- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Identifying Protein Interactions with Histone Peptides Using Bio-layer Interferometry





(*contributed equally to this work) Published: Vol 8, Iss 18, Sep 20, 2018 DOI: 10.21769/BioProtoc.3012 Views: 9216
Reviewed by: Anna VangoneAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2018

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics









Abstract
Histone post-translational modifications (PTMs) regulate numerous cellular processes, including gene transcription, cell division, and DNA damage repair. Most histone PTMs affect the recruitment or exclusion of reader proteins from chromatin. Here, we present a protocol to measure affinity and interaction kinetics between histone peptides and the recombinant protein using Bio-layer interferometry.
Keywords: Histone post-translational modificationBackground
Eukaryotic chromatin structure is broadly divided into euchromatin and heterochromatin (Cheung and Lau, 2005), with heterochromatin structure further subdivided depending on the combination of histone post-translational modifications (PTMs). These PTMs alter not only the chromatin conformation but also establish direct regulatory roles in gene expression and protein recruitment (Felsenfeld and Groudine, 2003; Allshire and Madhani, 2017). Myriad combinations of histone PTMs–including acetylation, phosphorylation, methylation, ubiquitination, biotinylation, sumoylation, and proline isomerization, collectively known as the “histone marks”–can be found, particularly on the unstructured N-terminal tail protruding from the nucleosomal core (Guetg and Santoro, 2012). These PTMs regulate numerous cellular processes, including gene transcription, cell division, and DNA damage repair (Suganuma and Workman, 2011), through the activities of different “readers” or effector proteins (Musselman et al., 2012). Thus, large efforts have been made to identify the readers for histone modifications.
Studying the interactions between reader proteins and their target histone PTMs using conventional methods (e.g., surface plasmon resonance [SPR] and SPR imaging [SPRi] biosensors) often requires large amounts of substrates or complex, multistep experimental methods, and is complicated by the various method-specific limitations. These concerns preclude the ease and accuracy of quantifying the strength of an interaction (Phizicky and Fields, 1995; Berggard et al., 2007; Rowley and Corces, 2016; Wierer and Mann, 2016). In recent work, we employed bio-layer interferometry (BLI) octet methodology (Kamat and Rafique, 2017; Petersen, 2017) to elucidate the binding between fission yeast Swi6, the counterpart of the human heterochromatin protein 1, and dimethylated histone H3 lysine 9 (H3K9me2) in the presence or absence of a phosphorylation moiety on tyrosine 41 residue on the histone H3 N-terminus (Ren et al., 2018). BLI is an optical technique for real-time measurement of macromolecular interaction. This aim is achieved via the analysis of interference patterns of the white light that is reflected off the biosensor surface. In a typical BLI experiment, the ligand is immobilized on the biosensor tip and then allowed to interact with the analyte (for example, a protein). The binding of the analyte to the immobilized ligand will increase the thickness of the biological layer at the surface of the biosensor tip resulting in a shift in the interference pattern, which is then documented in real time.
Akin to SPRi biosensors, BLI sensors do not entail isotopic labeling. Label-free biosensors offer a significant advantage when studying PTM-reader interactions, as small modification groups–such as a label–could affect the binding affinity of specific reader proteins. The use of label-free biosensors thus avoids any bias generated using labels with a different molecular mass. Although BLI technology has some limitations where small-sized molecules are concerned, its high flexibility and robustness for the simultaneous analysis of multiple (96) independent analyte-ligand pairs offer significant advantages. The disposable biosensors also allow for ad hoc replacement and real-time re-loading of the analytes or ligand substrate arrays (Abdiche et al., 2008). The BLI technology uses a non-fluidic system of dipping the sensors into a well plate instead of delivering the sample liquid to the sensor. This change in sample delivery not only increases the robustness of the assay but also decreases the operating costs (Nirschl et al., 2011).
Here, we present a simple, quick, and highly sensitive method for the detection of protein interactions using a synthesized peptide as bait and a recombinant protein in the BLI octet approach, in the determination of interaction kinetics of a histone PTM (H3K9me2) with its reader protein (Swi6). The protocol below describes detailed procedures for bacterial expression, extraction, and purification of recombinant protein Swi6, followed by the set-up of BLI octet, detection of Swi6 interaction with biotinylated peptide (H3K9me2), and subsequent processing and analysis of the readout data.
Materials and Reagents
- 96-well black, flat-bottomed, polypropylene sample microplate (Geiner Bio One International, catalog number: 655209 )
- Centrifuge tubes 50 ml, screw cap (Greiner Bio One International, catalog number: 227261 )
- Microcentrifuge tubes 1.5ml (Eppendorf, catalog number: 0030120086 )
- Ni-NTA spin column (QIAGEN, catalog number: 31014 )
- E. coli BL21 (DE3) cells (Merck, Novagen®, catalog number: 69450-3 )
- pET32a plasmid (Merck, Novagen®, catalog number: 69015 )
- 1 M IPTG (store at -20 °C) (Thermo Fisher Scientific, catalog number: R0392 )
- 1 mg/ml DNase I (store at -20 °C) (Sigma-Aldrich, catalog number: DN25 )
- 1 mg/ml RNase A (store at -20 °C) (Thermo Fisher Scientific, catalog number: EN0531 )
- 10 mg/ml Lysozyme (dissolved in 10 mM Tris-HCl, pH 8.0, store at -20 °C) (Sigma-Aldrich, catalog number: L6876 )
- 100 mg/ml Carbenicillin (stock at -20 °C) (Thermo Fisher Scientific, GibcoTM, catalog number: 10177012 )
- 1x PBS (dilution from 10x PBS) (Vivantis, catalog number: PB0344-1L )
- 2-Morpholinoethanesulfonic acid sodium salt (MES Na) (Merck, catalog number: 1.06197.0100 )
- Bromophenol blue (Sigma-Aldrich, catalog number: B8026 )
- Bovine serum albumin (BSA) (Sigma-Aldrich, catalog number: A2153 )
- Glycerol (QRec, CAS: 56-81-5)
- Biotin-labeled Histone peptides (Biotin labeling is done during synthesis) (Mimotopes) dissolved in 10% acetonitrile (50 μg/μl) (Merck, catalog number: 100029 )
- Imidazole (Sigma-Aldrich, catalog number: I2399 )
- KCl (Sigma-Aldrich, catalog number: P9541 )
- KH2PO4 (Sigma-Aldrich, catalog number: P9791 )
- Na2HPO4 (QRec, CAS: 10028-24-7)
- NaCl (Merck, catalog number: 1.06404.0500 )
- NaCl* (Sigma-Aldrich, catalog number: S9888 )
*Note: An alternative brand of NaCl is used for Recipes 1-3; 5-7. - NaH2PO4 (Sigma-Aldrich, catalog number: S3139 )
- NaOH (Fisher Scientific, catalog number: S318 )
- Sodium dodecyl sulfate (SDS) (Sigma-Aldrich, catalog number: L3771 )
- Tris (Vivantis, catalog number: PR0612 ) or Tris base (Sigma-Aldrich, catalog number: T1503 )
- Tryptone (BD, catalog number: 211705 )
- Tween-20 (Sigma-Aldrich, catalog number: P1379 )
- Yeast extract (BD, catalog number: 288620 )
- β-mercaptoethanol (store at 4 °C) (Sigma-Aldrich, catalog number: M6250 )
- LB liquid medium (see Recipes)
- Phosphate buffer saline (10x PBS) (see Recipes)
- Lysis buffer (store at room temperature) (see Recipes)
- 2x Sample buffer (store at room temperature) (see Recipes)
- Wash buffer (store at 4 °C) (see Recipes)
- Elution buffer (store at 4 °C) (see Recipes)
- Exchange buffer (store at 4 °C) (see Recipes)
- MES Assay buffer (see Recipes)
- PBS Assay buffer (see Recipes)
- TBS Assay buffer (see Recipes)
Equipment
- Biosensors/Streptavidin (SA) Tray (PALL, FortéBio®, catalog number: 18-5019 )
- FortéBIO® Octet RED96 System (PALL)
- Heat block (Bio Laboratories, Elite dry bath incubator)
- High-speed refrigerated centrifuge (TOMY DIGITAL BIOLOGY, model: MX-305 )
- NanoDrop spectrophotometer (DeNovix, model: DS-11 )
- PIPETMAN® Classic Pipettes P20, P200, P1000 (Gilson, catalog numbers: F123600 , F123601 , F123602 )
- PierceTM Protein concentrator 10K MWCO (Thermo Fisher Scientific, catalog number: 88517 )
- Shaker incubator (Eppendorf, New BrunswickTM, model: Innova® 44 incubator shaker, catalog number: M1282-0002)
- Sonifier (Emerson Electric, model: Branson ultrasonic sonifier® 450A )
Software
- Data acquisition software for Octet RED96 (v9.0) was purchased from PALL FortéBIO® Co. The software can be installed in other computers with Windows 7 (or above) operating system (OS) for data analysis. This software is however incompatible with MacIntosh OS.
