- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Quantitative ChIP-seq by Adding Spike-in from Another Species
Published: Vol 8, Iss 16, Aug 20, 2018 DOI: 10.21769/BioProtoc.2981 Views: 22046
Reviewed by: Neelanjan BoseSabine Le SauxAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2018
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

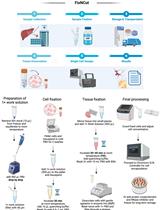
Abstract
Chromatin immunoprecipitation followed by sequencing (ChIP-seq) is a routine procedure in the lab; however, epigenome-wide quantitative comparison among independent ChIP-seq experiments remains a challenge. Here, we contribute an experimental protocol combined with a computational workflow allowing quantitative and comparative assessment of epigenome using animal tissues.
Keywords: EpigenomeBackground
Chromatin and epigenetic complexes that modify histones regulate the accessibility of DNA to transcriptional machinery, thereby permitting direct control of gene expression. To characterize the epigenomic feature of histone modification, chromatin immunoprecipitation followed by sequencing (ChIP-seq) has become a widely used method. However, traditional ChIP-seq protocols are not inherently quantitative and therefore prohibit direct comparison between samples derived from distinct cell types or cells that have been through different genetic or chemical perturbation. Despite the fact that several in silico normalization methods have been proposed to overcome this disadvantage, an experiment-based strategy is still lacking. In 2014, Orlando et al. (2014) developed a method, called ChIP with reference exogenous genome (ChIP-Rx), which utilizes a constant amount of reference or ‘‘spike-in’’ epigenome for cell-based comparison among epigenomes. In current protocol, we have refined this method by using the percentage of mapped spike-in reference epigenome. And we have successfully applied this protocol in direct comparison between two or more ChIP-seq datasets from animal tissues.
Materials and Reagents
- Consumables
- Pipette tips
- 1.5 ml microcentrifuge tube
- 15 ml microcentrifuge tube
- 1.5 ml Bioruptor Microtubes (Diagenode, catalog number: C30010016 )
- 10 cm Petri-dish
- Glass Dounce tube
- Biological materials
- Mouse Neuro-2a cells
- Drosophila (1,000 embryos [30 min-1 h after egg-laying], 30 larvae [96 h after egg-laying], and 30 pupae [7 d after egg-laying])
Starting material: Cells were cultured at 37 °C, 5% CO2, and saturated humidity in complete LG-DMEM (Thermo Fisher Scientific, Life Technologies, catalog number: 11995-065 ) containing 10% FBS (Sigma-Aldrich, catalog number: 12003C ), seeded at 1.0 x 106 cells/cm2 in a 10 cm Petri-dish. Flies were cultured in standard Drosophila media (Recipe 1) at 25 °C with 60% humidity in a 12 h light and 12 h dark cycle unless otherwise specified.
- Reagents
ChIP reagents
- Formaldehyde, 37% (weight/volume) (Sigma-Aldrich, catalog number: 252549 ), stored at room temperature (20-25 °C)
- Glycine (Sigma-Aldrich, catalog number: G7403 ), stored at room temperature (20-25 °C)
- PBS buffer, 20x (Sangon Biotech, catalog number: B548117 ), stored at room temperature (20-25 °C)
- RIPA buffer (Sigma-Aldrich, catalog number: R0278 ), stored at 4 °C
- Tris buffer, 1 M, PH 8.0 (Sangon Biotech, catalog number: B548127 ), stored at room temperature (20-25 °C)
- Sodium chloride, 5 M (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM9759 ), stored at room temperature (20-25 °C)
- Triton X-100 (Sigma-Aldrich, catalog number: T8787 ), stored at 4 °C
- EDTA, 0.5 M, pH 8.0 (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM9260G ), stored at room temperature (20-25 °C)
- SDS, 10% (weight/volume) (Sangon Biotech, catalog number: B548118 ), stored at room temperature (20-25 °C)
- cOmplete proteinase inhibitors cocktail tablets (Roche Diagnostics, catalog number: 11697498001 ), stored at 4 °C
