma-Aldrich, catalog number: 746398 )
- LB agar plate containing Ampicillin at 100 μg/ml (homemade, see Recipes)
- Lysogeny broth (LB) medium supplemented with 0.3 M NaCl (homemade, see Recipes)
- Infection
- 1.5 ml Eppendorf safe-lock tubes (Eppendorf, catalog number: 0030120086 )
- Semi-micro disposable cuvettes (BRAND, catalog number: 759105 )
- Parafilm® M sealing tape (Sigma-Aldrich, Parafilm, catalog number: P7543 )
- Pipetting reservoir, 25 ml (Thermo Fisher Scientific, catalog number: 10717964 )
- Filter tips, 200 µl (Sorenson Bioscience, catalog number: 035230 )
- Gentamicin solution (Sigma-Aldrich, catalog number: G1397 ) (Keep the stock solution of 50 mg/ml at -20 °C)
- NaCl (Sigma-Aldrich, catalog number: 746398 )
- KCl (Thermo Fisher Scientific, catalog number: 13305 )
- CaCl2 (Sigma-Aldrich, catalog number: 449709 )
- MgCl2 (Sigma-Aldrich, catalog number: M8266 )
- Glucose (Sigma-Aldrich, catalog number: G8270 )
- HEPES (Sigma-Aldrich, catalog number: H3375 )
- Heat-inactivated Fetal Bovine Serum (Sigma-Aldrich, catalog number: F7524 ) (see Recipes for heat-inactivation)
- 20x EM medium (homemade, see Recipes)
- 1x EM medium (see Recipes)
- Fixation
- Staining
Equipment
- Single-channel pipettes (Eppendorf, model: Research® plus )
- Multichannel Pipette, 30 to 300 μl (Thermo Fisher Scientific, catalog number: 4661030N )
- Pipet Filler (Thermo Fisher Scientific, catalog number: 9501 )
- Inverted Microscope (Motic, model: AE2000 )
- Laminar hood (Thermo Fisher Scientific, model: 1300 Series Class II Type A2 )
- Incubator set at 37 °C, 5% CO2 (Thermo Fisher Scientific, catalog number: NC0689918 )
- Centrifuge 5810/5810 R (Eppendorf, model: 5810/ 5810 R )
- Incubator with orbital shaker (INFORS, Multitron no series 112569-3)
- Water Bath (JULABO, model: TW12, catalog number: 9550112 )
- Thermomixer compact (Eppendorf, model: ThermoMixer® C )
- Photometer (Eppendorf, AG 22331)
- Centrifuge 5424/ 5424 R (Eppendorf, model: 5424/5424 R )
- Fume hood (SORBONNE ASPRIL/1000)
- Inverted widefield microscope equipped with a 20x/0.5NA air objective, an automatic programmable XY-stage, and a focusing system (Nikon), a CoolSnap2 camera (Roeper Scientific), a mercury lamp and the following filter cubes: DAPI (Excitation filter: 387/11, Emission filter: 447/60), FITC (Excitation filter: 482/35, Emission filter: 536/40), TRITC (Excitation filter: 543/22, Emission filter: 593/40) Cy5 (Excitation filter: 628/40, Emission filter: 692/40)
- 250 ml Erlenmeyer flask
- Electroporation Systems (Bio-Rad Laboratories, model: Gene Pulser XcellTM )
- Double boiler: 2 L beaker filled with 600 ml water
- Microwave
Software
- NIS-Elements Microscope Imaging Software (Nikon)
- Icy (Copyright 2011 Institut Pasteur, Icy is free software under the terms of the GNU General Public License, http://icy.bioimageanalysis.org)
- R (R is free software under the terms of the GNU General Public License, https://www.r-project.org/). The code available in the data analysis part has been written with R, version 3.4.2
- GraphPad Prism version 7.00 for Windows, GraphPad Software (La Jolla California USA, www.graphpad.com)
Procedure
Note: HeLa cells and bacteria should be handled under a sterile laminar BSL-II hood, unless otherwise mentioned. This protocol describes fluorophore-expressing Salmonella infection of HeLa cells but can be adapted for other cell lines and pathogens. In particular, non-fluorescent pathogens can also be labeled by immunofluorescence.
- Culture of bacteria 2 days before infection
- Take out a vial of glycerol stock of the bacteria SL1344-pGG2 (dsRed) and SL1344-pM965 (GFP) stored at -80 °C.
- Strike the bacteria on an LB agar plate containing the appropriate antibiotics (here ampicillin) to allow the formation of single colonies with both strains.
- Incubate the bacteria at 37 °C overnight.
- The next day, store the plate at 4 °C. The Petri dishes can be kept up to one week (Figure 1, see Note 1).
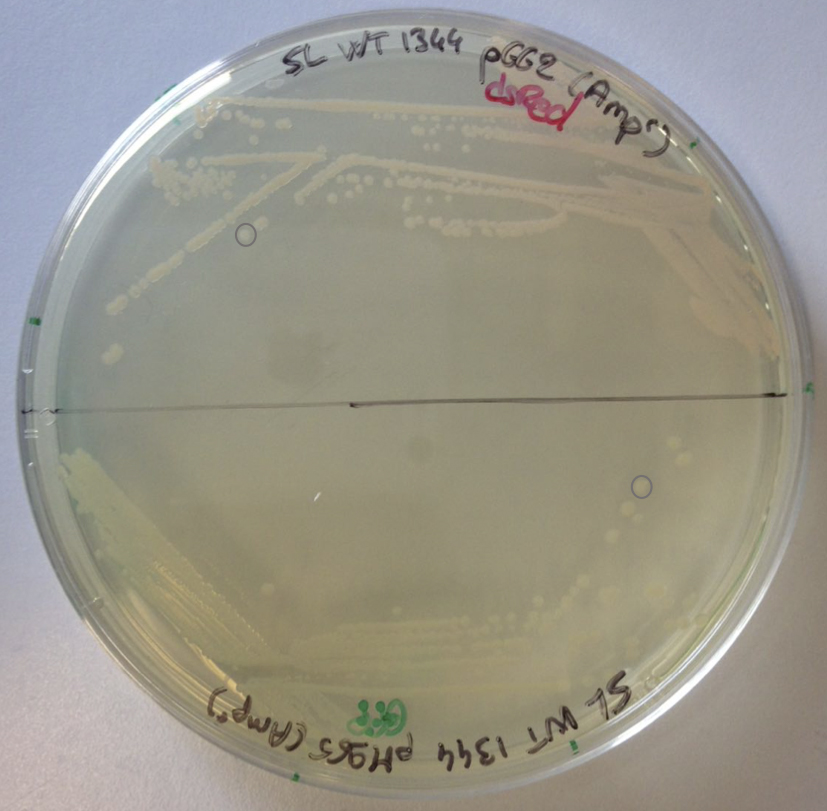
Figure 1. Bacterial culture on Petri dish. SL1344-pGG2 (dsRed) and SL1344-pM965 (GFP) are struck on a Petri dish containing the antibiotics (ampicillin). Individual colonies are circled in grey.
- Culture of bacteria 1 day before infection
- Fill two Falcon 14 ml polypropylene round-bottom tubes with 3 ml of LB medium supplemented with 0.3 M NaCl and ampicillin at 50 μg/ml.
- Pick 3 colonies of one strain from the Petri dish using the same inoculation loop and add them into a first tube. Repeat this step with the second strain in a second tube.
- Incubate at 37 °C under agitation (220 rpm) in a 45°-tilted rack overnight (see Note 2).
- Culture of cells 1 day before infection
- Use a water-bath to pre-warm the medium (trypsin, DMEM + 10% FBS, DPBS 1x) at 37 °C.
- Take a tissue culture flask of 75 cm2 containing HeLa cells at about 80% confluency based on microscopic observation of the cell density.
- Wash the cells with 5 ml of warm PBS 1x.
- Add 5 ml of warm 0.05% trypsin-EDTA and incubate the cells for 5 min in an incubator at 37 °C, 5% CO2.
- Detach the remaining attached cells by carefully hitting the flask on the side and moving the medium along the surface of the flask.
- Check the efficiency of the trypsin action under the microscope: the cells should be round and fully detached from the surface of the flask. Incubate longer if necessary.
- Stop the trypsin action by adding 5 ml DMEM + 10% FBS.
- Resuspend the cells and transfer all the content of the flask into a 15 ml Falcon® tube.
- Centrifuge the cells for 4 min at 200 x g using Centrifuge 5810/5810 R (see Note 3).
- Discard the supernatant.
- Resuspend the cell pellet in warm DMEM + 10% FBS medium.
- Transfer 15 μl of the cell suspension into a cell counter chamber.
- Count the cells in the cell counter chamber using a bench microscope and a cell counter.
- Calculate the initial cell concentration (Ci).
- Calculate the volume of cells needed (Vi) to obtain a final concentration (Cf) of 1.5 x 104 cells/ml in a final volume (Vf) of 2 ml. Use the following formula:
Vi = Cf x Vf/Ci - Dilute the cells in DMEM + 10% FBS medium in a new Falcon® tube.
- Take a 96-well cell culture microplate and label the plate map. In the following part of the protocol, we consider using 16 wells of the plate (see Note 4, Figure 2).
- Add 100 μl of the cell dilution per well.
- Add 100 μl of PBS (1x) in the wells surrounding the cells (see Note 4, Figure 2).
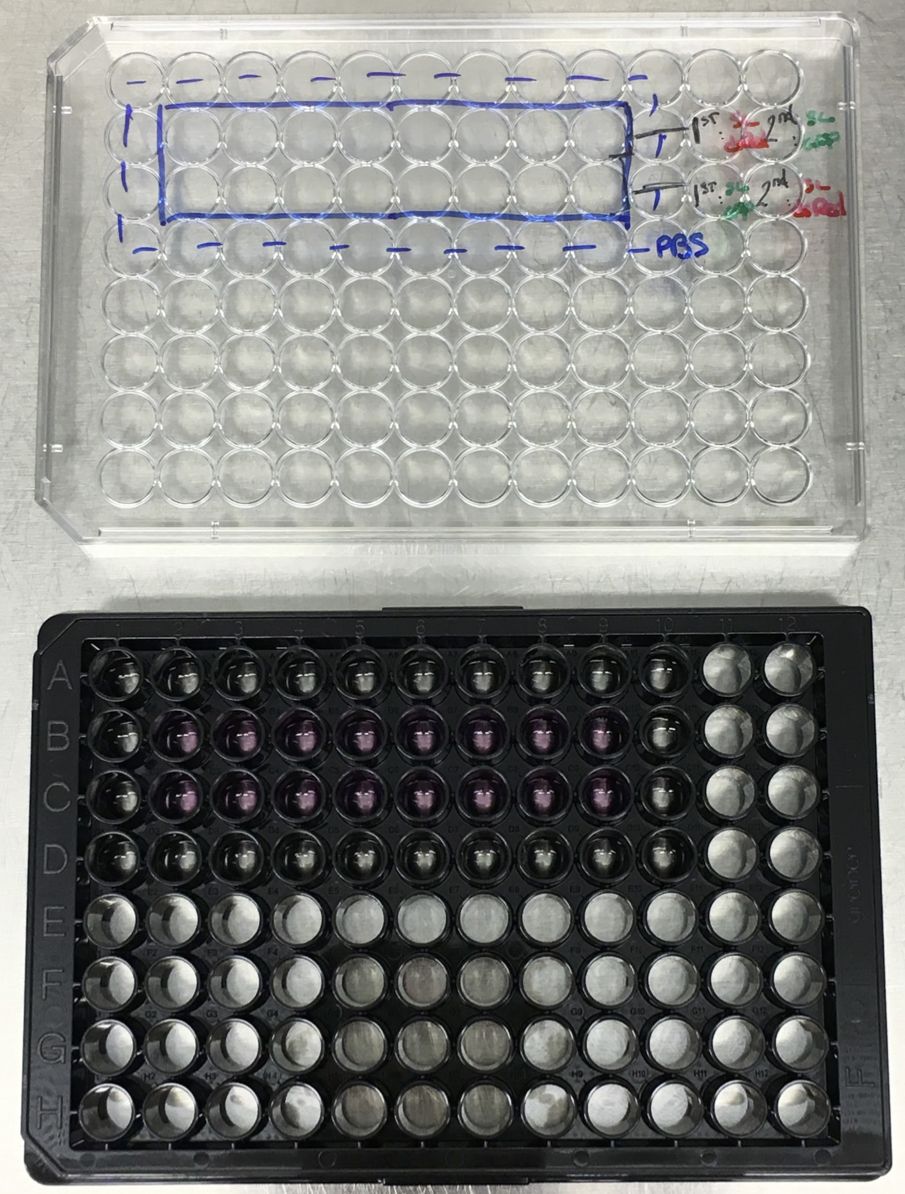
Figure 2. Cell culture in 96-well plate. The cells occupy the wells B2 to B9 and C2 to C9. The surrounding wells are filled with PBS 1x (see Note 4). - Keep the 96-well plate in the incubator at 37 °C, 5% CO2 for 24 h.