Procedure
The coding sequence of the protein of interest is cloned into the pET32a plasmid (Merck, Novagen®) and transformed into E. coli BL21 (DE3) cells.
- Recombinant protein expression
- Grow E. coli BL21 (DE3) cells in 500 ml LB media containing 100 μg/ml of carbenicillin overnight at 30 °C, in a shaker incubator.
- Induce protein expression with 1 mM IPTG for 4.5 h at 30 °C.
Note: For verification of protein expression, take 1 ml of cell culture and spin at 2,300 x g for 1 min to collect the cell pellet. Add 100 μl of 2x Sample buffer, and heat at 95 °C for 5 min to prepare the protein sample for SDS-PAGE analysis. - Collect the cell pellet in 50 ml tubes by spinning at 9,000 x g for 2 min at 4 °C and store the cell pellet at -80 °C.
- Protein extraction and purification
The pET32a vector can provide a 6x His-tag on the N- or C-terminus of a recombinant protein. Below, we present the purification of a 6x His-tagged recombinant protein from E. coli BL21 (DE3) cell lysates using the Ni-NTA spin column (QIAGEN), with some optimization from the manufacturer’s handbook.- Thaw the cell pellet on ice and resuspend cells in 10 ml (1/50 to 1/100 of cell culture volume) of Lysis buffer.
Note: Keep buffers, cell suspension and protein at 0-4 °C for the duration of extraction and purification. - Incubate cells on ice for 30 min with gentle vortexing.
- Sonicate on ice using two 30 sec bursts at 400 W with 2 min cooling between bursts.
- Centrifuge at 9,000 x g for 30 min at 4 °C to pellet the cell debris. Transfer the supernatant to a new 50 ml tube.
Note: If the supernatant is murky, repeat this step once. - Prepare the Ni-NTA column for purification by equilibrating the column with 600 μl of Lysis buffer. Centrifuge at 890 x g, 4 °C for 2 min with the lid open.
- Transfer the supernatant containing the soluble fraction of the recombinant protein to the Ni-NTA column for purification. Up to 600 μl of lysate can be loaded each time. Centrifuge for 5 min at 270 x g, 4 °C. Discard flow-through (soluble His-tagged protein will now be bound to Ni-NTA column).
Note: Take 20 μl of lysate for SDS-PAGE analysis. Add in 20 μl of 2x Sample buffer, and heat at 95 °C for 5 min before storing at -20 °C. - Wash the column twice with Wash buffer. Centrifuge for 2 min at 890 x g, 4 °C.
- Elute the 6x His-tagged protein twice with 300 μl of Elution buffer. Collect the eluate (eluted target protein) by centrifuging at 890 x g for 2 min, 4 °C.
- Repeat Steps B5-B8 for remaining lysates. Consolidate eluates for the next step (Step B10).
- Load the eluate (target protein) into the top chamber of a Pierce Protein Concentrator (10K MWCO) to exchange the Elution buffer and concentrate the protein.
- Immediately top up the eluate (target protein) with Exchange buffer to 5 ml. Centrifuge at 9,000 x g, 4 °C until the sample volume is less than 500 μl. Remove and discard the flow-through in the bottom collection chamber. Repeat this step at least 5 times to ensure the final imidazole concentration is lower than 0.1 mM.
Note: The Exchange buffer pH is determined by the isoelectric point of the recombinant protein. - Use a pipette to gently aspirate the concentrated protein within the top chamber. Transfer the concentrated protein into a microcentrifuge tube. Assess the protein concentration with NanoDrop.
- Dilute the protein to 100 μM with Exchange buffer and store at -80 °C.
Note: To confirm protein integrity, take 20 μl of the recombinant protein for SDS-PAGE analysis. Add in 20 μl of 2x Sample buffer, and heat at 95 °C for 5 min before storing at -20 °C.
- Thaw the cell pellet on ice and resuspend cells in 10 ml (1/50 to 1/100 of cell culture volume) of Lysis buffer.
- Bio-layer interferometry binding assay
We performed bio-layer interferometry binding on a FortéBIO® Octet RED96 instrument (PALL).- Turn on the instrument in advance for at least 15 to 20 min to warm up the lamp. Set the working temperature to 30 °C to pre-warm the sample plate holder (Figure 1).

Figure 1. Octet RED96 machine. Switch power on to warm up Octet RED96 machine. When the blue indicator light is illuminated as shown, the equipment is ready for use. - Prepare the Assay buffer
Note: A buffer test needs to be conducted to determine the appropriate Assay buffer for each protein. - Pre-wet the streptavidin sensors in the Assay buffer for at least 10 min at room temperature, to remove the sucrose coating on the sensors (Figure 2).

Figure 2. Preparation of sensors. Detach the green sensors holder from the plate holder (blue), followed by the addition of assay buffer (250 μl per well) into plate wells (black) (A). Finally, fit the green sensor holder (with sensor tips) onto the plate holder (B). Caution should be exercised not to touch the sensor tips. Verify the sensor tips are immersed and pre-wet in the assay buffer. - Set up the experimental design in the Octet data acquisition software (v9.0, PALL) using the experiment wizard tab (Figure 3). The tab menu includes:
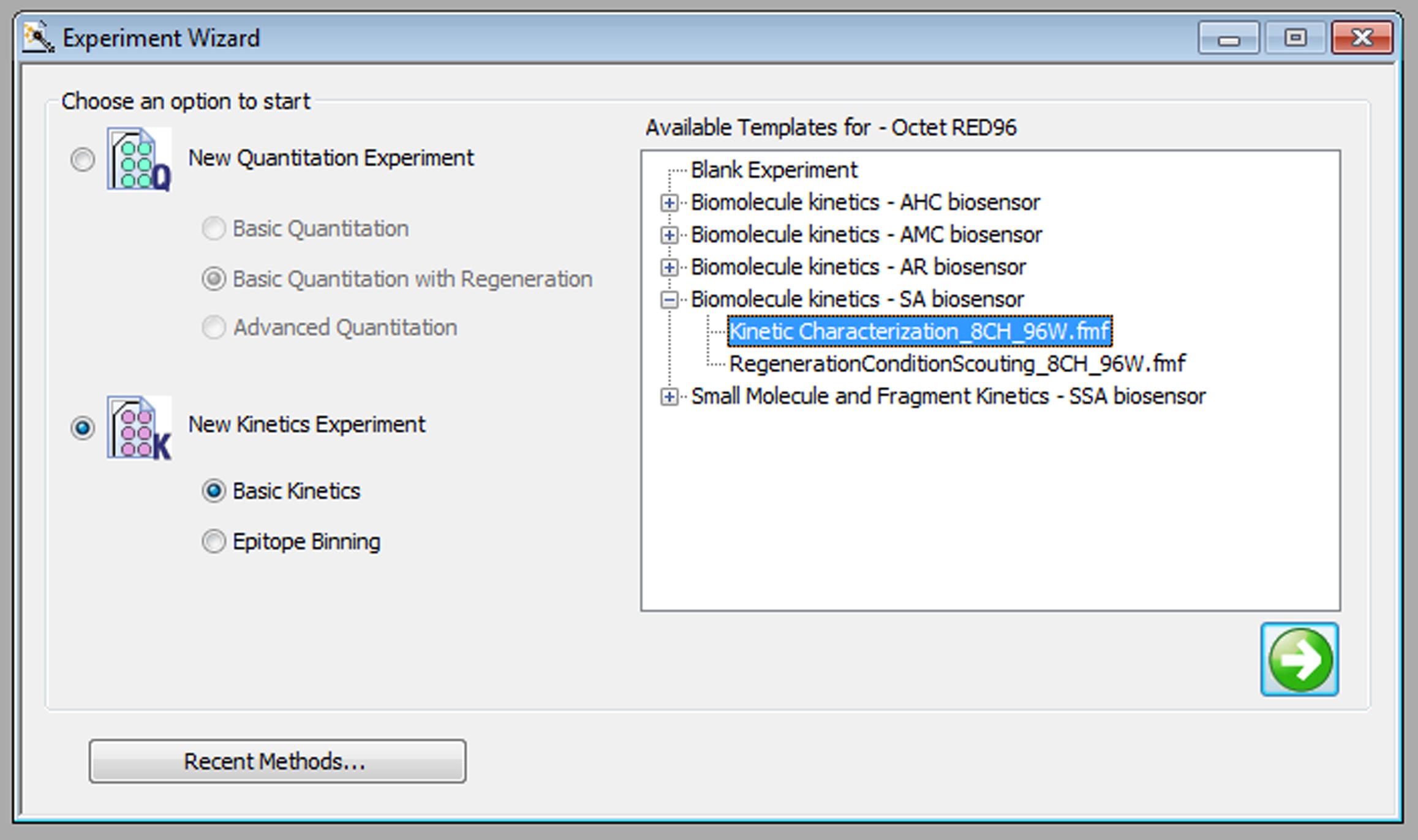
Figure 3. Startup of experimental design in Octet data acquisition software. In the Octet data acquisition software window, select ‘New Kinetics Experiment’ (‘Basic Kinetics’ option) as shown. Under available templates, select ‘Kinetic Characterization_8CH_96W.fmf’ under ‘Biomolecule Kinetics - SA biosensor’ if Streptavidin biosensors are used. Click on the green arrow button to enter the program.- Plate definition (Figure 4). Assign the content of each column, including the buffer, biotin-labeled histone peptide, and binding protein to be tested. Enter the concentration of the binding protein in each well. In addition, a reference well with Assay buffer only needs to be included in the same column.