- Dynabeads Protein G (Thermo Fisher Scientific, InvitrogenTM, catalog number: 10004D ), stored at 4 °C
- Anti-trimethyl Histone H3 (Lys27) antibody (Merck, catalog number: 07-449 ), stored at -20 °C
- Sodium bicarbonate (Sangon Biotech, catalog number: A100865 ), stored at room temperature (20-25 °C)
- Proteinase K (Sangon Biotech, catalog number: A600451 ), stored at -20 °C
- RNase A, 10 mg/ml (Thermo Fisher Scientific, catalog number: EN0531 ), Stored at -20 °C
- PCR purification kit (QIAGEN, catalog number: 28106 ), stored at room temperature (20-25 °C)
- Qubit dsDNA HS assay kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: Q32854 ), stored at 4 °C (Warning: protect from light)
- Trypsin (Thermo Fisher Scientific, catalog number: 25300054 )
- Liquid nitrogen
- Glycine, 2.5 M (see Recipes)
- ChIP Wash Buffer (see Recipes)
- ChIP Final Wash Buffer (see Recipes)
- ChIP Elution Buffer (see Recipes)
- Agencourt AMPure XP (SPRI beads; Beckman Coulter, catalog number: A63881 ), stored at 4 °C
- NEBNext Ultra DNA Library Prep Kit for Illumina (New England Biolabs, catalog number: E7370L ), stored at -20 °C
- NEBNext Multiplex Oligos for Illumina (New England Biolabs, catalog number: E7335L ), stored at -20 °C
- Gel extraction kit (QIAGEN, catalog number: 20021 ), stored at room temperature (20-25 °C)
- Sodium hydroxide solution, 10 M (Sigma-Aldrich, catalog number: 72068 )
Equipment
- Bioruptor Pico (Diagenode)
- DynaMag-2 magnet (Thermo Fisher Scientific, catalog number: 12321D )
- Mortar and pestle
- 1 ml glass homogenizer
- Water bath
- Rotate
- Centrifuge
- Vortexing
- QuantStudio 6 Flex real-time PCR system (Thermo Scientific, USA)
- Qubit 2.0 Fluorometer (Life Technologies)
- Agilent Bioanalyzer 2100
- Illumina Miseq System
- Illumina Nextseq 550 System
Software
- Quality check of sequence reads: FastQC v0.11.7
(http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) - Read mapping: Bowtie2-2.2.9 (Langmead and Salzberg, 2012)
- Determination of the normalization factor: deeptools-2.2.4 (Ramirez et al., 2014)
- Quantitative analyses: Bwtool (Pohl and Beato, 2014)
- Peak calling: homer-v4.8.3 (Heinz et al., 2010)
- Peak visualization: IGV-2.3.31 (Robinson et al., 2011)
- J-circos-V1 (An et al., 2015)
Procedure
- Crosslinking and lysis of mammalian cells and fly tissues
- Mouse Neuro-2a cells
- Grow mouse Neuro-2a cells to a level of ~106 cells per 10 cm Petri-dish. For this experiment, use ~106 cells. Aspirate and discard the medium. Wash two times each with 4 ml 1x PBS pre-warmed in a 37 °C water bath. Add 1 ml trypsin (Thermo Fisher Scientific, USA) and incubate at 37 °C for 2 min. Quench by adding 3 ml standard cell culture medium. Collect the mixture into a 15 ml centrifuge tube.
- Add 108 μl of 37% formaldehyde (Sigma, USA) to a final concentration of 1% (weight/volume) for crosslinking. Rotate at room temperature for 10 min.
- Stop crosslinking with 200 μl of 2.5 M glycine (Sigma, USA) for 5 min at room temperature. Centrifuge at 1,000 x g for 3 min at 4 °C. Discard the supernatant.
- Resuspend the pellet by adding 1 ml ice-cold 1x PBS with 1x cOmplete proteinase inhibitor (Roche, Germany), and transfer it into a 1.5 ml microcentrifuge tube.
- Centrifuge at 1,000 x g for 3 min at 4 °C. After removing the supernatant, wash the pellet two times each with 1 ml ice-cold 1x PBS containing 1x cOmplete proteinase inhibitor (Roche, Germany).
- Add 1 ml of 1x RIPA buffer (Sigma, USA) supplemented with 1x cOmplete proteinase inhibitor (Roche, Germany). Incubate at 4 °C for 30 min.
- Fly tissues
For developmental stages, use 1,000 embryos (30 min-1 h after egg-laying), 30 larvae (96 h after egg-laying), and 30 pupae (7 d after egg-laying). For adult stage, use 100 dissected fly muscles and 200 fly heads at each time point.- Collect samples into a 1.5 ml microcentrifuge tube and immediately freeze samples in liquid nitrogen.
- Grind tissues into fine powder using mortar and pestle pre-cooled with liquid nitrogen.