- Infection
- Make a subculture for each bacterial strain by diluting 150 μl of the overnight bacterial culture in 3 ml of LB medium supplemented with 0.3 M NaCl and 50 μg/ml ampicillin (dilution 1:21).
- Incubate the subcultures for 3 h in a 45°-tilted rack of an incubator at 37 °C with an orbital shaker at 220 rpm (see Note 5).
Important: Steps D1 and D2 should be repeated 3 h later to prepare the subculture necessary for the second infection. - Under a laminar hood, prepare 4 ml of “Gent100” solution containing EM medium supplemented with 10% FBS and gentamicin at 100 μg/ml.
Note: Mix 400 μl of FBS with 8 μl of gentamicin at 50 mg/ml and complete with 3,592 μl of EM. - Under a laminar hood, prepare 2 ml of “Gent10” solution containing EM medium supplemented with 10% FBS and gentamicin at 10 μg/ml.
Note: Mix 200 μl of FBS with 0.8 μl of gentamicin at 50 mg/ml and complete with 1,799.2 μl of EM. - Use the water-bath to prewarm the EM medium, the Gent100 and Gent10 solutions to 37 °C.
- Use the thermomixer to pre-warm four empty Eppendorf tubes at 37 °C.
- At the end of the 3 h of incubation, dilute 100 μl of the bacterial subculture in 400 μl of LB medium (volume final 500 μl, dilution 1:5), transfer the dilutions into two semi-micro disposable cuvettes, seal them with parafilm and measure the OD using the photometer. The OD of the dilutions should be around 0.4, corresponding to the late log phase of the bacterial growth.
- Prepare the HeLa cells for the infection by removing the DMEM + 10% FBS medium from the cell wells and add 100 μl of EM medium per well. Keep the plate at 37 °C, 5% CO2 until the infection.
- Transfer 500 μl of the two bacterial subculture from Step D2 into warmed one empty Eppendorf tube each.
- Centrifuge the Eppendorf tubes using Centrifuge 5424/5424 R for 1 min, at 7,600 x g.
- Discard the supernatant and resuspend the bacterial pellet in 500 μl warm EM medium.
- Centrifuge again the Eppendorf tubes (washing step) for 1 min, at 7,600 x g.
- Discard the supernatant and resuspend the bacterial pellet in 500 μl warm EM.
- Dilute 100 μl of the bacterial suspension in 400 μl of EM medium (final volume 500 μl, dilution 1:5). Transfer the dilutions into two semi-micro disposable cuvettes and measure the OD using the photometer.
- Calculate the dilution for a final multiplicity of infection (MOI) of 30, using the following calculation method:
- Wanted concentration of bacteria (Cf):
30 x (3.5 x 104 cells/well) = 1.05 x 106 bacteria/well
= 1.05 x 107 bacteria/ml - Initial volume to collect (Vi) for a final volume (Vf) of 0.5 ml:
Vi = (Cf x Vf)/Ci
Vi = (1.05 x 107 x 0.5)/(OD x 5 x 109)
- Wanted concentration of bacteria (Cf):
- Prepare the two dilutions in the two remaining empty warm Eppendorf tubes for SL1344-pGG2 (dsRed) and SL1344-pM965 (GFP) completing the volume with warm EM (see Note 6).
- Remove the medium from each well of the 96-well plate using the multichannel pipette.
- Add 50 μl of the bacterial dilution into each well according to the map of the plate (Figure 3).
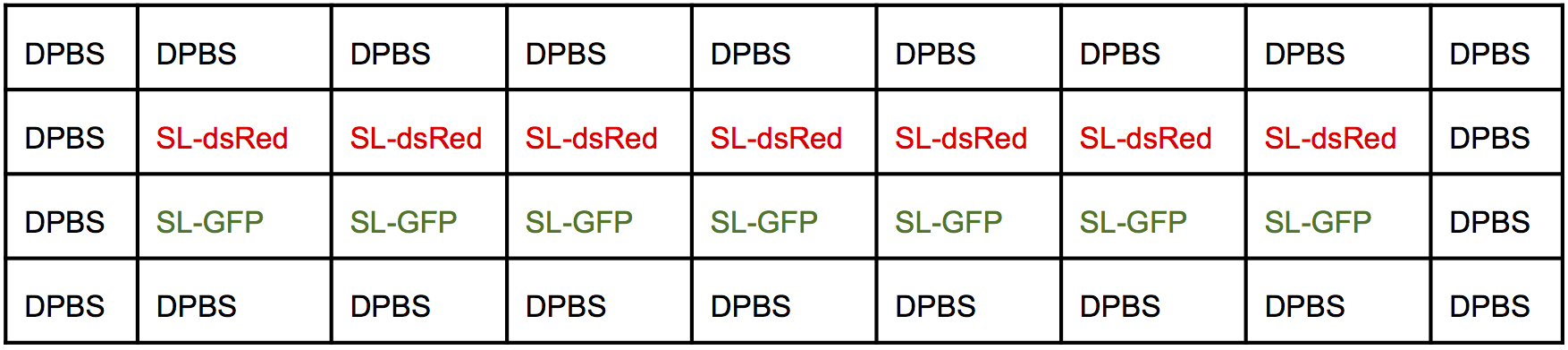
Figure 3. Infection map of a 96-well plate. First round of infection. - Incubate the 96-well plate for 30 min in an incubator at 37 °C, 5% CO2.
- Using the multichannel pipette and the pipetting reservoir, wash each well 3 times using 100 μl of warm EM each (Figure 4).
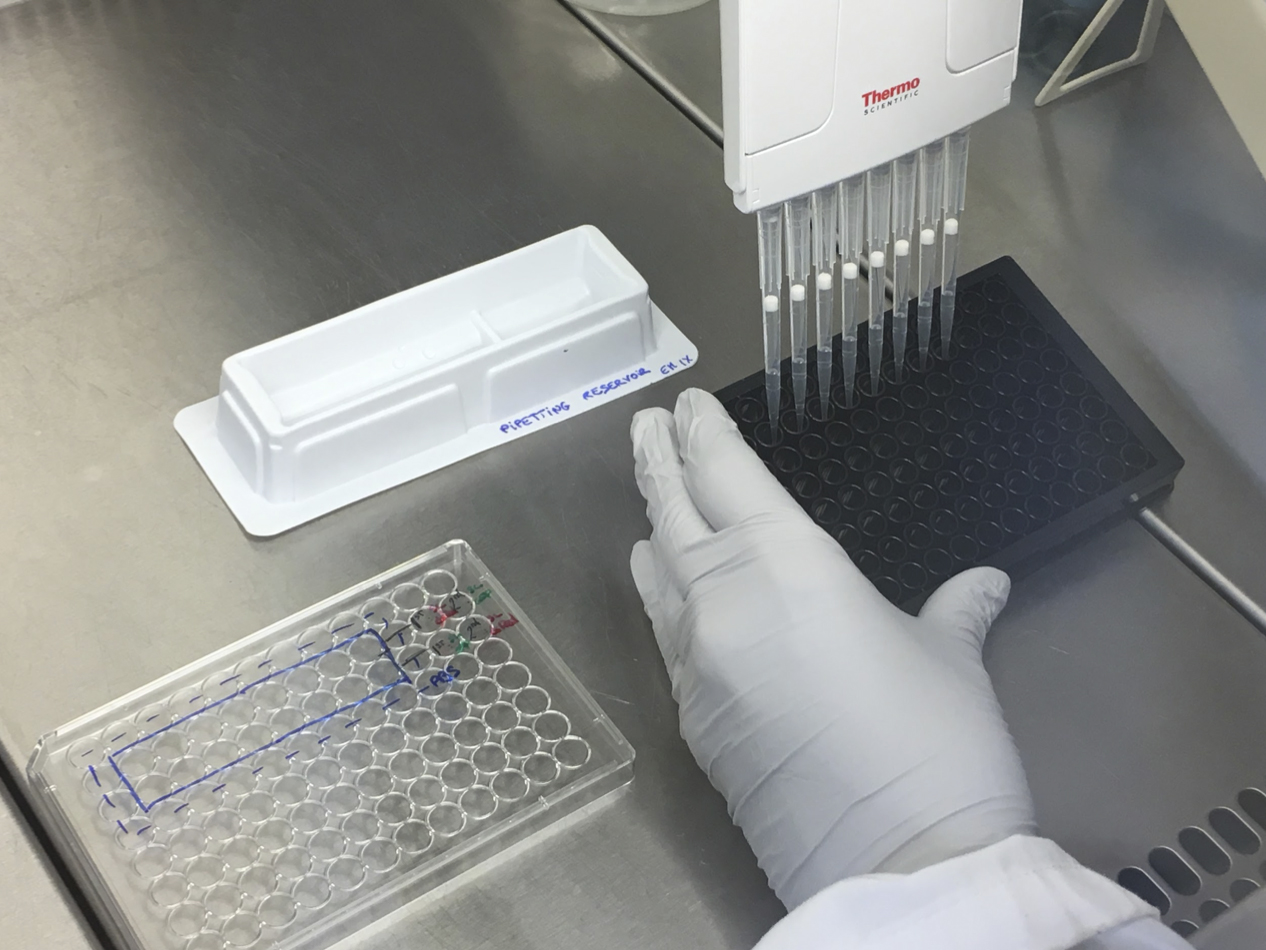
Figure 4. Washing step using the multichannel pipette and the pipetting reservoir - Change the medium to 100 μl of Gent100 solution in each well.
- Incubate the cells for 1 h in an incubator at 37 °C, 5% CO2.
- Change the medium to 100 μl of Gent10 solution in each well (see Note 7).
- Incubate the cells for 1 h in an incubator at 37 °C, 5% CO2.
- Wash the cells twice each with 100 μl of warm EM medium to remove the remaining gentamicin.
- Proceed to the second infection following the Steps D6 to D22 and using the second infection map (Figure 5).
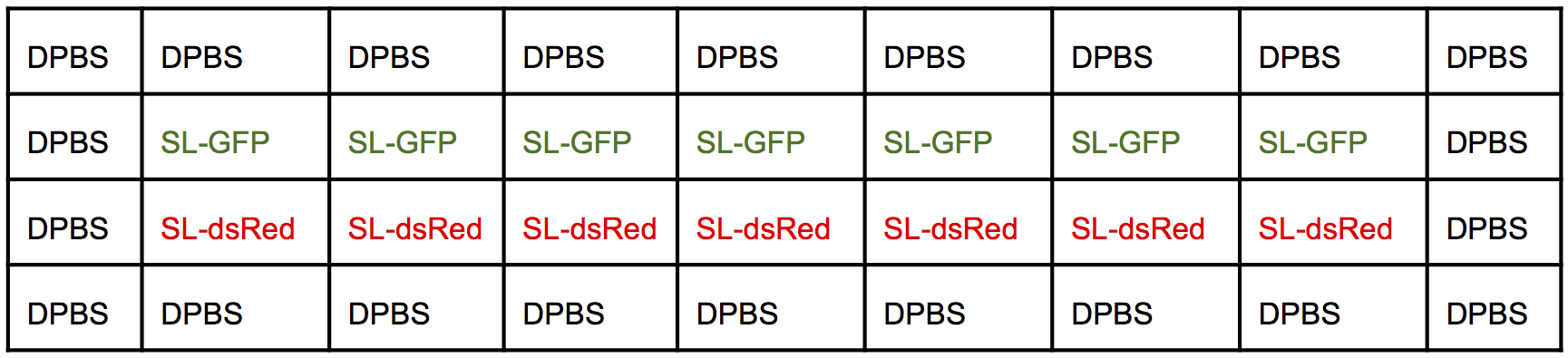
Figure 5. Infection map of a 96-well plate. Second round of infection.
- Fixation (under fume hood)
- Prepare 700 μl of 4% PFA by diluting the 16% PFA in DPBS 1x.
- Remove the medium from each well of the 96-well plate and add 40 μl of 4% PFA into each well.
- Incubate the plate for 15 min at room temperature (RT). From this step, protect the plate from light using aluminum foil.
- Remove the PFA and wash 3 times each with 100 μl of DPBS using the multichannel pipette and pipetting reservoir.
- Staining
- Dilute CellMask at 1:1,000 and DAPI at 1:1,000 in 1 ml of DPBS to make a “staining mix”.
- Add 40 μl of the staining mix per well and incubate for 20 min at RT.
- Wash the wells 3 times with 100 μl of DPBS using the multichannel pipette and the pipetting reservoir.