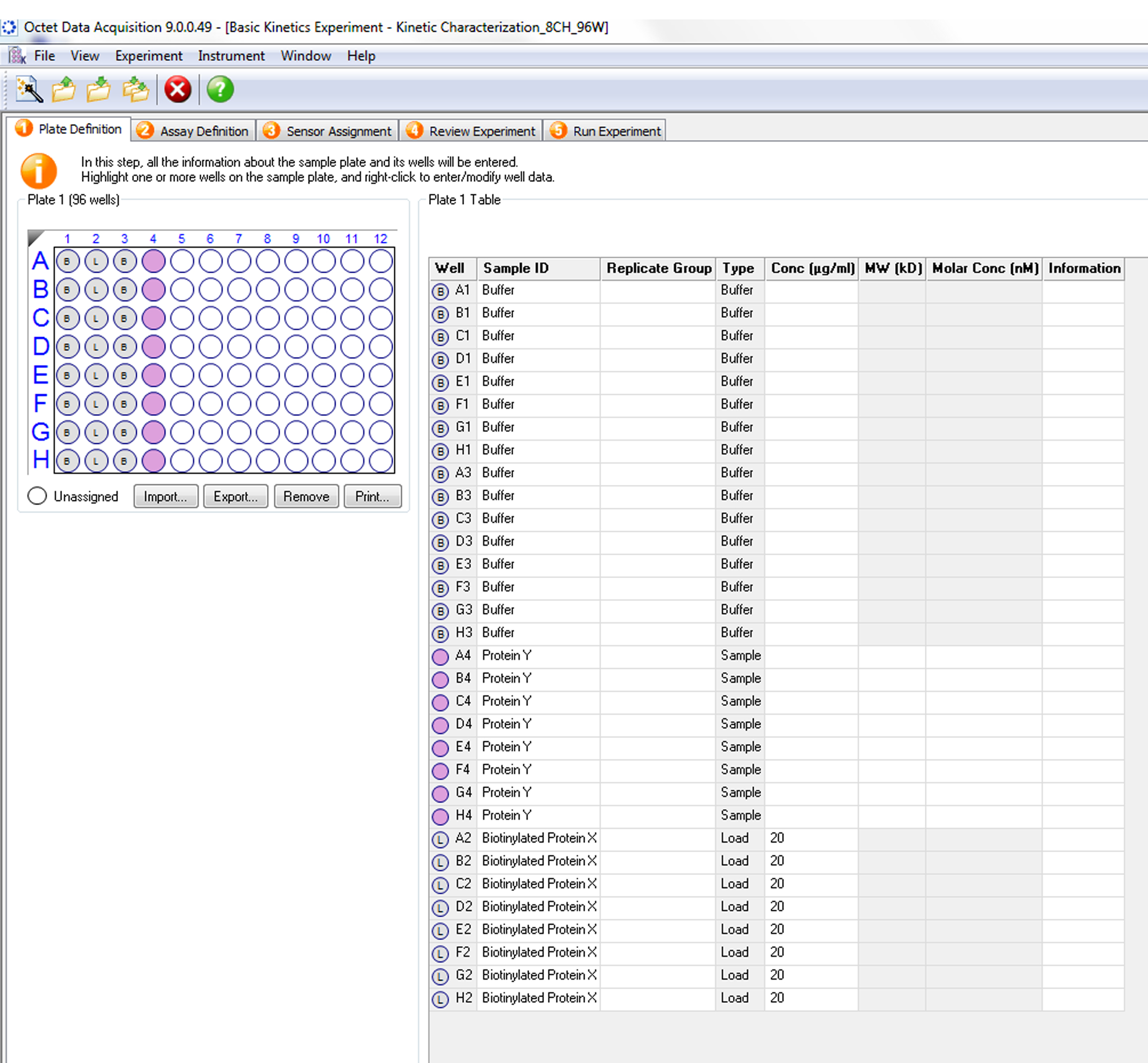
Figure 4. Plate definition. Select the wells on the layout of the sample plate. Right click on each column to assign the content of each well in the plate layout (top left hand corner), ‘B’: Buffer, ‘L’: Load, ‘Pink Circles’ denote position where the samples are located. Enter additional identifier information (for example, buffer composition) in the table on the right. - Assay definition (Figure 5). Usually, 5 steps are included in the binding kinetics assay between 2 objects: baseline 1, loading; baseline 2, association and disassociation. To generate a stable initial signal, in baseline 1 step, sensors need to be soaked in Assay buffer for at least 120 sec. The loading step immobilizes the biotin-labeled histone peptides on the sensors. The threshold function can be used to set the amount of peptide loaded onto the sensors. Sensors are then assigned to the Assay buffer again for at least 120 sec to establish another stable baseline signal (baseline 2) before the association step. During the association step, the sensors with immobilized peptides are assigned to the wells containing different concentrations of the protein of interest. Adjust the time of the association step so that the well with the highest protein concentration can reach the equilibrium binding signal. Finally, assign sensors to the Assay buffer again for a disassociation of 5 min.

Figure 5. Assay definition. Assigning each specific step from the drop-down menu under ‘Step Name’ in ‘Step Data List’ to each column in the plate assay. A. Assignment of baseline step and wells. Select ‘Baseline’ in the ‘Step Data List’, and double click on the first column. B. Assignment of loading step and wells. Select ‘Loading’ under ‘Step Data List’, and then double click on the second column of wells. C. Assignment of second baseline step and wells. Select ‘Baseline’ in ‘Step Data List’, and select ‘Baseline2’, then double click on the third column of wells. D. Assignment of association step and wells. Select ‘Association’ from ‘Step Data List’, and double click on the fourth column of wells. E. Assignment of dissociation step and wells. Select ‘Dissociation’ from ‘Step Data List,’ and double click on the third column of wells as above. Sensor type can be changed according to the tag of the immobilized ligand by selecting ‘Sensor Type’ from ‘Assay Steps List’. - Sensor assignment. Select the location of pre-wetted sensors on the sensor tray (Figure 6).