- Resuspend the fine powder in 1.2 ml 1x PBS. Add 32.4 μl of 37% formaldehyde (Sigma, USA). Incubate at room temperature for 10 min. To quench formaldehyde, add 60 μl of 2.5 M glycine (Sigma, USA). Centrifuge at 5,000 x g for 5 min at 4 °C. After removing the supernatant, wash the pellet three times with 1 ml ice-cold 1x PBS containing 1x cOmplete proteinase inhibitor (Roche, Germany).
- Add 1 ml of 1x RIPA buffer (Sigma, USA) supplemented with 1x cOmplete proteinase inhibitor (Roche, Germany). Homogenize the tissue pellet with a glass Dounce tube, and transfer it into a 1.5 ml microcentrifuge tube. Rotate at 4 °C for 1 h.
- Mouse Neuro-2a cells
- Sonication
- Split the lysate into 250 μl aliquots for four pre-chilled 1.5 ml Bioruptor microtubes (Diagenode, Belgium).
- For mouse Neuro-2a cells, sonicate at 4 °C using Bioruptor Pico (Diagenode, Belgium) for 6 cycles with 15 sec on and 15 sec off. For fly tissues, sonicate at 4 °C using Bioruptor Pico (Diagenode, Belgium) for 15 cycles with 30 sec on and 30 sec off.
- Transfer sonicated samples into a 1.5 ml microcentrifuge tube.
- Centrifuge at 12,000 x g for 20 min at 4 °C, and transfer the supernatant to a new tube.
- Store sonicated samples at -80 °C.
- Determination of chromatin size and concentration
- Use 30 μl aliquot of chromatin sample from the sonicated lysate, and add 90 μl of 1x RIPA buffer (Sigma, USA).
- Add 1 μl RNase A (Thermo Scientific, USA), and incubate at 37 °C for 30 min to remove RNA.
- To reverse crosslinking, add 5.04 μl of 5 M NaCl (Thermo Scientific, USA), and incubate at 65 °C for 4 h.
- Add 1.5 μl of 0.5 M EDTA (Thermo Scientific, USA) and 1.2 μl of 20 mg/μl proteinase K (Sangon Biotech, China) at 55 °C for 2 h.
- Isolate DNA by PCR purification kit (QIAGEN, Germany), and elute DNA in 30 μl of Milli-Q water.
- Quantify DNA by Qubit dsDNA HS assay kit (Thermo Scientific, USA). A concentration of about 2-3 ng/μl DNA (~30 μl) would be expected from 200 fly heads.
- Examine the chromatin DNA on a 1.5% agarose gel to visualize average size. The optimal size range is between 100 bps and 300 bps. If the chromatin is not in that range, adjust sonication conditions by adding more pulses and repeat Step C3.
- Chromatin immunoprecipitation
- According to the DNA mass (the volume of fly sample x the concentration of fly sample measured in Procedure C), add 5% (weight/weight) of the mouse epigenome to the fly sample, and mix well. Save 1% (volume/volume) of the sample to a new tube as ChIP input control, and freeze at -20 °C until the elution step.
- Add 3 μg antibody, and rotate at 4 °C for 5 h.
- Place a magnetic stand (Thermo Scientific, USA) on ice. Add 30 μl Dynabeads (Thermo Scientific, USA) to a 1.5 ml microcentrifuge tube, and wash three times with 1x RIPA buffer (Sigma, USA). Collect beads using magnetic stand (Thermo Scientific, USA), and remove supernatant by aspiration.
- Add samples (Fly + Mouse Neuro-2a + antibody) to the pre-washed Dynabeads (Thermo Scientific, USA). Gently mix overnight on a rotator at 4 °C.
- Apply ChIP sample to an ice-cold magnetic stand (Thermo Scientific, USA). Remove supernatant by aspiration.
- Wash the beads one time with 1x RIPA buffer (Sigma, USA).
- Wash the beads two times with ChIP Wash Buffer.
- Wash the beads one time with ChIP Final Wash Buffer.
- Elution, crosslinking reversal and DNA isolation
- Resuspend the beads in 120 μl of ChIP Elution Buffer. Incubate at 65 °C for 30 min.
- Add 1 μl RNase A (Thermo Scientific, USA), and incubate at 37 °C for 30 min to remove RNA. At the same time, dilute input sample with 1x RIPA buffer (Sigma, USA) to 120 μl, and incubate with 1 μl RNase A (Thermo Scientific, USA) at 37 °C for 30 min.