- Keep the plate at 4 °C with 100 μl of DPBS per well, protected from light (with an aluminum foil) until acquisition at the microscope (see Note 8).
- Automatic microscopic image acquisition
- Acquire the images of the 96-well plate using the Nikon inverted widefield microscope equipped with a 20x/0.5NA air objective, and the NIS software.
- Using JOBS Explorer, set an automatic acquisition pipeline such as presented in Figures 6 to 12:
- Give a name to the pipeline and select the type of storage as .nd2 (Figure 6).
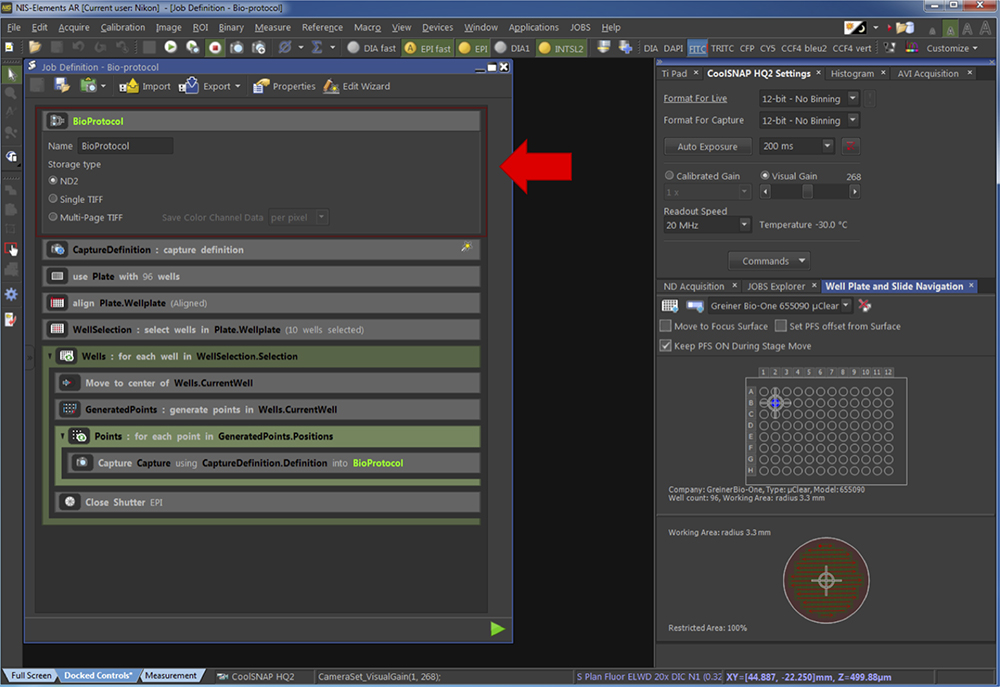
Figure 6. Creation of an automatic pipeline of acquisition. The red arrow points at the part of the pipeline related to the Step G2a. - Define the channel to capture (Figure 7). The time of exposure appears identical as in the “CoolSNAP HQ2 Settings” panel. It needs to be adapted for each experiment.
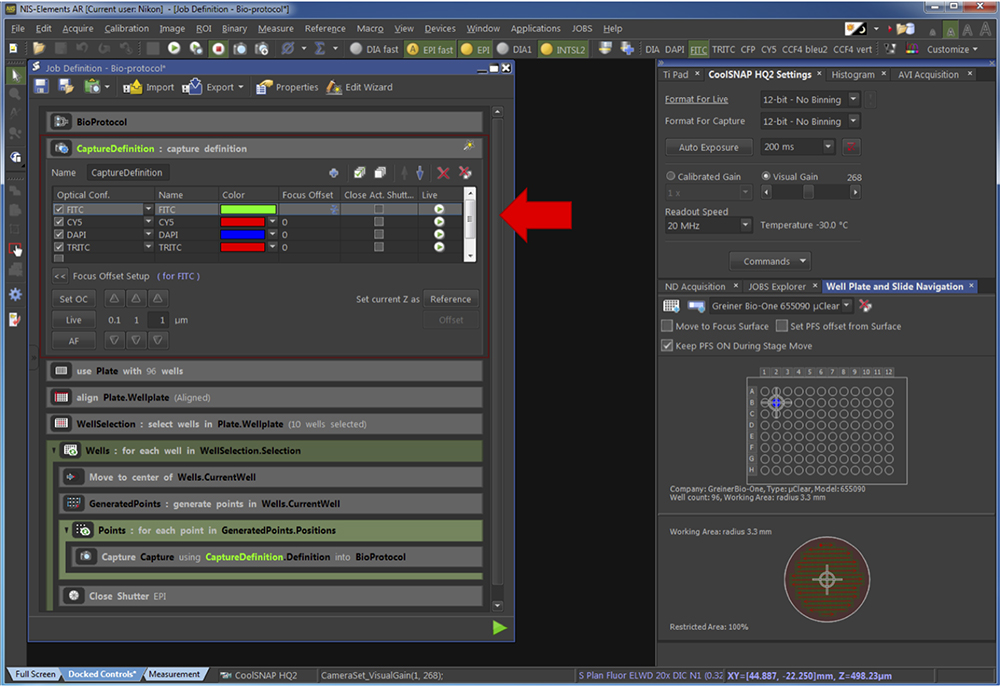
Figure 7. Definition of the parameters used for image capture. The red arrow points at the part of the pipeline related to the Step G2b. - Select the type of plate used by selecting it from a list of reference clicking on “Select from DB…” or by defining the plate characteristic by clicking on “Custom Well Plate” (Figure 8).
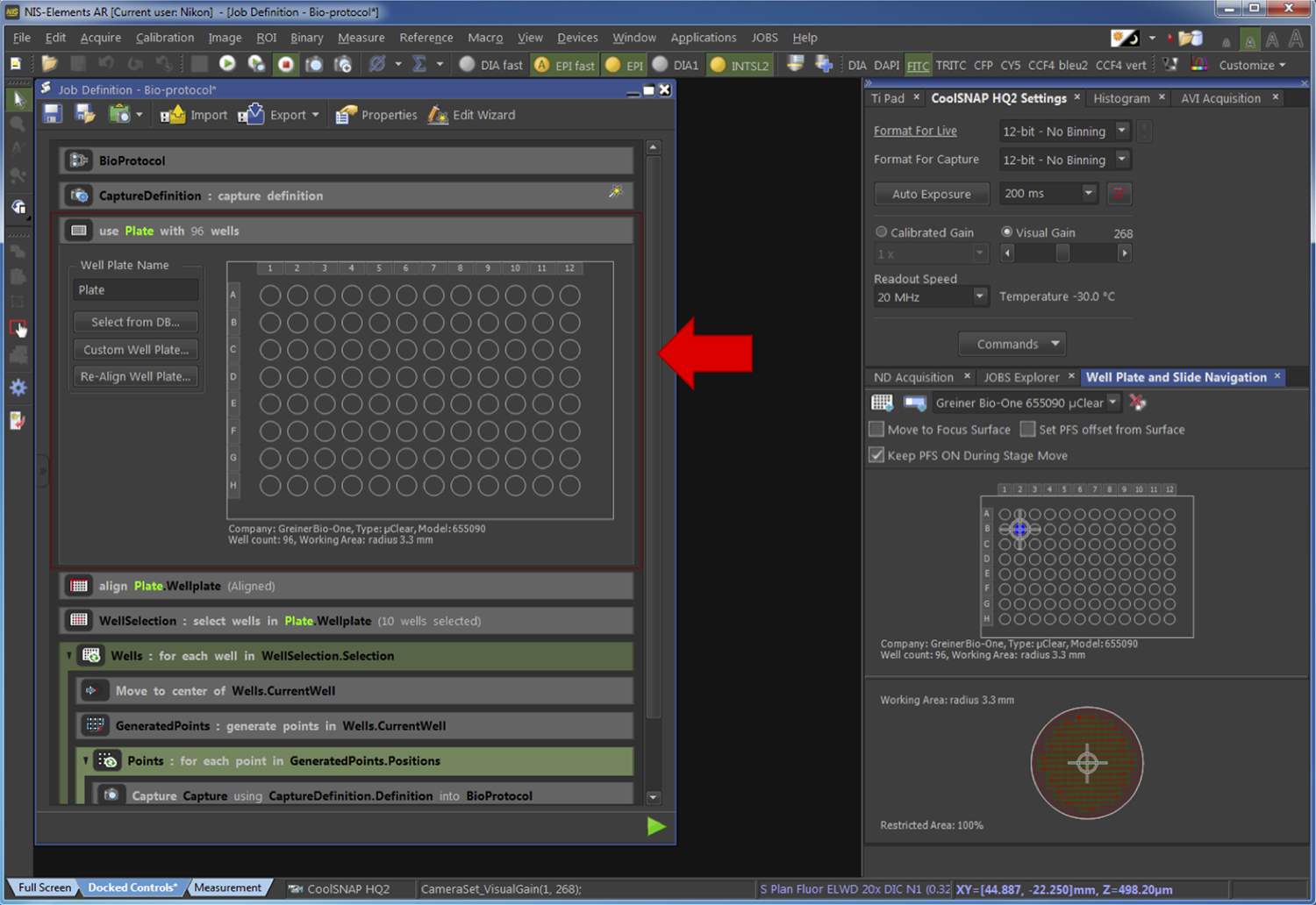
Figure 8. Selection of the type of plate used. The red arrow points at the part of the pipeline related to the Step G2c. - Align the plate by defining the border of 3 wells of the plate (Figure 9).
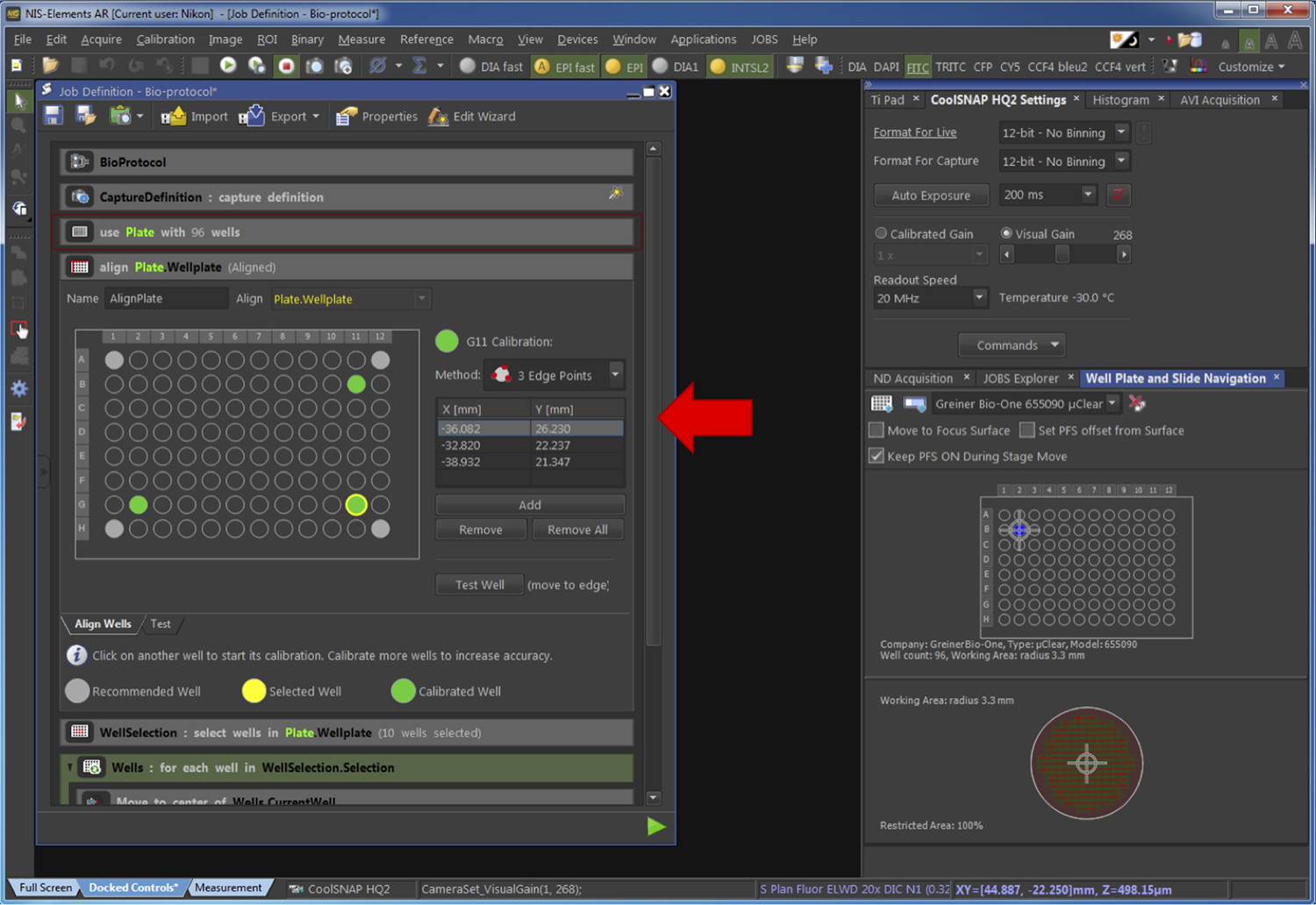
Figure 9. Alignment of the plate. The red arrow points at the part of the pipeline related to the Step G2d. - Select the wells of the plate to be acquired (Figure 10).