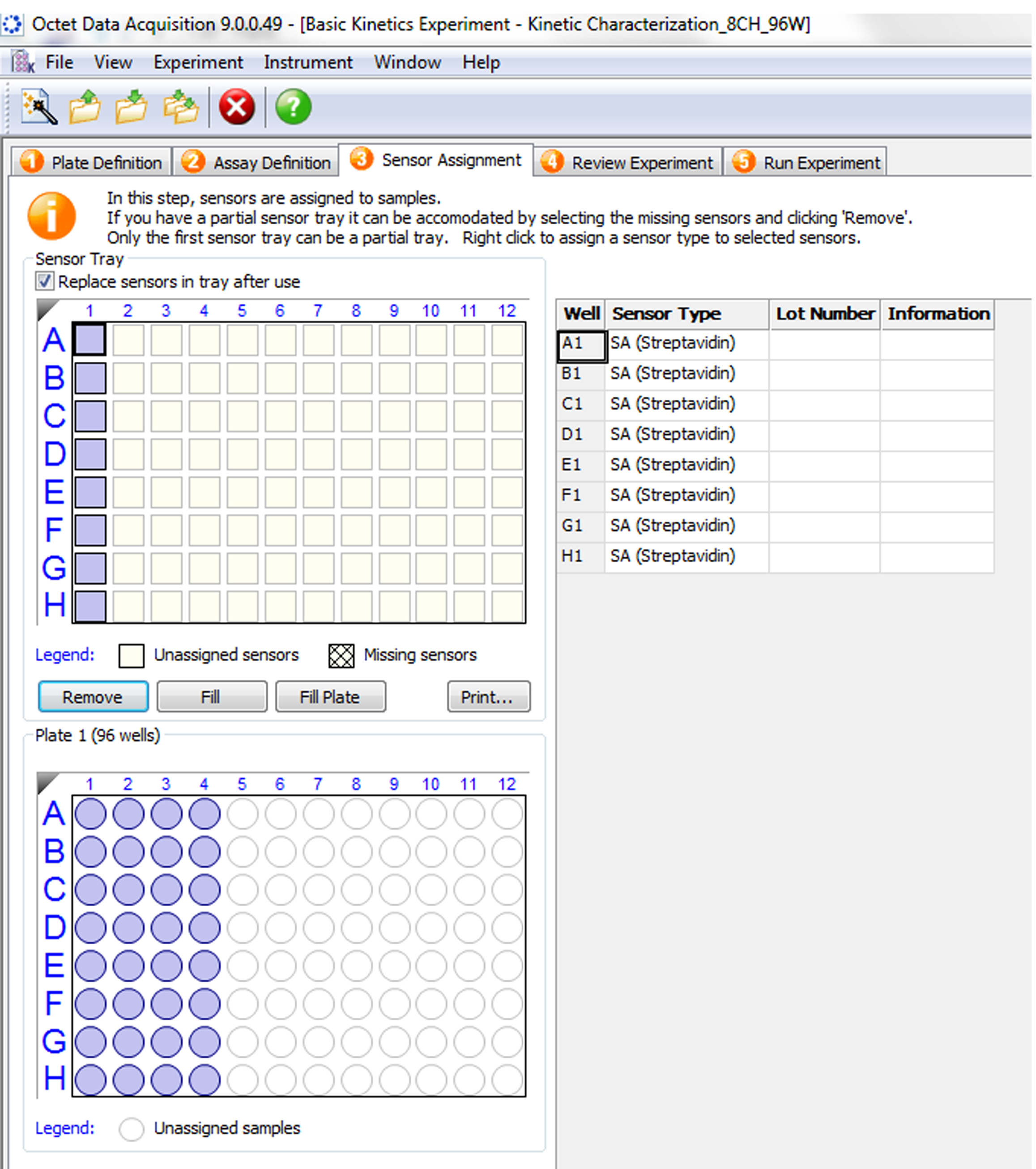
Figure 6. Assignment of pre-wet sensors location. Select the sensors (purple squares, top left) according to the location of the sensors on the sensor tray. - Review the experiment (Figure 7).
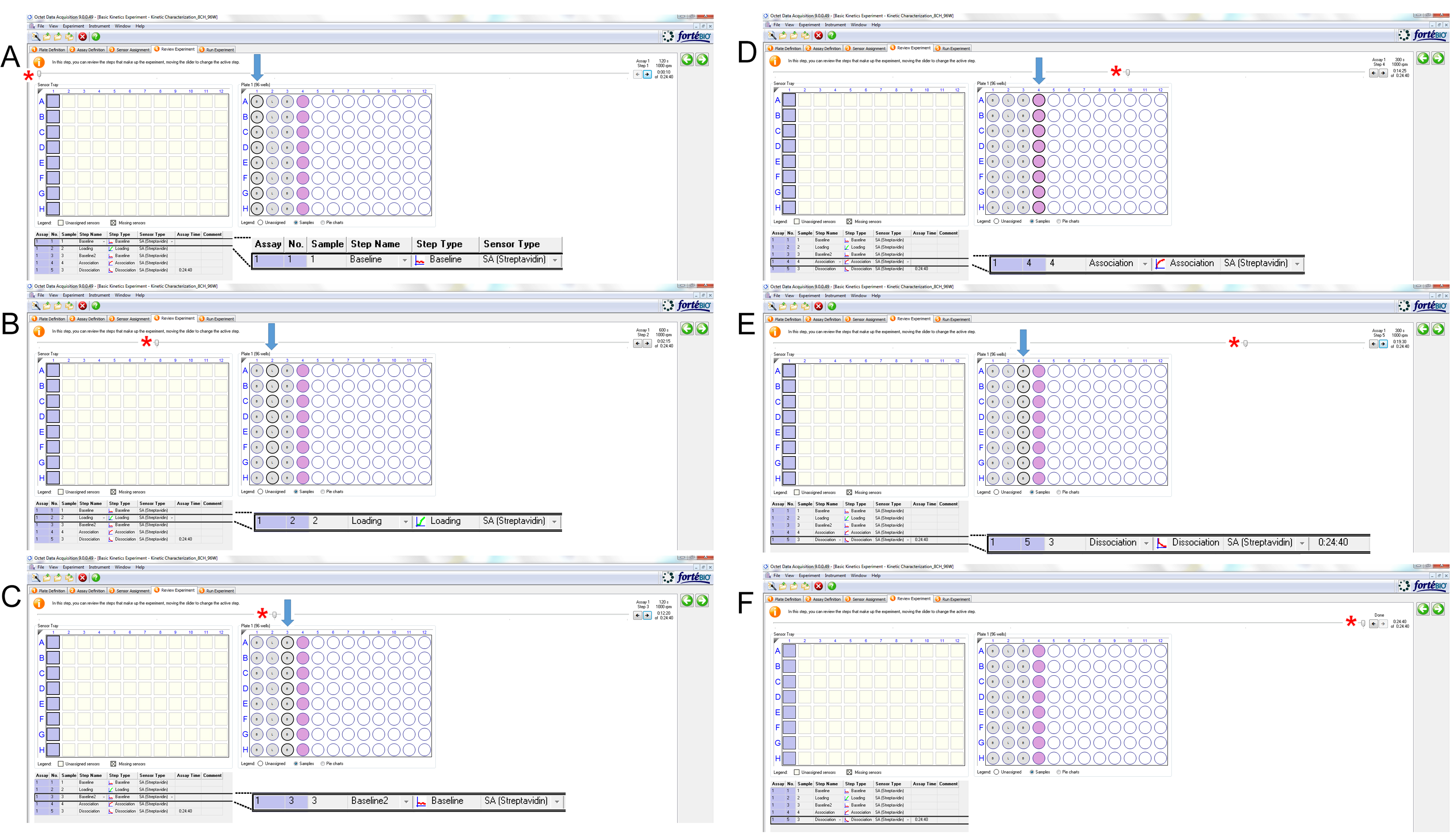
Figure 7. Review of experiment. Simulated movement of sensors (squares) to pre-determined locations on the assay plate. Squares on the left represent starting position of the sensors. The movement of the sensors from the docking position (square, top left) to different positions are shown in A-E: A. Baseline determination. B. Loading of peptide. C. Baseline 2 determination. D. Association (of peptide with target protein). E. Dissociation (of protein from peptide). F. End of procedure. Red asterisks, position of the level that indicates the progress of the procedure; blue arrows, positions of the sensors on the sample plate (A-E, top right). The window on the bottom left shows the exact step being executed, which is also enlarged on the bottom right corner.