- To reverse crosslinking, add 5.04 μl of 5 M NaCl (Thermo Scientific, USA), and incubate at 65 °C for 4 h.
- Add 1.5 μl of 0.5 M EDTA (Thermo Scientific, USA) and 1.2 μl of 20 mg/μl proteinase K (Sangon Biotech, China) at 55 °C for 2 h.
- Isolate DNA by PCR purification kit (QIAGEN, Germany), and elute DNA in 50 μl water.
- Quantify DNA by Qubit dsDNA HS assay kit (Thermo Scientific, USA). A concentration of about 0.15-0.3 ng/μl DNA (~50 μl) would be expected from one experiment of immunoprecipitation.
- Quality control for ChIP experiment
- Performing ChIP-qPCR assays
- Design primers to yield PCR product between 100 bps and 200 bps.
- Dilute input DNA with RIPA buffer to the same concentration with IP DNA.
- Set up real-time PCRs in triplicate with SYBR selected master mix (Thermo Scientific, USA), and the same volume of diluted input DNA and IP DNA.
- Mix the samples by vortexing for 2 sec and precipitate samples by brief centrifugation.
- Perform real-time PCR with the QuantStudio 6 Flex real-time PCR system (Thermo Scientific, USA) using cycling conditions as shown in the flowing table:

- Calculate the fold difference between the experimental sample and negative control.
- Preparing high-throughput sequencing library
- Use 5-10 ng of DNA harvested by ChIP experiment to generate sequencing library using NEB DNA library prep kit (NEB, USA).
- Check the quality of libraries with Bioanalyzer 2100 (Agilent, USA).
- Perform quantification by qRT-PCR with a reference to a standard library.
- Pool the libraries together in equimolar amounts to a final 2 nM concentration.
- Denature the normalized libraries with 0.1 M NaOH (Sigma, USA).
- Sequence the pooled libraries on the Miseq/Next-seq platform (Illumina, USA) with single end 100 bps.
- Performing ChIP-qPCR assays
Data analysis
The following procedures showed the detailed steps for data analysis (Figure 1).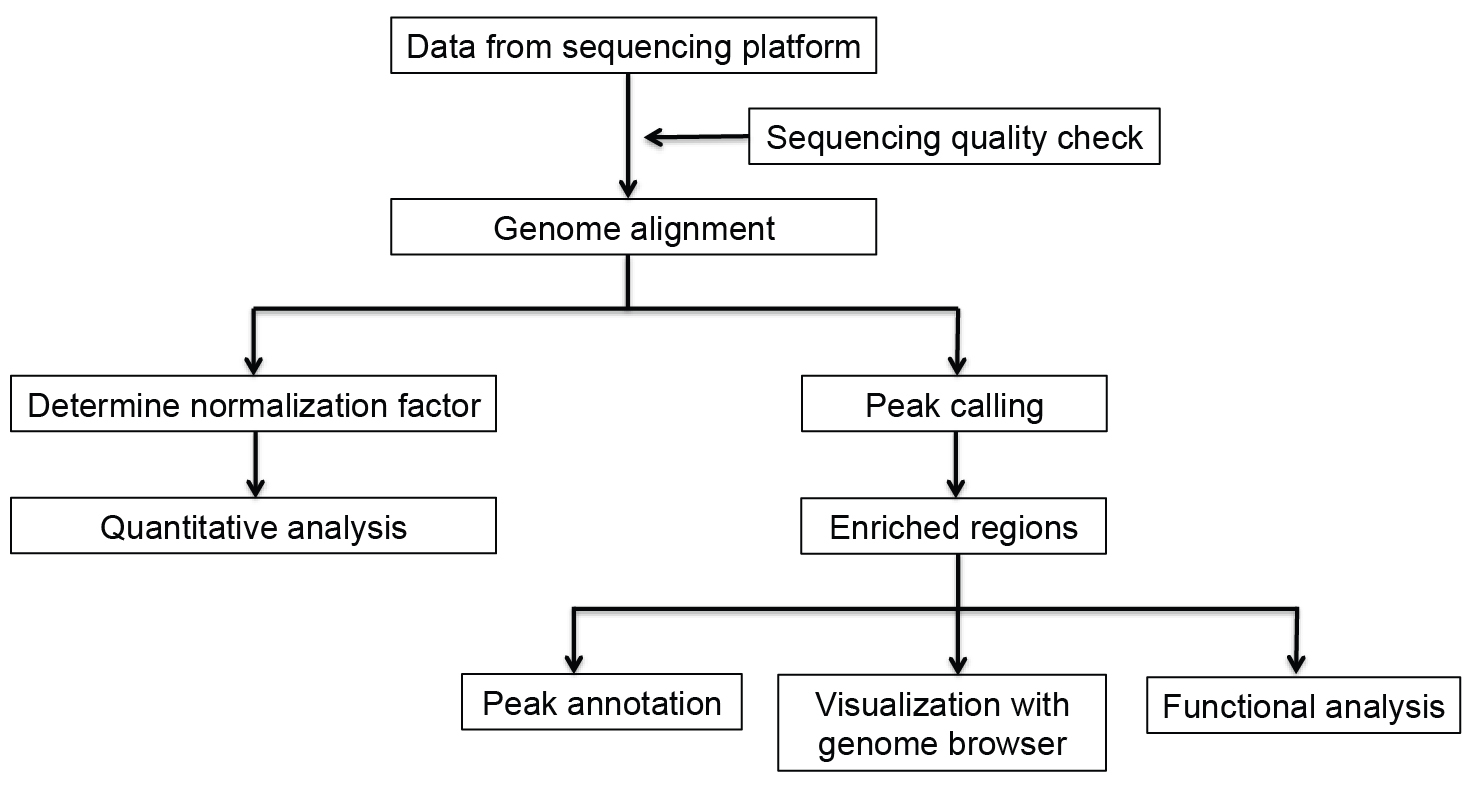
Figure 1. Overview of ChIP-seq analysis
- Sequence quality check
Use FastQC to assess the read quality by importing data from FastQ files. - Read mapping
Map Sequence reads to the reference genome dm6 (Drosophila) or mm10 (mouse), respectively with Bowtie2-2.2.9 by default parameters.
># Map Sequencing reads to the reference genome mm10