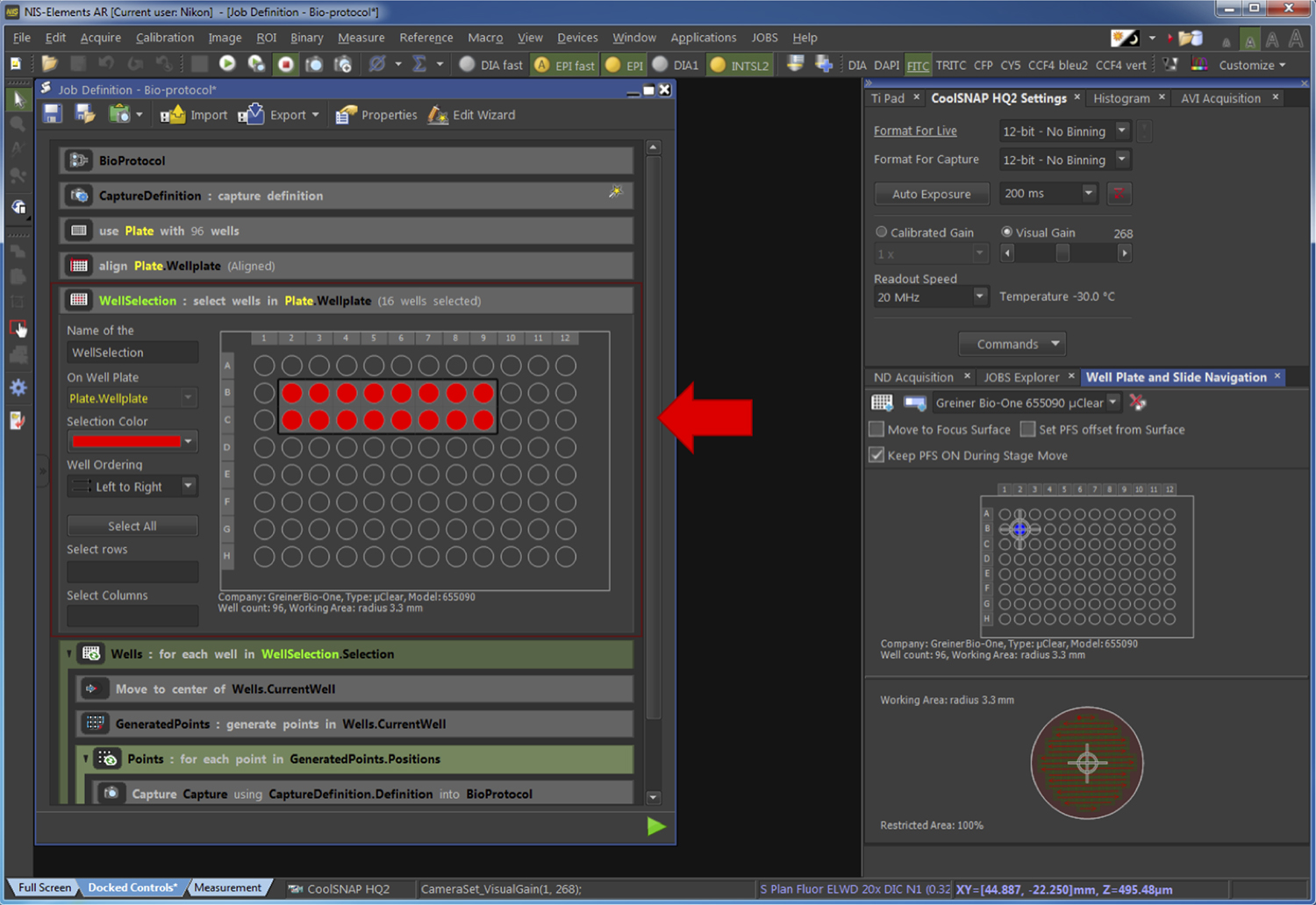
Figure 10. Selection of the wells to be acquired. The red arrow points at the part of the pipeline related to the Step G2e. - Generate the fields of view to be acquired (Figure 11).
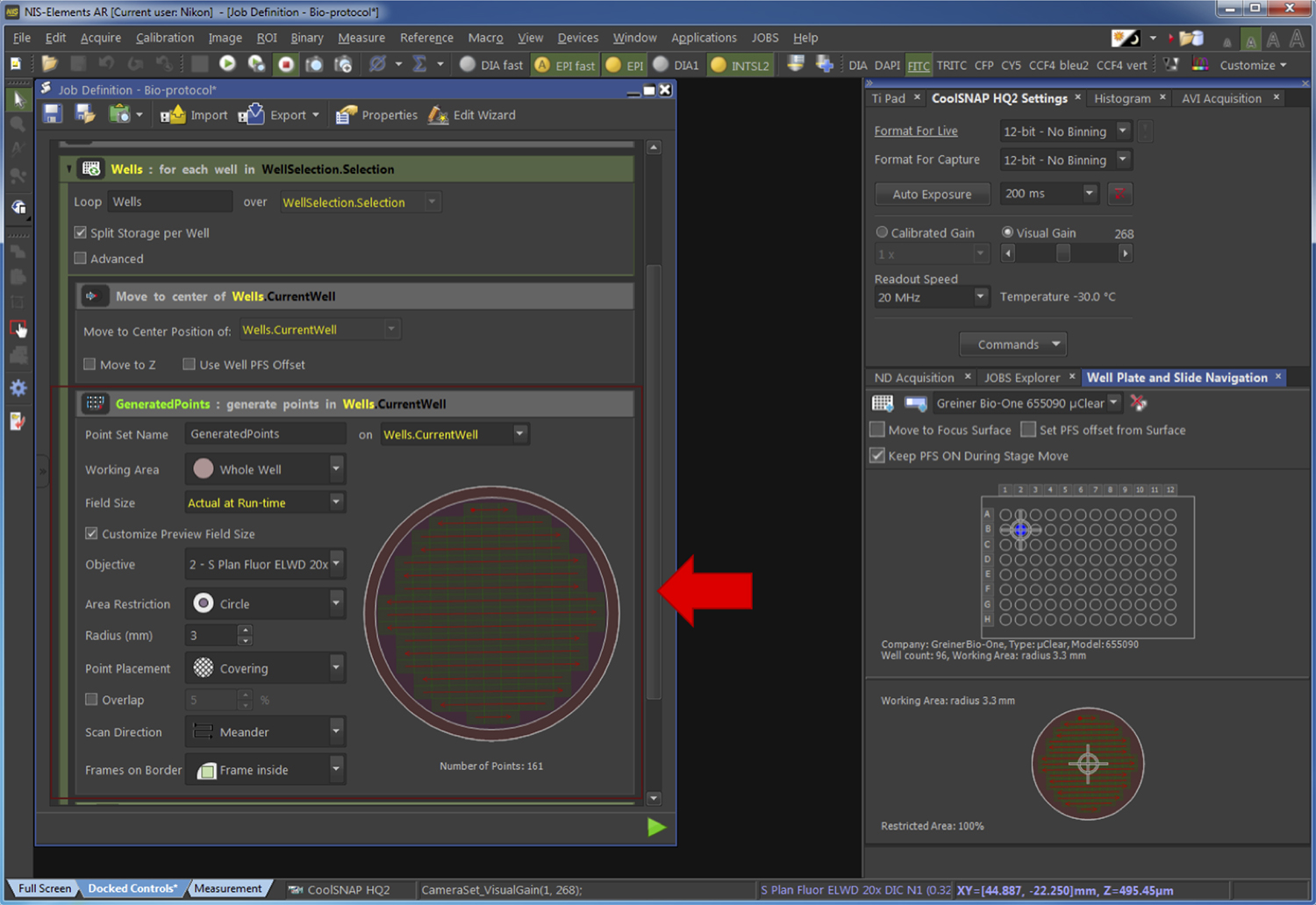
Figure 11. Generation of the fields to be acquired. The red arrow points at the part of the pipeline related to the Step G2f. - Select the parameters of image capture as presented in Figure 12.
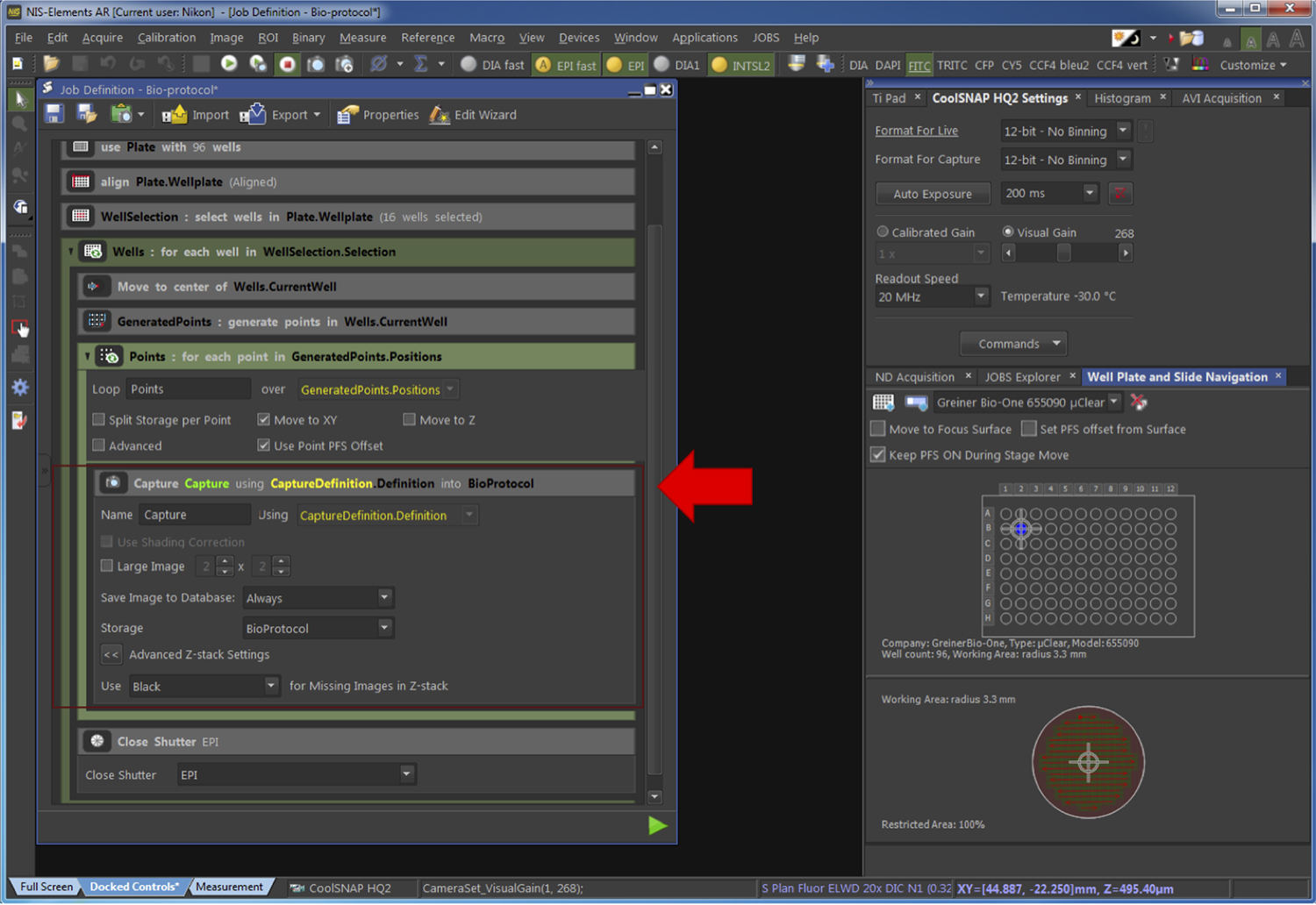
Figure 12. Definition of the capture process. The red arrow points at the part of the pipeline related to the Step G2g. - Run the acquisition. The time of acquisition varies depending on the number of wells and the number of positions per well to be acquired and commonly lasts for several hours. The files are saved automatically. An example of the obtained images is provided in Figure 1 of the associated publication (Voznica et al., 2018).