- Plate definition (Figure 4). Assign the content of each column, including the buffer, biotin-labeled histone peptide, and binding protein to be tested. Enter the concentration of the binding protein in each well. In addition, a reference well with Assay buffer only needs to be included in the same column.
- Dilute the biotin-labeled histone peptides (Mimotope, Australia) to 5 μg/ml with Assay buffer and add 250 μl of each peptide into the corresponding wells.
- Prepare a serial dilution of the purified recombinant protein with Assay buffer and transfer 250 μl of protein solution into each well corresponding to the design in Step C3.
- Carefully put the sensor tray on the sensor platform (left side inside the machine), and the sample tray onto the plate holder on the right. Ensure that both trays are in the correct orientation and stably set (Figure 8).

Figure 8. Arrangement of sensor tray and sample tray in Octet RED96. The sensor tray is placed on the left while the sample tray on the right. Close the door of the Octet RED96 machine. - Start the assay (Figure 9). Click ‘Go’ to start assay.
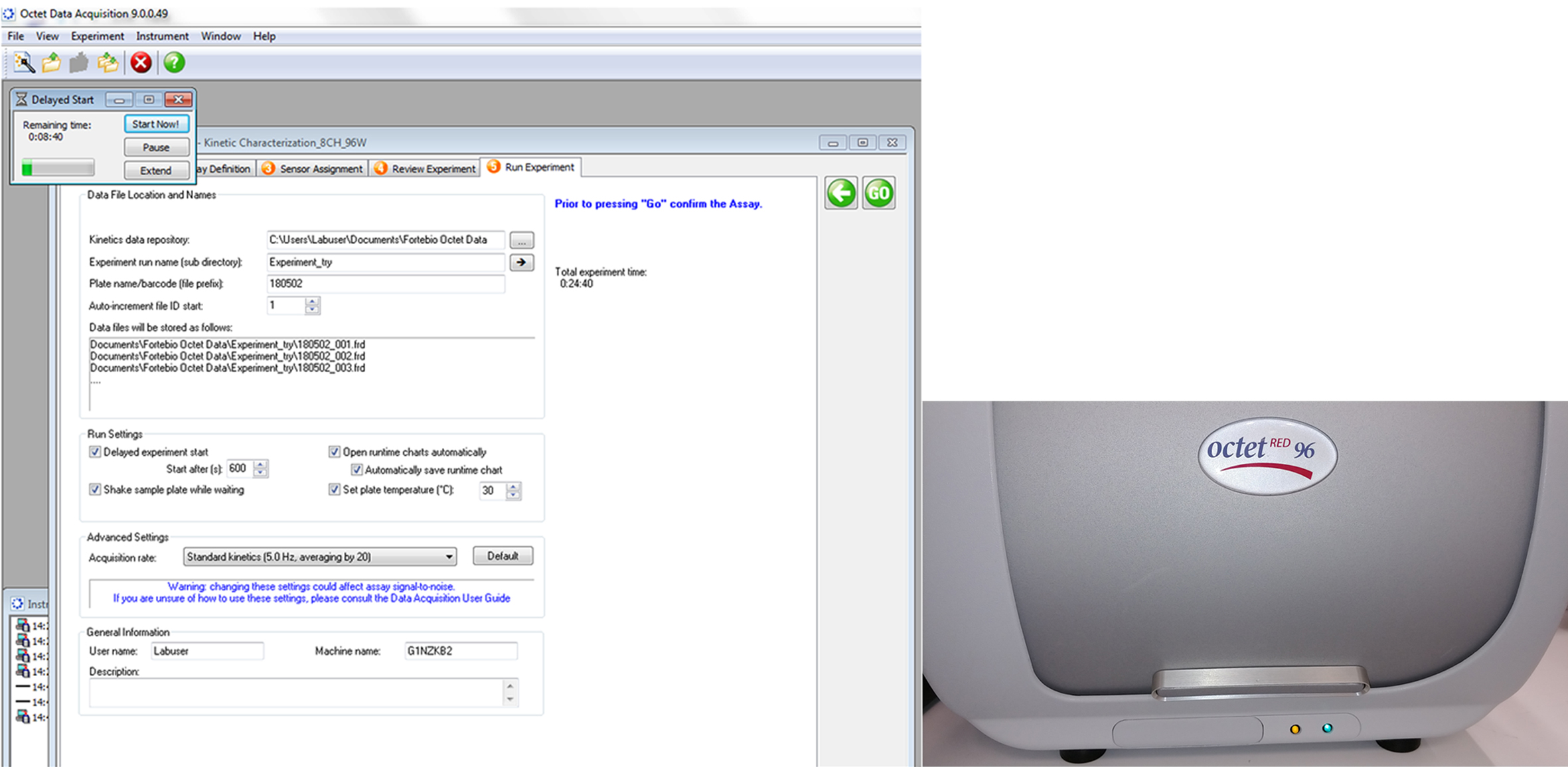
Figure 9. Window depicting properties of assay and run settings. Assay start at ‘Go’ (left). Note that orange indicator light on machine will light up (active mode) (right).
- Turn on the instrument in advance for at least 15 to 20 min to warm up the lamp. Set the working temperature to 30 °C to pre-warm the sample plate holder (Figure 1).
Data analysis
In this protocol, we demonstrate the data analysis in the Octet data analysis software (v9.0, PALL).
- Once the assay is complete, open the Octet data analysis software and load the experiment folder.
- Conduct data processing in the “Processing” tab. This usually includes 5 steps:
- In the “data selection” box, click “sensor selection” to assign a reference well (Figure 10).
- Tick the “subtraction” box to subtract the reference data from the raw protein-peptide interaction data (Figure 11).
- Tick the “align Y axis” box to align data of all samples to Y = 0. Select “baseline” and choose the time range for the baseline level. This time range needs to be at the stage where the baseline signal is stabilized, usually from the last 10 sec of baseline 1 step (Figure 11).
- Tick “inter-step correction” and select align to the “association.” This step diminishes the signal shifts between the association and dissociation steps.
- Select “Savitzky-Golay filtering” function and start the data process.
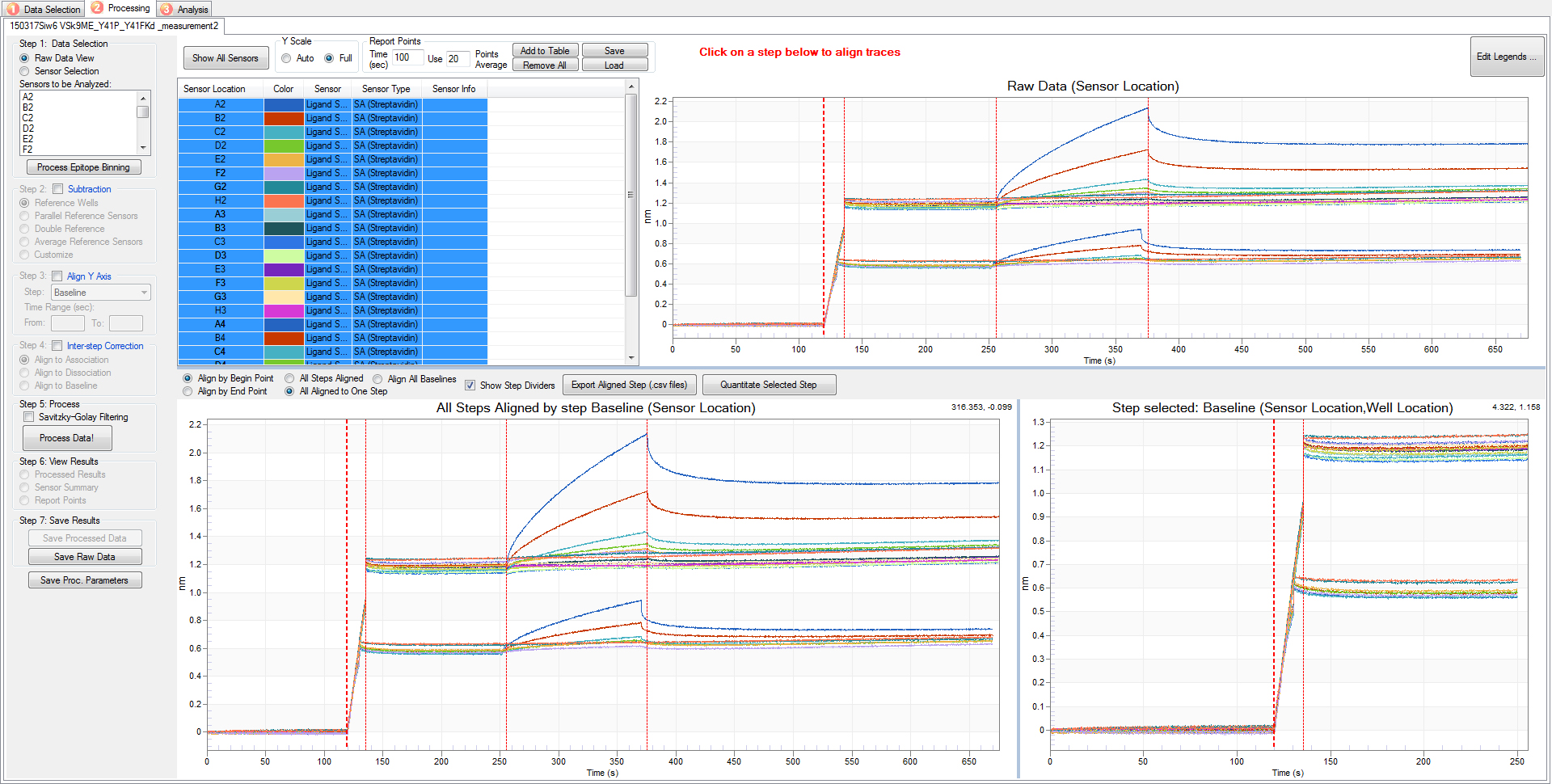
Figure 10. Screen capture of the raw data from the BLI assay. The X-axis indicates the timeline of the experiment. The Y-axis shows the signal shift at each time point. Red vertical lines indicate the transition point of 5 different steps of the experiment: baseline 1, loading, baseline 2, association, and dissociation.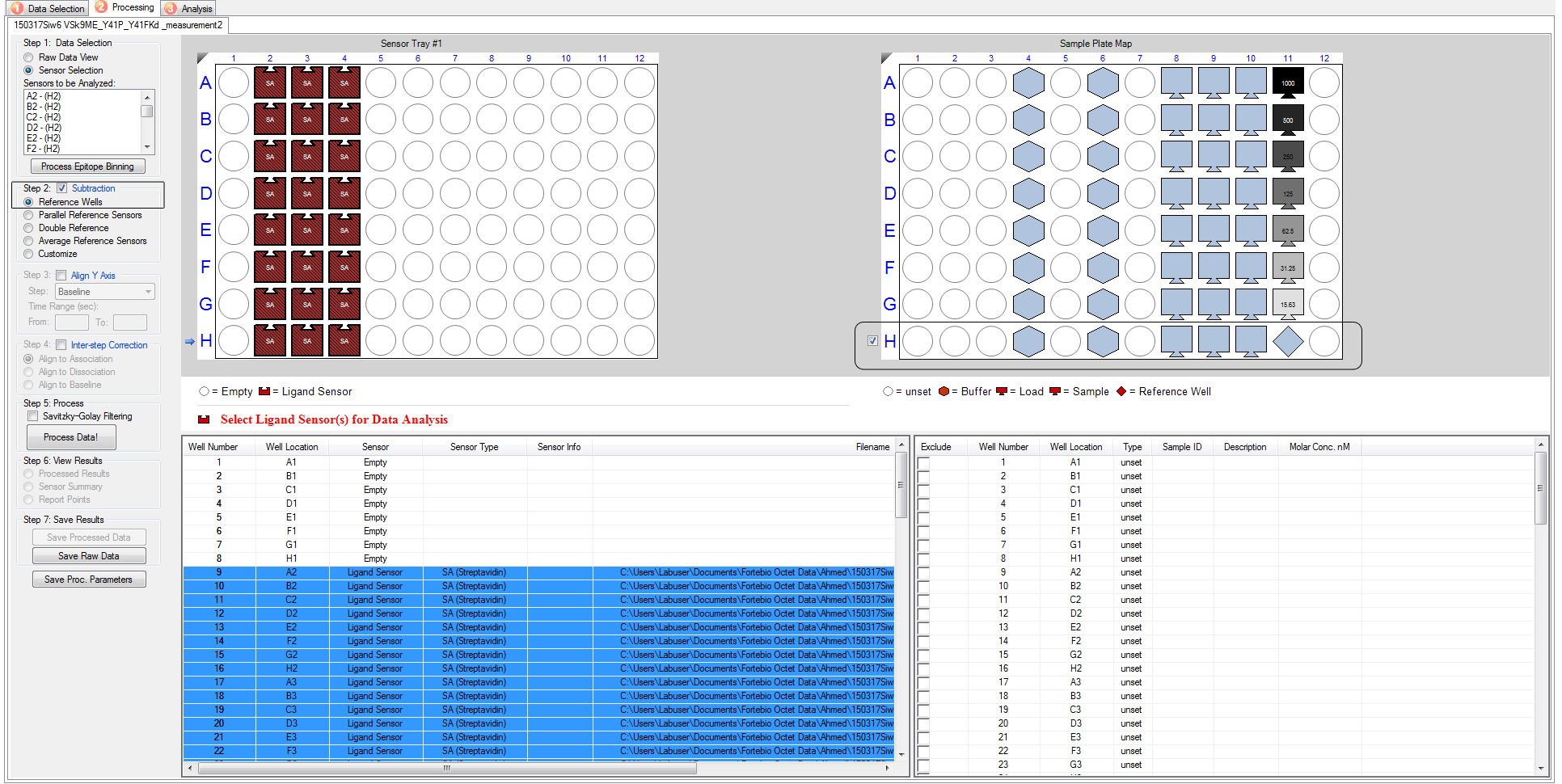
Figure 11. Screen capture of the BLI data process. “Subtraction” box and reference wells are indicated in square boxes. Reference wells have an association well (H 11) with no protein in the Assay buffer.
- After data processing, further analyze the processed data and obtain the dissociation constant (KD) in the “Analysis” tab.
- In the “step to analyze” box, choose “association and dissociation” and model 1:1.
- In the “fitting” box, select the global fitting grouped by color and Rmax unlinked by sensor (Figure 12). Click the “fit curves” button to start the fitting. Non-linear regression fitting was used to analyze the data and derive the dissociation constants (KD). Goodness of the data fitting is validated by R2 and χ2 values.

Figure 12. Processed BLI data. Upper left, raw data; upper right, data after reference well subtraction; bottom left, data after aligning the Y-axis to baseline; bottom right, data after aligning the X-axis to the association step.
- For result viewing, fitting curves can be grouped in different ways. For example, in Figure 13, we group graphs by loading sample ID, so that curves with the same loading sample ID (same peptide name) will be included in the same graph.