- Give a name to the pipeline and select the type of storage as .nd2 (Figure 6).
Data analysis
- Automatic image segmentation
Icy is open-source software for image processing developed by the BioImage Analysis team at Institut Pasteur (de Chaumont et al., 2012) and is available online (http://icy.bioimageanalysis.org). Its graphic interface, called “Protocols”, enables to create pipelines for image processing.
The integral pipeline described below can be downloaded here. To obtain precise results, all detection parameters need to be adjusted between experiments to balance potential variation between acquisitions (see Note 9). An example of the obtained image segmentation is provided in Figure S5 of the associated publication (Voznica et al., 2018).
Each processing step is represented as a “Block” that can be linked together (Figure 13). Each identified object is called “Region of Interest” (ROI). An output from one block, for instance a list of ROIs, is generally used as an input for another block (see Note 10).- General architecture of the pipeline and iterations
- Use “Folder” block to set the folder of origin that contains the image sets. Its output goes to “File batch” block.
- Use “File batch” block to execute the pipeline over all image sets contained in the original folder. “File batch” contains the blocks “Append file name” and “Sequence series batch.” “Sequence series batch” contains blocks for all following steps of analysis.
- Use “Append file name” block to store the name of the file. This is used for naming the output file.
- Use “Sequence series batch” to execute image processing on each acquired field of view, further called “image series”. Each image series contains 4 channels: the DAPI channel containing nucleus, the FITC channel containing GFP-Salmonella, the TRITC channel containing dsRed-Salmonella and the Cy5 channel containing HeLa cells labeled with CellMask.
- Use “Integer” block to store the number of the image series to be analyzed. This is used to identify cells from the same image.
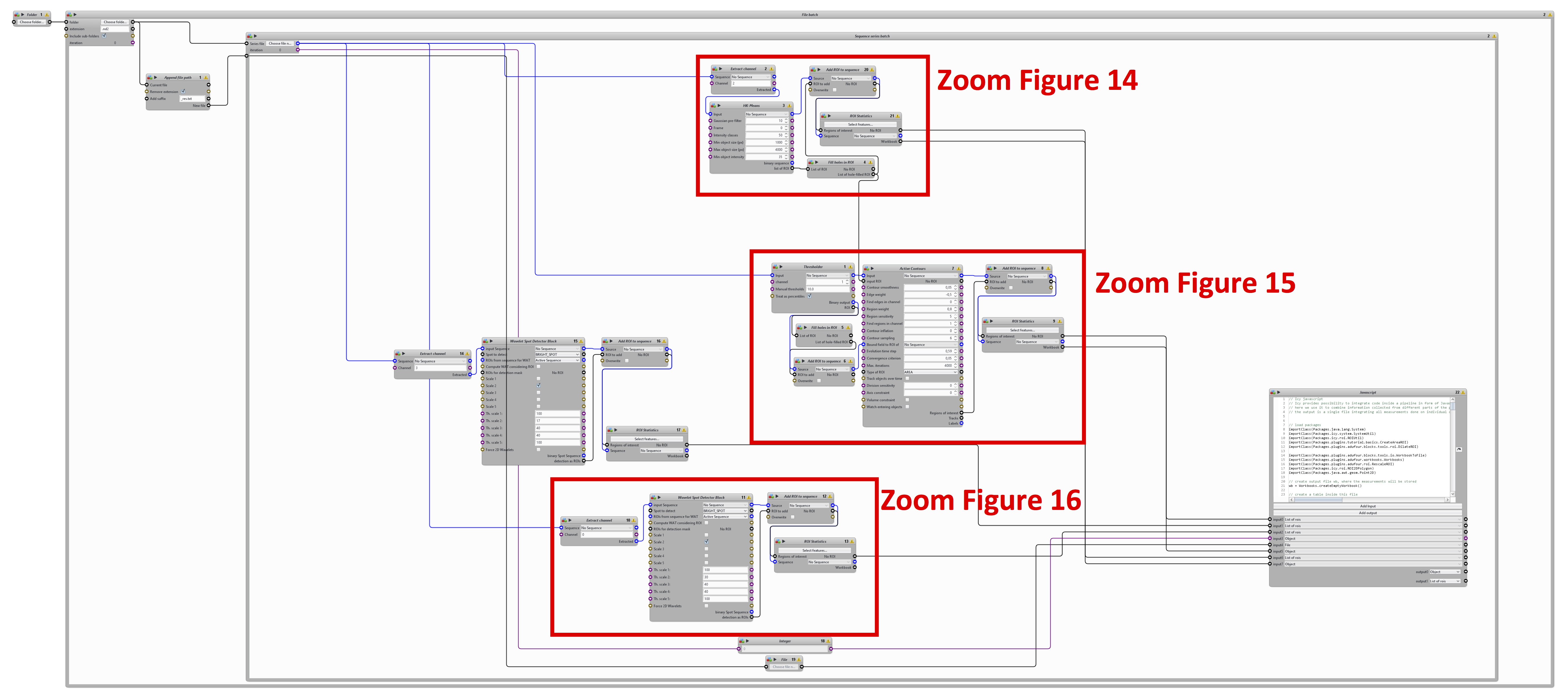
Figure 13. General architecture of the pipeline - Identification of individual nuclei from the DAPI channel (Figure 14)
- Use “Extract channel” block to extract the DAPI channel from image series. Its output goes to “HK-means” block.
- Use “HK-means” block to segment image into ROI (here nuclei) within a certain size range.
- Use “Fill holes in ROI” block to obtain complete nuclei without holes. The ROI output is used for the blocks “Add ROI to sequence” and “Active contours” that are further used to segment individual cells.
- Use “Add ROI to sequence” to convert newly created ROIs to a binary image. This binary image is used as the input of the block “ROI statistics”.
- Use “ROI statistics” to measure different features of these nuclei (XY-position, etc.).
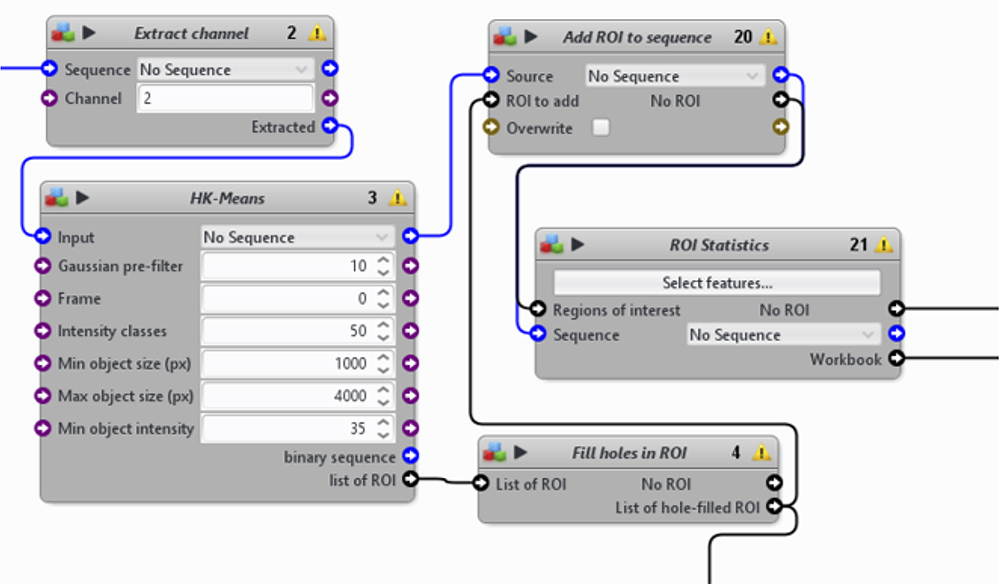
Figure 14. A first section from the automatic image-processing pipeline. From DAPI channel extraction to the measurements of nuclei statistics. - Identification of individual cells from the Cy5 channel (Figure 15)
- Use “Active Contours” block to segment individual cells. The algorithm uses the ROI previously detected for the nuclei as initial cell ROI, which is expanded until the detection of the cell border.
- The “Active Contours” block requires a binary image with a determined threshold. Use “Thresholder” block to create a filtered binary image from the Cy5 channel and to identify cellular ROIs. Complete the binary image with a “Fill Holes in ROI” and “Add ROI to sequence” blocks. This image is used as the input for the “Bound field to ROI” step of the “Active Contours” block.
- Adapt the different detection parameters to the image (see Note 11).
- Use the blocks “add ROI to sequence” and “ROI statistics” to measure different features of these cells (XY-position, size, shape, etc.).
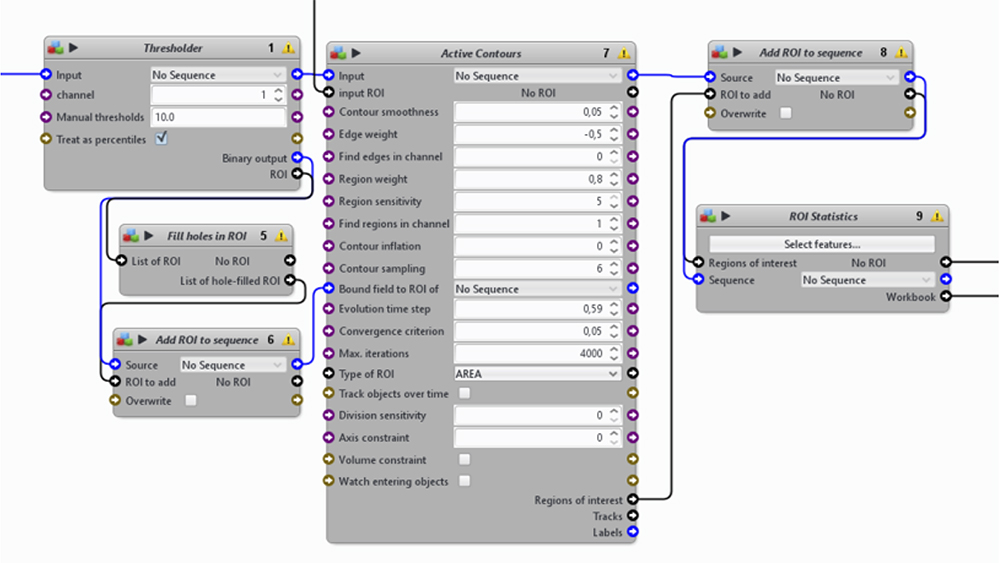
Figure 15. A second section from the automatic image-processing pipeline. From nuclei to single cells. - Identification of individual bacterium from either the FITC or TRITC channel to detect GFP- or dsRed-Salmonella, respectively (Figure 16).
- Use 2 “Extract channel” blocks to extract the FITC and TRITC channels from image series.
- Use 2 “Wavelet Spot Detector” blocks to identify individual bacterium within a certain size range.
- Use 2 “add ROI to sequence” and 2 “ROI statistics” blocks to measure different features of the GFP- or dsRed-Salmonella (XY-position, size, etc.).
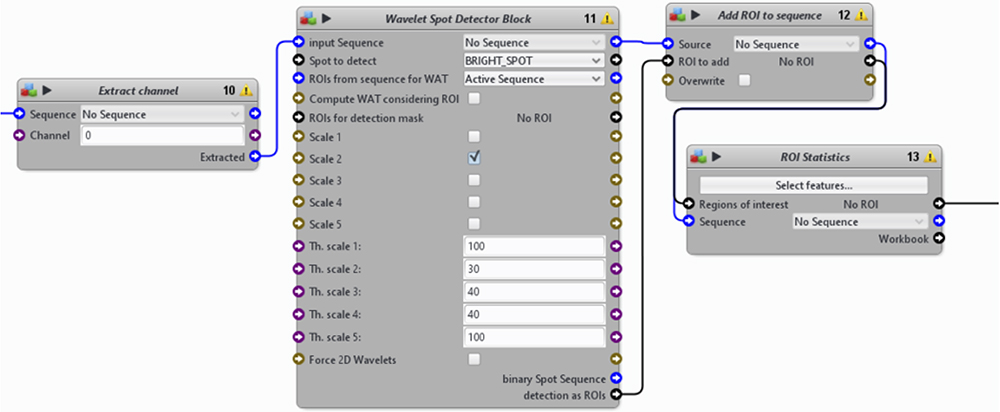
Figure 16. A third section from the automatic image-processing pipeline. From the FITC or TRITC channel extraction to the measurement of bacterial ROI. - More cellular features can be obtained by combining information from nuclei, cells, and bacteria. Icy provides the possibility to integrate a Javascript code to the pipeline inside a “Javascript” block, which enables the manipulation of ROIs in a less rigid manner.