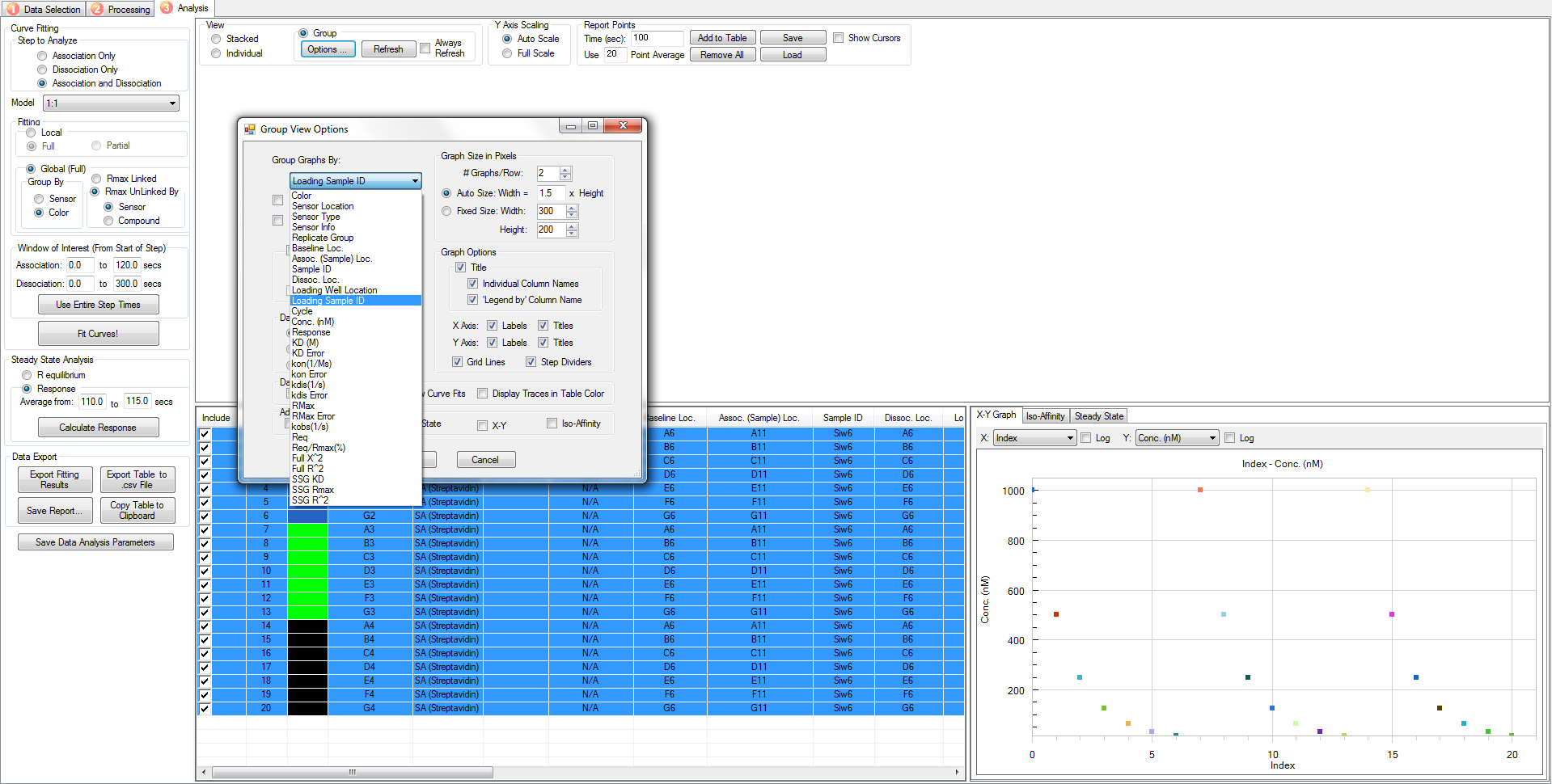
Figure 13. Screen capture of data analysis surface. An example of a graph with data sorted according to loading sample ID. - Save and export the data file. Quantitative results, including KD value, R2 and χ2 can be found in the “result table” worksheet of the Excel report.
Notes
- Problem: Recombinant protein cannot be stably expressed
Solution: Some proteins are easily degraded during expression. Several optimizations can be made to minimize protein degradation during expression.- Reduce the concentration of IPTG to 0.5 mM (Larentis et al., 2014).
- Conduct the protein expression at a lower temperature range (from 16 °C to 26 °C) over a longer induction time (24 h-32 h) (de Groot and Ventura, 2006; San-Miguel et al., 2013).
- Change the growth media to Terrific Broth (Tartof and Hobbs, 1987).
- Pre-cool the buffers and maintain the working temperature at 0 °C to 4 °C, as this could help to minimize degradation during protein extraction and purification.
- Add protease inhibitor tablets (CompleteTM, Mini, EDTA-free Protease inhibitors, Roche) to the cell lysate after sonication step.
- Problem: Additional bands in the purified protein
Solution: After protein purification, take 5 μl of the protein, mix with 2x Sample buffer, denature at 95 °C and use SDS-PAGE gel to check the purity. If additional bands can be observed using Coomassie blue stain, further changes can be made to remove the non-specifically bound proteins.- Increase the imidazole concentration to 20-50 mM in Wash buffer (Crowe et al., 1996; Bornhorst and Falke, 2000).
- Add 0.1% NP-40 into the Wash buffer.
- If unspecific bands are smaller in size compared to target protein, use a larger MWCO (>10K) (Molecular Weight Cut-Off) for the protein concentrator.
- Use gel chromatography for purification.
- Problem: Protein shows high background binding in the Assay buffer
Solution: Assay buffer optimization is a critical step required before the BLI assay. The non-specific interaction between the protein and the sensors (without immobilization of the biotinylated peptides) needs to be minimized. Below are several buffers that can be tested (see Recipes section):- 20 mM Tris pH 8.0, 200 mM NaCl
- 20 mM Tris pH 8.0, 200 mM NaCl, 0.1% BSA, 0.1% Tween-20
- TBS assay buffer (see Recipes)
- PBS assay buffer (see Recipes)
- MES assay buffer (see Recipes)
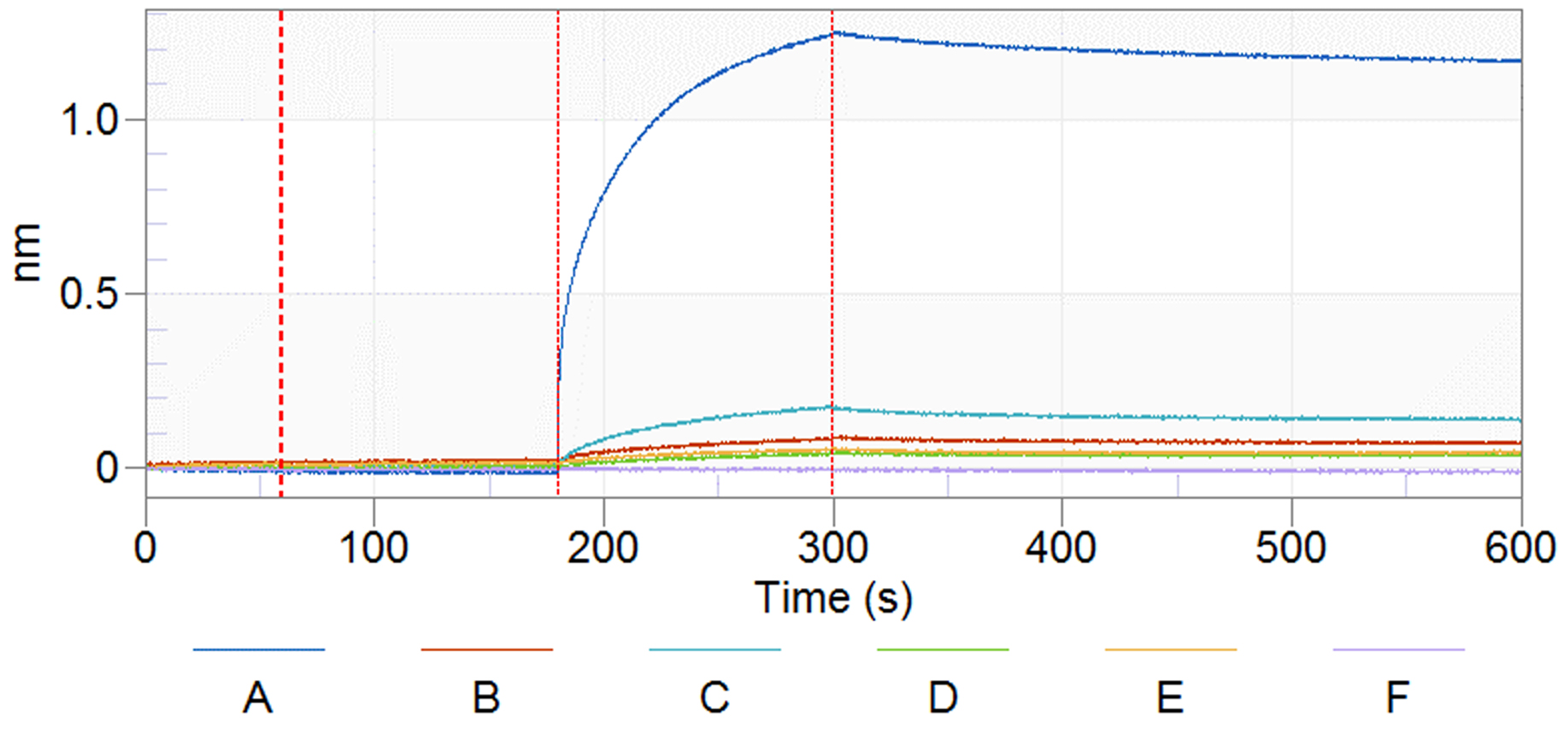
Figure 14. Results from a buffer test. The X-axis indicates the timeline of the experiment. The Y-axis shows the signal shift for each time point. Red vertical lines indicate the transition point of 4 different steps of the experiment: baseline, loading, association, and dissociation. No peptides are added in the loading wells. Components of buffer A to E are stated in the text. F, reference well loaded with H2O.