- Use the “Javascript” block with the pre-integrated code from our pipeline to obtain the number of bacteria per cell and the number of neighboring cells (infected or not).
- The inputs of the block are the different lists of the detected ROIs, the measurements on these ROIs, the number of image series extracted from the “Integer” block, and the name of the image set file extracted from the “File” block.
- The output is a single spreadsheet (.txt) integrating all measurements done on individual cells from one experimental well.
- General architecture of the pipeline and iterations
- Statistical analysis of the cell feature using R code
- From Icy output files to the final R dataset
The data obtained with the automatic measurement of Icy are computed and analyzed with the open-source R software. All the lines in the grey boxes correspond to a code written in R language. These lines can be directly copied and pasted into the R software (R Core Team, 2013).
The following part describes how an R dataset is prepared from the output file obtained with Icy. Each line of the table corresponds to one cell, referred latter as “cell of interest,” and each column corresponds to the value of one of the measured variables.
Such a dataset contains all the variables measured from the analysis of images of one experimental well, plus some more elaborated variables obtained from the raw variables.
The following step (a) describes how the data are loaded from the variables measured with Icy. The step (b) describes how the cells on the border of the image are filtered. The steps (c) and (d) describe how more elaborated variables are added as new columns of the dataset. The step (e) describes how the final variables used for the modeling are selected. The step (f) describes how the data from each well are combined to create the final dataset that will be used in the modeling.- Load one of the .txt output files from the Icy analysis and check the number of variables.
#change working directory to the directory containing Icy output files
setwd(dir="path/to/data_set/")
#load a dataset obtained from one well and store it in the table p1
p1 <- read.table("....txt", header=TRUE, skip=1, na.strings=c("”))
#check the number of measured variables (columns) for each cell (the number is 45)
ncol(p1) - Add more variables to be used in the model such as square root of circularity, local cell density, and non-infected neighboring cells. Then, add manually the variable “delay”, which is not included in the Icy output.
#add new columns to the table p1: they are used to store variables (square root of the circularity: "sqrt-circ", number of cells within 100 μm of the given cell: "Cells_100_microns", number of non-infected neighboring cells: "neighbor_non_inf1", delay between the first and the second infection: "delay")
p1 <- cbind(p1, 0, 0, 0, 0)
names(p1)[46:49] <- c("sqrt_circ", "Cells_100_microns", "neighbor_non_inf1", "delay")
#compute the square root of circularity for each cell using the formula: sqrt(4 x Cell Area / Cell Perimeter^2). The measured cell area is stored in the variable called “interior“ and the cell perimeter is stored in the variable called “contour“. The result is stored in the “sqrt_circ” column.
for (i in 1:nrow(p1)) {
p1$sqrt_circ[i] <- sqrt(4 * pi * p1$interior[i] / (p1$contour[i] * * 2))
}
#compute the local cell density as the number of surrounding cells within the vicinity of 100 μm of the cell of interest. The result is stored it in the “Cells_100_microns” column.
for (i in 1:nrow(p1)) { #i is the index of the cell of interest
#set the number of cells within 100 μm to minus 1: the cell of interest is within its own 100 μm vicinity
p1$Cells_100_microns[i]<- -1
#subselect all the cells from the same image and store the data in a matrix t
t <- p1[(p1$img == p1$img[i]), ]
for (j in 1:nrow(t)) { #j is the index of a cell in the same image
#check for each cell of the image whether it is within the vicinity of 100 μm (i.e. 312.5 pixels in our conditions) from the cell of interest by using the X and Y coordinates.
if (sqrt((p1$X[i] - t$X[j]) ^ 2 + (p1$Y[i] - t$Y[j]) ^ 2) < 312.5) {
#add one to the counter if this is true
p1$Cells_100_microns[i] <- p1$Cells_100_microns[i] + 1
}
}
}
#compute the number of non-infected neighboring cells: it is the difference between the number of neighboring cells (stored in the column “bystanders”) and the number of infected neighboring cells (stored in the column “neighborinf1”). The result is stored it in the “neighbor_non_inf1” column.
p1$neighbor_non_inf1 <- as.numeric(as.character(p1$bystanders)) – as.numeric(as.character(p1$neighborinf1))
#convert the variable “neighborinf1” to numerical value
p1$neighborinf1 <- as.numeric(as.character(p1$neighborinf1))
#set the delay between the first and the second infection for this experiment (1 for 1 hour, 2 for 2 hours and 3 for 3 hours). The result is stored it in the “delay” column.
p1$delay <- 1 #if the value here is 1 - Cells on the border of images present a bias as not all the variables can be measured accurately for these cells. Filter them.
###After taking into account cells on the border of image for the local cell density, remove them from the dataset to reduce bias
#use the estimation of dsRed expressing bacteria (stored in the 7th column): javascript in Icy sets this variable to ‘NA’ when the cell of interest is in contact with the border of the image
p1 <- p1[which(p1[, 7] != 'NA'), ] - Add variables linked with the infection. Using estimates of the number of bacteria obtained in Icy, create variables representing the intensity of the first infection and the presence of the second one.
###add variables indicating the intensity of the first infection (low intensity: "low_first", intermediate intensity: "mid_first", and high intensity: "high_first")
#load the package plyr containing function mutate that enables to create new columns by combining existing columns
require("plyr") (Wickham, 2011).
###estimate the number of intracellular bacteria in individual cells
#Icy output contains the number of pixels at high intensity in FITC and TRITC channels (green_surface and red_surface, respectively). Divide this variable by an average number of pixels corresponding to one bacteria. Here 25 pixels correspond to one bacteria.
p1$green_est <- as.numeric(as.character(p1$green_surface))/25
p1$red_est <- as.numeric(as.character(p1$red_surface))/25
#set the three variables to 0 if the estimated number of total GFP expressing bacteria is lower than 0.7; this correspond to noise on the original image and not to bacteria.
#set the variable "low_first" to 1 if the estimated number of total GFP expressing bacteria is between 0.7 and 2.5.
#set the variable "mid_first" to 1 if the estimated number of total GFP expressing bacteria is between 2.5 and 8.5.
#set the variable "high_first" to 1 if the estimated number of total GFP expressing bacteria is higher than 8.5.
p1 <- cbind(mutate(p1, low_first=as.numeric(p1$green_est > 0.7 & p1$green_est < 2.5), mid_first=as.numeric(p1$green_est > 2.5 & p1$green_est < 8.5), high_first=as.numeric(p1$green_est > 8.5)))
###add a variable indicating the presence of the second infection
#set the variable "second_inf_bool" to 1 if the estimated number of total dsRed expressing bacteria is higher than 0.7
p1 <- cbind(mutate(p1, second_inf_bool=as.numeric(p1$red_est > 0.7)))
###save complete datasets for each well
save(p1, "complete_p1.Rdata") - The table now contains over 50 variables. Select the variables to be used in the model (this can be adapted depending on the purpose of the analysis). Here, we select only 10 variables as described below:
- second_inf_bool: boolean variable that takes the value 1 if the cell of interest contains bacteria from the second wave of infection, and 0 otherwise.
- low_first: boolean variable that takes the value 1 if the number of bacteria from the second infection is between 1 and 2, and 0 otherwise.
- med_first: boolean variable that takes the value 1 if the number of bacteria from the second infection is between 3 and 8, and 0 otherwise.
- high_first: boolean variable that takes the value 1 if the number of bacteria from the second infection is 9 or more, and 0 otherwise.
- cell_perim: continuous variable that takes the value of the perimeter of the cell of interest.
- sqrt_circularity: continuous variable that takes the value of the square root of the circularity of the cell of interest.
- neighbor_inf1: discrete variable that takes the value of the number of neighboring cells that were infected during the first wave of infection.
- neighbor_non_inf1: discrete variable that takes the value of the number of neighboring cells that were not infected during the first wave of infection.
- Cells_100_microns: discrete variable that takes the value of the number of cells within 100 μm of the cell of interest
- delay: categorical variable that takes the value of the delay (in hour) between the first and the second wave of infection.
##select and save the variables to be used in the model.
p1 <- cbind(p1$second_inf_bool, p1$low_first, p1$mid_first, p1$high_first, p1$contour, p1$sqrt_circ, p1$neighborinf1, p1$neighbor_non_inf1, p1$Cells_100_microns, p1$delay)
#rename columns
colnames(p1) <- c("second_inf_bool”, “low_first”, “mid_first”, “high_first”, “cell_perim”, “sqrt_circ”, “neighborinf1”, “neighbor_non_inf1”, “Cells_100_microns”, “delay”)
#convert p1 to data frame, a data structure in R
p1 <- as.data.frame(p1)
#convert the perimeter from pixels to micrometers
p1$cell_perim <- p1$cell_perim / 3.125
#save the dataset with selected variables
save(p1, "subselect_p1.Rdata") - Combine the datasets acquired from single wells to create the final dataset used for further analysis.
###combine datasets
#load the datasets (p1, p2, …, px) with selected variables from individual wells
load("subselect_p1.Rdata") #load p1 dataset
load("subselect_p2.Rdata") #load p2 dataset
load("subselect_px.Rdata") #load px dataset
#create the final dataset
final_dataset <- rbind(p1, p2, ..., px)
#save the final dataset
save(final_dataset, "final_dataset.Rdata")
- Load one of the .txt output files from the Icy analysis and check the number of variables.
- Visualizing the distribution of variables in infected and non-infected cells
Build plots comparing the distribution of a variable for infected and non-infected cells. These plots correspond to Figure 5A of our published article (Voznica et al., 2018). Here, we use the variable of infected neighboring cells (“neighinf1”) as an example, but the script is adaptable to other variables.- Load the complete dataset containing the data from all wells.
#change the working directory to the folder containing the data file
setwd(dir="path/to/data_file/")
#load the data set and stock it in the matrix GthenR– the name indicate that the infection was performed with GFP (Green) bacteria first, and then dsRed (Red) bacteria.
load('final_dataset.Rdata') #load dataset called final_dataset
GthenR <- final_dataset #Rename it to GthenR - Separate the data into two groups. The first group corresponds to the cells infected during the second infection, called “infected cells”. The second group corresponds to the cells not infected during the second infection, called “non-infected cells”.
###prepare a vector containing the number of infected neighboring cells of infected cells
#subselect all the data of infected cells
Inf2 <- as.data.frame(GthenR[GthenR$second_inf_bool == 1, ])
#subselect the data on the number of infected neighboring cells
Inf2 <- Inf2$neighborinf1
Inf2 <- unlist(Inf2)
#filter extreme values for the purpose of plotting: here we take out all observations with more than 11 neighboring cells, representing less than 1% of the dataset. This number should be adapted for each variable.
Inf2 <- Inf2[Inf2 < 11]
###follow the same procedure for the number of infected neighboring cells of non-infected cells
#subselect all the data of non-infected cells
NonInf2 <- as.data.frame(GthenR[GthenR$second_inf_bool == 0, ])
#subselect the data on the number of infected neighboring cells
NonInf2 <- NonInf2$neighborinf1
NonInf2 <- unlist(NonInf2)
#filter extreme values for the purpose of plotting, similarly to the infected cells.
NonInf2 <- NonInf2[NonInf2 < 11] - Prepare the histograms of the distribution of infected neighboring cells in infected and non-infected cells. The final plot is a density plot summing to 100%.
#create the first histogram of infected neighboring cells with the data from infected cells; stock this histogram in the variable h_inei_inf
h_inei_inf <- hist(Inf2, breaks=seq(0, 10, length.out=12), freq=TRUE)
#create the second histogram with the data from non-infected cells; stock it in the variable h_inei_noninf
h_inei_noninf <- hist(NonInf2, breaks=seq(0, 10, length.out=12), freq=TRUE)
#transform the two histograms into density plot: divide the number of observations of each value by the total number of observations
h$density = h$counts / sum(h$counts) * 100
addin$density = addin$counts / sum(addin$counts) * 100
#the columns now sum to 100% - Plot the distribution to obtain a figure similar to Figure 17.