Non-specific binding may also be observed when the concentration of the test protein is too high. Concentration screening can be used to determine the optimum concentration of protein for the assay without showing a background interaction with the unloaded sensors. The optimum protein concentration for the BLI assay is variable for each protein. However, lower concentrations of proteins are recommended to minimize the non-specific binding signal from the sample matrix.
Recipes
- LB medium (1 L)
10 g Tryptone
10 g NaCl*
5 g Yeast extract
Adjust to pH 7.0, autoclave - Phosphate buffer saline (10x PBS, 1 L)
80 g NaCl*
2 g KCl
14.4 g Na2HPO4
2.4 g KH2PO4
Adjust pH to 7.4 and autoclave or filter sterilize
For 1x PBS, dilute 100 ml of 10x PBS with 900 ml of deionized H2O and autoclave or filter sterilize - Lysis buffer
50 mM NaH2PO4
300 mM NaCl*
10 mM Imidazole
Adjust to pH 8.0 using NaOH; filter sterilize and store at room temperature
Pre-chill to 4 °C and add in lysozyme (1 mg/ml), DNase I (5 μg/ml) and RNase A (10 μg/ml) before use - 2x Sample buffer
62.5 mM Tris-HCl pH 6.8
25% (v/v) glycerol
2% (w/v) SDS
0.01% (w/v) bromophenol blue
Store at room temperature
Add 1/20 (v/v) of β-mercaptoethanol before use - Wash buffer
50 mM NaH2PO4
300 mM NaCl*
20 mM Imidazole
Adjust to pH 8.0
Filter sterilize, store and use at 4 °C - Elution buffer
50 mM NaH2PO4
300 mM NaCl*
500 mM Imidazole
Filter sterilize, store and use at 4 °C - Exchange buffer
200 mM NaCl*
50 mM Tris-HCl
Adjust to pH 7.5 with HCl
Filter sterilize, store and use at 4 °C - MES Assay buffer (pH 6.0)
20 mM 2-Morpholinoethanesulfonic acid sodium salt (MES Na)
200 mM NaCl
Adjust to pH 6.0
0.1% BSA
0.1% Tween-20
Filter sterilize, store and use at 4 °C - PBS Assay buffer (pH 7.4)
1x PBS (dilution from 10x PBS Vivantis)
Note: 1x PBS solution contains 137 mM NaCl, 2.7 mM KCl and 10 mM Phosphate.
0.1% BSA
0.1% Tween-20
Filter sterilize, store and use at 4 °C - TBS Assay buffer (pH 8.5)
20 mM Tris
200 mM NaCl
Adjust to pH 8.5
0.1% BSA
0.1% Tween-20
Filter sterilize, store and use at 4 °C
Acknowledgments
This work was supported by a Singapore Ministry of Education Tier 1 grant (Reference: R-183-000-389-112) awarded to E.S.C. We thank Rebecca Jackson for critically editing this manuscript.
Competing interests
The authors declare no competing interests.
References
- Abdiche, Y., Malashock, D., Pinkerton, A. and Pons, J. (2008). Determining kinetics and affinities of protein interactions using a parallel real-time label-free biosensor, the Octet. Anal Biochem 377(2): 209-217.
- Allshire, R. C. and Madhani, H. D. (2018). Ten principles of heterochromatin formation and function. Nat Rev Mol Cell Biol 19(4): 229-244.
- Berggard, T., Linse, S. and James, P. (2007). Methods for the detection and analysis of protein-protein interactions. Proteomics 7(16): 2833-2842.
- Bornhorst, J. A. and Falke, J. J. (2000). Purification of proteins using polyhistidine affinity tags. Methods Enzymol 326: 245-254.
- Cheung, P. and Lau, P. (2005). Epigenetic regulation by histone methylation and histone variants. Mol Endocrinol 19(3): 563-573.
- Crowe, J., Masone, B. S. and Ribbe, J. (1996). One-step purification of recombinant proteins with the 6xHis tag and Ni-NTA resin. Methods Mol Biol 58: 491-510.
- de Groot, N. S. and Ventura, S. (2006). Effect of temperature on protein quality in bacterial inclusion bodies. FEBS Lett 580(27): 6471-6476.
- Felsenfeld, G. and Groudine, M. (2003). Controlling the double helix. Nature 421(6921): 448-453.
- Guetg, C. and Santoro, R. (2012). Formation of nuclear heterochromatin: the nucleolar point of view. Epigenetics 7(8): 811-814.
- Kamat, V. and Rafique, A. (2017). Designing binding kinetic assay on the bio-layer interferometry (BLI) biosensor to characterize antibody-antigen interactions. Anal Biochem 536: 16-31.
- Larentis, A. L., Nicolau, J. F., Esteves Gdos, S., Vareschini, D. T., de Almeida, F. V., dos Reis, M. G., Galler, R. and Medeiros, M. A. (2014). Evaluation of pre-induction temperature, cell growth at induction and IPTG concentration on the expression of a leptospiral protein in E. coli using shaking flasks and microbioreactor. BMC Res Notes 7: 671.
- Musselman, C. A., Lalonde, M. E., Cote, J. and Kutateladze, T. G. (2012). Perceiving the epigenetic landscape through histone readers. Nat Struct Mol Biol 19(12): 1218-1227.
- Nirschl, M., Reuter, F. and Voros, J. (2011). Review of transducer principles for label-free biomolecular interaction analysis. Biosensors (Basel) 1(3): 70-92.
- Petersen, R. L. (2017). Strategies using bio-layer interferometry biosensor technology for vaccine research and development. Biosensors (Basel) 7(4): 49.
- Phizicky, E. M. and Fields, S. (1995). Protein-protein interactions: methods for detection and analysis. Microbiol Rev 59(1): 94-123.
- Ren, B., Tan, H. L., Nguyen, T. T. T., Sayed, A. M. M., Li, Y., Mok, Y. K., Yang, H. and Chen, E. S. (2018). Regulation of transcriptional silencing and chromodomain protein localization at centromeric heterochromatin by histone H3 tyrosine 41 phosphorylation in fission yeast. Nucleic Acids Res 46(1): 189-202.
- Rowley, M. J. and Corces, V. G. (2016). Capturing native interactions: intrinsic methods to study chromatin conformation. Mol Syst Biol 12(12): 897.
- San-Miguel, T., Perez-Bermudez, P. and Gavidia, I. (2013). Production of soluble eukaryotic recombinant proteins in E. coli is favoured in early log-phase cultures induced at low temperature. Springerplus 2(1): 89.
- Suganuma, T. and Workman, J. L. (2011). Signals and combinatorial functions of histone modifications. Annu Rev Biochem 80: 473-499.
- Tartof, K. D. and Hobbs, C. A. (1987). Improved media for growing plasmid and cosmid clones. Focus 9: 2-12.
- Wierer, M. and Mann, M. (2016). Proteomics to study DNA-bound and chromatin-associated gene regulatory complexes. Hum Mol Genet 25(R2): R106-R114.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Ren, B., Sayed, A. M. M., Tan, H. L., Mok, Y. K. and Chen, E. S. (2018). Identifying Protein Interactions with Histone Peptides Using Bio-layer Interferometry. Bio-protocol 8(18): e3012. DOI: 10.21769/BioProtoc.3012.
Category
Molecular Biology > Protein > Protein-protein interaction
Microbiology > Microbial biochemistry > Protein > Interaction
Biochemistry > Protein > Modification
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.