#plot the distribution of infected neighboring cells of infected cells
#the attribute “col” defines the color of the distribution. Here the infected cells are in red. The forth parameter of “col” defines the color transparency. Here it is set to 50% so that overlapping distributions are visible.
plot(h, freq=FALSE, col=rgb(1, 0, 0, 0.5), main="Infected Neighbors distribution", xlab="Number of Neighbors", ylab="% cells", ylim=c(0, 60))
#add the distribution of infected neighboring cells of non-infected cells. Here the non-infected cells are in blue.
plot(addin, freq=FALSE, col=rgb(0, 0, 1, 0.5), add=TRUE)
#add a legend to the plot
legend("topright" , inset=.05, c("Infected Cells", "Non Infected Cells"), pch=c(22, 22), col=c(rgb(1, 0, 0), rgb(0, 0, 1)), bty="n", pt.bg=c(rgb(1, 0, 0), rgb(0, 0, 1)))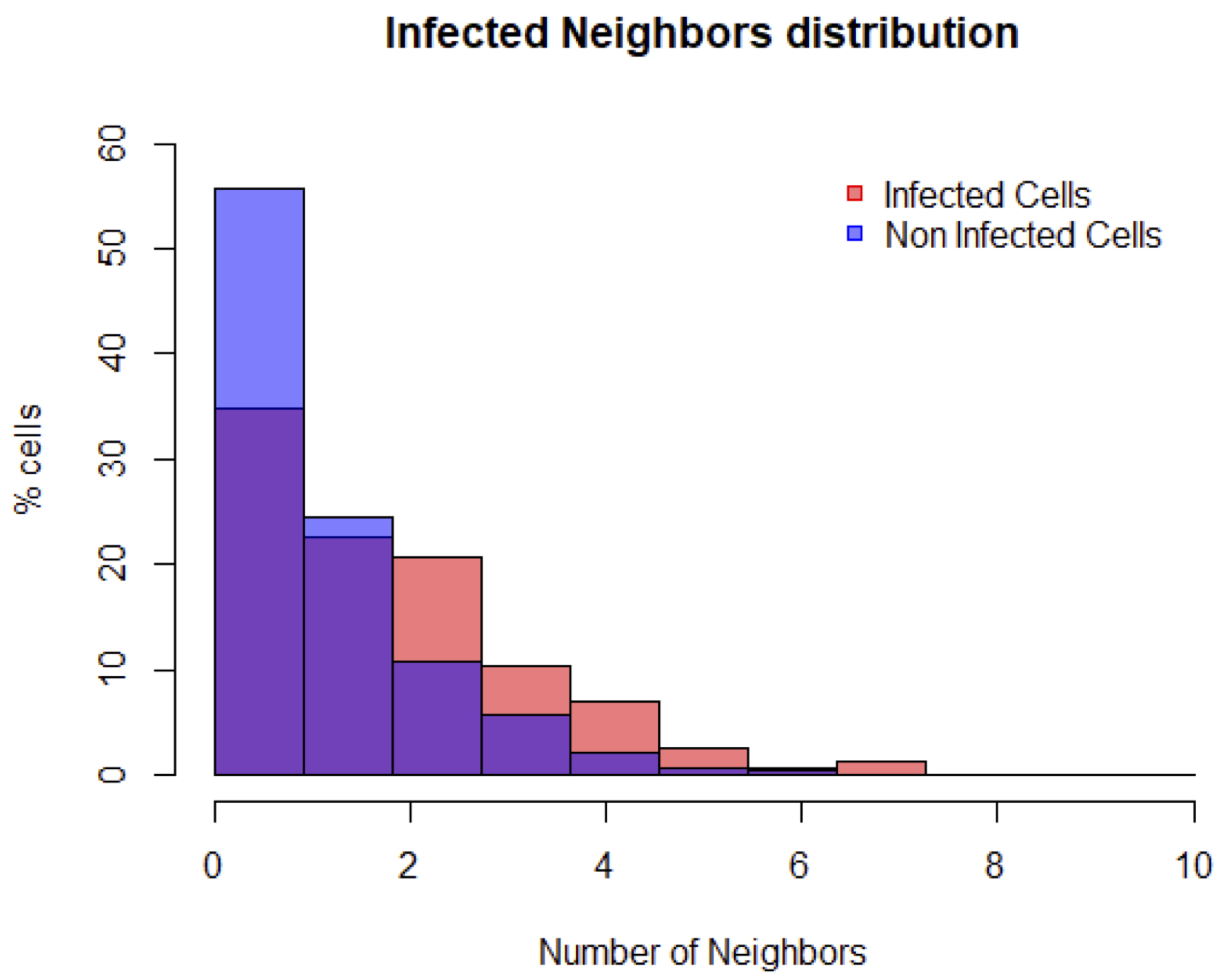
Figure 17. Visualization of the distribution of the number of infected neighboring cells for infected cells (in red) and non-infected cells (in blue)
- Load the complete dataset containing the data from all wells.
- Testing the involvement of cell features in the cell vulnerability to Salmonella infection
In order to test the involvement of different cell features in the cell vulnerability to Salmonella infection, build a trained model to predict the infected cells from their features, and test the accuracy (i.e., likelihood) of such model. Then, test a new model missing one of the parameters. By subtracting the likelihood between both models, quantify the improvement of the infection prediction and conclude on the involvement of the cell feature studied in the cell vulnerability. These quantifications correspond to Figure 5B of our published article (Voznica et al., 2018).- Load the complete dataset (see Note 12), and convert 2 of the variables to categorical variables.
#if necessary, reload the data set and stock it in the matrix GthenR (similarly to the step 2.a)
load('final_dataset.Rdata') #load dataset called final_dataset
GthenR <- final_dataset #Rename it to GthenR
#convert second_inf_bool and delay to categorical variables so that the model does not interpret their numerical value, but consider its value as an assignment to a group
GthenR$second_inf_bool <- as.factor(GthenR$second_inf_bool)
GthenR$delay <- as.factor(GthenR$delay) - Partition the data into training and testing datasets. Create two tables containing indexes for data partitioning. One table for the training dataset containing 9/10th of the total data, the other one for the testing dataset containing 1/10th of the total data. The repartition of the data between the training and the testing dataset is done with 100 different cuts.
#create a variable containing the number observations
observation_num <- nrow(GthenR)
#create a variable containing the number of observations used for training the model
train_num <- floor(observation_num * 0.9)
#create a variable containing the number of observations used for testing the model
test_num <- observation_num - train_num
#create a matrix that contains 100 sets of indexes of individual observations constituting the training set (ranged in rows)
samptrain <- matrix(nrow=100, ncol=train_num)
#create a matrix that contains 100 sets of indexes of individual observations constituting the testing set (ranged in rows)
samptest <- matrix(nrow=100, ncol=test_num)
#data partitioning: fill the rows with indexes for future data partition
for (i in 1:100) {
#create a vector containing all indexes of the data in a random order
indexes_random <- sample(observation_num)
#fill the i-th column of training set (“samptrain”) with the first “train_num” indexes. They are used to create the i-th training dataset
samptrain[i, ] <- indexes_random[1:train_num]
#fill the i-th column of testing set (“samptest”) with the remaining indexes. They are used to create the i-th test dataset
samptest[i, ] <- indexes_random[(train_num + 1):length(indexes_random)]
}
#save the matrices
write.table(samptrain, "samptrain.txt")
write.table(samptest, "samptest.txt") - Evaluate predicting capacities for the complete and partial models missing one of the variables.
###evaluate model prediction
#load package caret for modelling (Kuhn, et al., 2008).
require(“caret“)
#create a table where log likelihood estimate (LL) is stocked
LLall <- matrix(0, nrow=8, ncol=100)
#for each data partition, train the model and test its prediction capacities using log likelihood
for (t in 1:100) { #data partitioning and log likelihood estimate for the set number t
#create training set
GthenRtrain <- GthenR[samptrain[t, ], ]
#create testing set
GthenRtest <- GthenR[samptest[t, ], ]
###calculate log likelihood estimate of the complete model, i.e. containing all variables
#train the complete model on the training set called GthenRtrain
#precise that the model is a generalized linear model (method=“glm”) and that the link function is a logistic one as a binomial variable second_inf_bool (family=binomial(link=”logit”) is predicted
mod_fit <- train(second_inf_bool ~., data=GthenRtrain, method="glm", family=binomial(link="logit"))
#predict the probability (type=”prob”) of the second infection given the observations in the testing set called GthenRtest
model <- predict(mod_fit, newdata=GthenRtest, type="prob")
#calculate log likelihood estimate of the model. In this model, log likelihood corresponds to the sum of predicted (log-)probabilities of infection in cells for which the infection was observed and of predicted (log-)probabilities of no infection in cells for which no infection was observed. Calculate this estimate only for the test dataset that was not used for the training of the model.
LL <- sum((GthenRtest[, 1] == 1) * log(model[, 2])) + sum((GthenRtest[, 1] == 0) * log(model[, 1]))
#save the log likelihood estimate in a table
LLall[2, t] <- LL
###calculate log likelihood for partial models missing one of the variable ("cell_perim" or "sqrt_circularity" or "neighbor_inf1" or "neighbor_non_inf1" or "Cells_100_microns" or "delay")
for (i in 5:10) {
#preparing training set
GthenRtr <- GthenRtrain
#deleting the data of the training set corresponding to the omitted variable
GthenRtr[, i] <- NULL
#preparing testing set
GthenRte <- GthenRtest
#deleting data of the testing set corresponding to the omitted variable
GthenRte[, i] <- NULL
#train the partial model on the training dataset called GthenRtr
mod_fiti <- train(second_inf_bool ~., data=GthenRtr, method="glm", family=binomial(link="logit"))
#predict the probability of the second infection given the observations in the testing set called GthenRte
modelsi <- predict(mod_fiti, newdata=GthenRte, type="prob")
#calculate the log likelihood of the partial model
LL <- sum((GthenRte[, 1]==1) * log(modelsi[, 2])) + sum((GthenRte[, 1] == 0) * log(modelsi[, 1]))
#save corresponding log likelihood estimate
LLall[i-2, t] <- LL
}
}
###evaluate the capacities of prediction for the last partial model: this model does not contain the information on the intensity of first infection (low_first, med_first, high_first)
#prepare training set in the matrix GthenRtr_wo_i
GthenRtr_wo_i <- GthenRtrain
GthenRtr_wo_i[, c(2, 3, 4)] <- NULL #deleting low_first, med_first, high_first
#prepare testing set in the matrix GthenRte_wo_i
GthenRte_wo_i <- GthenRtest
GthenRte_wo_i[, c(2, 3, 4)] <- NULL #deleting low_first, med_first, high_first
#train the partial model on the training dataset called GthenRtr_wo_i
mod_fiti <- train(second_inf_bool ~., data=GthenRtr_wo_i, method="glm", family=binomial(link="logit"))
#predict the probability of the second infection given the observations in the testing set called GthenRte_wo_i
modelsii <- predict(mod_fiti, newdata=GthenRte_wo_i, type="prob")
#calculate the log likelihood estimate
LL_i <- sum((GthenRte[, 1] == 1) * log(modelsii[, 2])) + sum((GthenRte[, 1] == 0) * log(modelsii[, 1]))
#save the log likelihood estimate
LLall[2, t] <- LL_i
}
#save the table containing log likelihood estimates
write.table(LLall, "LL_results.txt") - Plot the data from the LL_results.txt file obtained using GraphPad Prism (Figure 18)
- Perform a statistical paired t-test between the complete and the partial model.
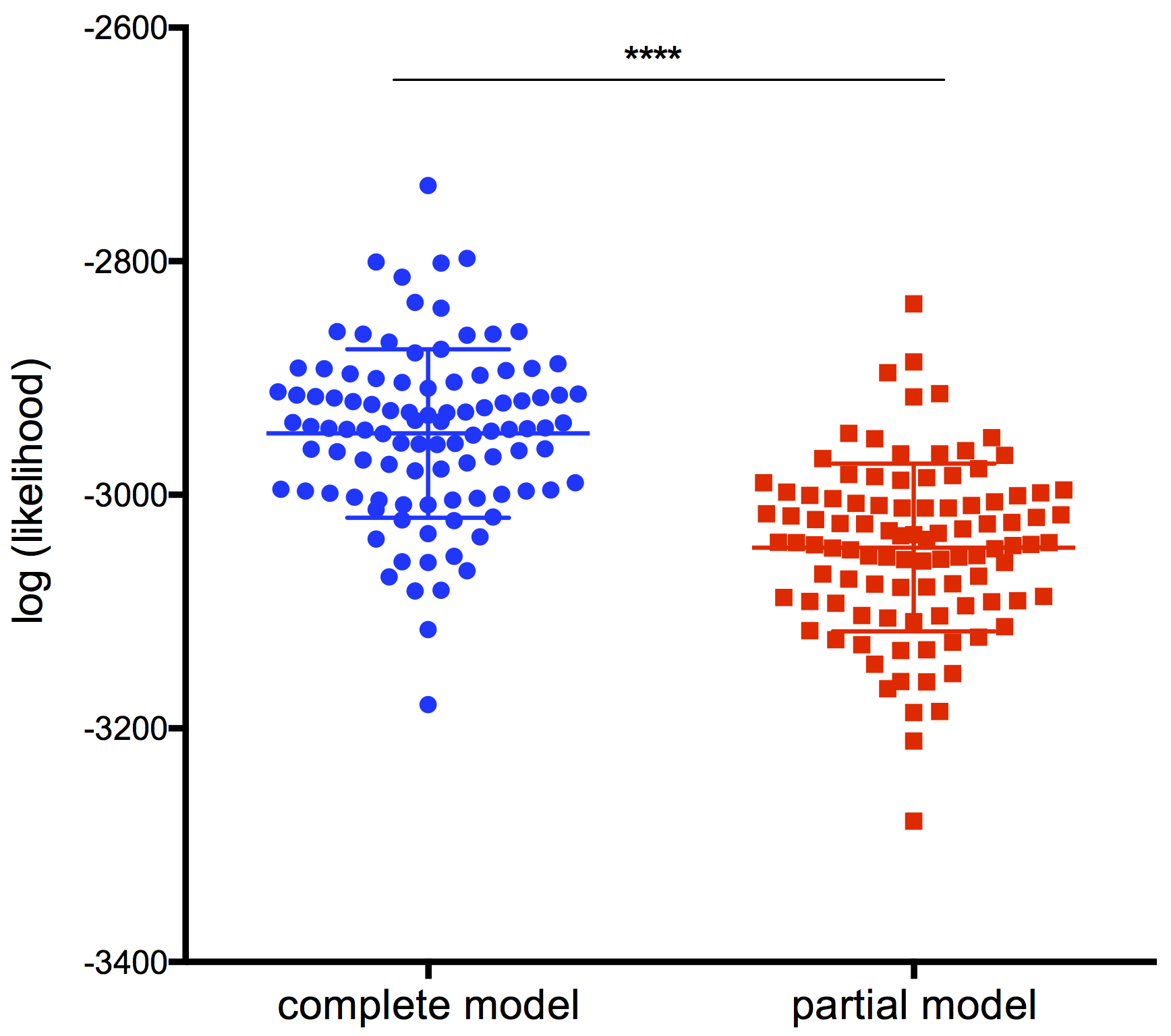
Figure 18. Comparison of the log (likelihood) of the complete model versus the partial model, lacking the information on the intensity of the first infection (****, P-value < 0.0001)
- Load the complete dataset (see Note 12), and convert 2 of the variables to categorical variables.
- From Icy output files to the final R dataset
Notes
- The colonies display a "red" or "green" color.
- The inclination of the tube is important to aerate the samples.
- Remember to equilibrate the centrifuge properly.
- The adjacent wells at the periphery of the designed map should be filled with 100 μl of warm DPBS to increase the reproducibility or the infection between each well (see Figure 2).
- The inclination of the tube is important to aerate the samples.
- Do not vortex any tube containing Salmonella, as it would break the flagella and drastically decrease the efficiency of infection.
- The decrease of the gentamicin concentration after 1 h of incubation helps to prevent the incorporation of gentamicin into the cells.
- The plate can be kept at 4 °C for a few days before acquisition.
- The parameters that need to be adapted between experiments are in the blocks "HK-means", "Active Contours" and "Wavelet Spot Detector".
- The number on the top right corner of each block indicates the order in which the blocks are executed in the pipeline.
- The proper calibration of the "Active Contours" block parameters determines the success of image analysis. They should be adapted by trial and error. A "Display" block can be used to follow the segmentation process visually.
- It is necessary to reload the dataset only if the Steps B2 and B3 of the analysis (Data analysis section) are not done on the raw.
Recipes
- Heat inactivation of the Fetal Bovine Serum
- Aliquot the serum into 50 ml labeled Falcon tubes
- Heat inactivate the aliquots in the water bath for 30 min at 56 °C
- Cool down at RT and store at -20 °C for up to several weeks
- Salmonella transformation
- Dilute 500 μl of an overnight culture of SL1344 in 50 ml of LB in a 250 ml Erlenmeyer flask
- Incubate the subcultures for 3 h in a 45°-tilted rack of an incubator at 37 °C with an orbital shaker at 220 rpm
- Transfer the subculture into a cold (4 °C) 50 ml Falcon® tube
- Centrifuge at 3,200 x g for 10 min at 4 °C using Centrifuge 5810/5810 R
- Discard the supernatant and resuspend the pellet in 20 ml of ice-cold distilled water
- Repeat steps d and e
- Discard the supernatant and resuspend the pellet in 100 μl of distilled water
- Add 2 μl of DNA (here, pGG2 or pM965 plasmids)
- Transfer the bacteria with DNA into a cold (4 °C) electroporation cuvette
- Incubate for 10 min on ice
- Electroporate the bacteria using the Gene Pulser Electroporation Systems programmed for E. coli (program number 4)
- Add 1 ml of LB to the cuvette and transfer the bacteria to an Eppendorf tub
- Incubate for 1 h in an incubator at 37 °C with an orbital shaker at 220 rpm
- Spread 20 μl of the culture on a petri dish containing appropriate antibiotics corresponding to the resistance cassette encoded on the plasmid (here, ampicillin)
- Incubate at 37 °C overnight
- Select individual colonies and grow overnight in LB supplemented with the appropriate antibiotics (here, ampicillin at 100 μg/ml)
- Make glycerol stock and store at -80 °C
- Lysogeny Broth (LB) medium supplemented with 0.3 M NaCl (1 L)
- Weigh 10 g of Tryptone (BD)
- Weigh 5 g Yeast Extract (BD)
- Weigh 17.5 g NaCl (Sigma-Aldrich)
- Volume up to 1 L of solution with distilled water
- Adjust the pH to 7.4
- Autoclave for 20 min at 120 °C
- Store at RT
- LB agar (1 L)
- Weigh 10 g Tryptone (BD)
- Weigh 5 g Yeast Extract (BD)
- Weigh 5 g NaCl (Sigma-Aldrich)
- Agar Solidifying Agent (BD)
- Volume up to 1 L of solution with distilled water
- Adjust the pH to 7.4
- Autoclave for 20 min at 120 °C
- Store at RT
- LB agar plate containing ampicillin at 100 μg/ml
- Melt the LB agar in a double boiler for 13 min in the microwave at 700 W
- Wait 30 min at RT until the medium cools down; add ampicillin at 100 μg/ml and swirl the LB agar solution to ensure the ampicillin is well mixed
Note: It should be comfortable to touch with an ungloved hand, but still fairly hot. - Pour agar into round Petri plates
- Allow plates to sit overnight at RT and store at 4 °C
- EM medium 20x (1 L)
- Weigh 140.3 g NaCl (Sigma-Aldrich)
- Weigh 10.4 g KCl (Thermo Fisher Scientific)
- Weigh 4 g CaCl2 (Sigma-Aldrich)
- Weigh 1.52 g MgCl2 (Sigma-Aldrich)
- Weigh 18 g glucose (Sigma-Aldrich)
- Weigh 119.1 g HEPES (Sigma-Aldrich)
- Volume up to 1 L of solution with distilled water
- Adjust the pH to 7.4
- Autoclave for 30 min at 110 °C
- Store at 4 °C
- EM medium 1x
For 1 L of EM medium 1x, dilute 50 ml of EM medium 20x in 950 ml of distilled water
Acknowledgments
V.S. was supported by a Ph.D. fellowship from the University Paris Diderot allocated by the ENS Cachan, Université Paris-Saclay; and a grant from the FRM. J.E. is a member of the LabEx consortia IBEID and MilieuInterieur. J.E. also acknowledges support from the ANR (grant StopBugEntry and AutoHostPath) and the ERC (CoG EndoSubvert).
This protocol was originally performed within the article entitled “Identifying parameters of host cell vulnerability during Salmonella infection by quantitative image analysis and modelling” (Voznica et al., 2018).
Competing interests
Authors have no conflicts of interest or competing interests to declare.
References
- de Chaumont, F., Dallongeville, S., Chenouard, N., Herve, N., Pop, S., Provoost, T., Meas-Yedid, V., Pankajakshan, P., Lecomte, T., Le Montagner, Y., Lagache, T., Dufour, A. and Olivo-Marin, J. C. (2012). Icy: an open bioimage informatics platform for extended reproducible research. Nat Methods 9(7): 690-696.
- Frechin, M., Stoeger, T., Daetwyler, S., Gehin, C., Battich, N., Damm, E. M., Stergiou, L., Riezman, H. and Pelkmans, L. (2015). Cell-intrinsic adaptation of lipid composition to local crowding drives social behaviour. Nature 523(7558): 88-91.
- Fredlund, J., Santos, J. C., Stevenin, V., Weiner, A., Latour-Lambert, P., Rechav, K., Mallet, A., Krijnse-Locker, J., Elbaum, M. and Enninga, J. (2018). The entry of Salmonella in a distinct tight compartment revealed at high temporal and ultrastructural resolution. Cell Microbiol 20(4).
- Haraga, A., Ohlson, M. B. and Miller, S. I. (2008). Salmonellae interplay with host cells. Nat Rev Microbiol 6(1): 53-66.
- Knodler, L. A. (2015). Salmonella enterica: Living a double life in epithelial cells. Curr Opin Microbiol 23: 23-31.
- Kuhn, M. (2008). Building predictive models in R using the caret package. J Stat Softw 28(5): 1-26.
- LaRock, D. L., Chaudhary, A. and Miller, S. I. (2015). Salmonellae interactions with host processes. Nat Rev Microbiol 13(4): 191-205.
- Lelouard, H., Henri, S., De Bovis, B., Mugnier, B., Chollat-Namy, A., Malissen, B., Meresse, S. and Gorvel, J. P. (2010). Pathogenic bacteria and dead cells are internalized by a unique subset of Peyer's patch dendritic cells that express lysozyme. Gastroenterology 138(1): 173-184 e171-173.
- Liberali, P., Snijder, B. and Pelkmans, L. (2015). Single-cell and multivariate approaches in genetic perturbation screens. Nat Rev Genet 16(1): 18-32.
- Lorkowski, M., Felipe-Lopez, A., Danzer, C. A., Hansmeier, N. and Hensel, M. (2014). Salmonella enterica invasion of polarized epithelial cells is a highly cooperative effort. Infect Immun 82(6): 2657-2667.
- Misselwitz, B., Barrett, N., Kreibich, S., Vonaesch, P., Andritschke, D., Rout, S., Weidner, K., Sormaz, M., Songhet, P., Horvath, P., Chabria, M., Vogel, V., Spori, D. M., Jenny, P. and Hardt, W. D. (2012). Near surface swimming of Salmonella Typhimurium explains target-site selection and cooperative invasion. PLoS Pathog 8(7): e1002810.
- Misselwitz, B., Kreibich, S. K., Rout, S., Stecher, B., Periaswamy, B. and Hardt, W. D. (2011). Salmonella enterica serovar Typhimurium binds to HeLa cells via Fim-mediated reversible adhesion and irreversible type three secretion system 1-mediated docking. Infect Immun 79(1): 330-341.
- R Core Team (2013). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria.
- Santos, A. J., Meinecke, M., Fessler, M. B., Holden, D. W. and Boucrot, E. (2013). Preferential invasion of mitotic cells by Salmonella reveals that cell surface cholesterol is maximal during metaphase. J Cell Sci 126(Pt 14): 2990-2996.
- Snijder, B., Sacher, R., Ramo, P., Damm, E. M., Liberali, P. and Pelkmans, L. (2009). Population context determines cell-to-cell variability in endocytosis and virus infection. Nature 461(7263): 520-523.
- Stecher, B., Hapfelmeier, S., Muller, C., Kremer, M., Stallmach, T. and Hardt, W. D. (2004). Flagella and chemotaxis are required for efficient induction of Salmonella enterica serovar Typhimurium colitis in streptomycin-pretreated mice. Infect Immun 72(7): 4138-4150.
- Vonaesch, P., Cardini, S., Sellin, M. E., Goud, B., Hardt, W. D. and Schauer, K. (2013). Quantitative insights into actin rearrangements and bacterial target site selection from Salmonella Typhimurium infection of micropatterned cells. Cell Microbiol 15(11): 1851-1865.
- Voznica, J., Gardella, C., Belotserkovsky, I., Dufour, A., Enninga, J. and Stevenin, V. (2018). Identifying parameters of host cell vulnerability during Salmonella infection by quantitative image analysis and modeling. Infect Immun 86(1): e00644-17.
- Watson, K. G. and Holden, D. W. (2010). Dynamics of growth and dissemination of Salmonella in vivo. Cell Microbiol 12(10): 1389-1397.
- Wickham, H. (2011). The split-apply-combine strategy for data analysis. J Stat Softw 40(1): 1-29.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Voznica, J., Enninga, J. and Stévenin, V. (2018). High-throughput Microscopic Analysis of Salmonella Invasion of Host Cells. Bio-protocol 8(18): e3017. DOI: 10.21769/BioProtoc.3017.
Category
Microbiology > Microbe-host interactions > In vitro model
Microbiology > Microbial cell biology > Cell imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.