>nohup bowtie2 -x / seqlib/igenome/Mus_musculus/UCSC/mm10/Sequence/Bowtie2Index/genome -U {sample}.fastq.gz -S {sample}_mm10.sam --no-unal &
># Map Sequencing reads to the reference genome dm6
>nohup bowtie2 -x / seqlib/igenome/Drosophila_melanogaster/UCSC/dm6/Sequence/Bowtie2Index/genome -U {sample}.fastq -S {sample}_dm6.sam --no-unal &
># For sample in ChIP_dm6, do the following
># Convert file from sam to bam
>samtools view -Sb {sample}_dm6.sam > {sample}_dm6_nonSorted.bam
># Sort BAM file
>samtools sort {sample}_dm6_nonSorted.bam -o {sample}_dm6_Sorted.bam
># Create index files
>samtools index {sample}_dm6_Sorted.bam - Determination of the normalization factor
For quantitative comparison, we derive of the scale factor for each sample using the percentage of mapped reads from mouse genome to total reads. Details are as follows:- Combine the number of mapped reads from Drosophila and mouse genomes as total mapped reads for each sample.
Let:
α = the spike-in scale factor
β = the histone modification level
γ = the percentage of input mouse reads in total input mapped reads
Nm = the number of mouse mapped reads (in millions) in IP sample
Nd = the number of Drosophila mapped reads (in millions) in IP sample - Calculate spike-in scale factor as follows.
α = γ/Nm - Calculate histone modification level as follows.
β = Nd x α - Normalize the dm6 mapped reads to the scale factor using deeptools-2.2.4 function bamCoverage with 10 bp bin size.
># For sample in ChIP_dm6, do the following
>bamCoverage -b {sample}_ChIP_dm6_Sorted.bam -o {sample}_ChIP_dm6_scaleFactor.bw –scaleFactor α -bs 10 -p 2 –v
- Combine the number of mapped reads from Drosophila and mouse genomes as total mapped reads for each sample.
- Quantitative analysis
Calculate the ChIP intensity for each gene or region using the Bwtool function bwtool summary with default parameters.
>bwtool summary {gene}.bed {sample}_ChIP_dm6_scaleFactor.bw {sample}_{gene}_summary.xls –header - Peak calling
Identify Peak regions by homer-v4.8.3 function findPeaks with parameter "-style histone -F 2 -size 3000 -minDist 5000".
>makeTagDirectory {sample}_ChIP_tag -fragLength 200 {sample}_ChIP.sam -single
>makeTagDirectory {sample}_input_tag -fragLength 200 {sample}_input.sam -single
>findPeaks {sample}_ChIP_tag/ -style histone -o {sample}_size3K_Peaks.xls -i {sample}_input_tag/ -F 2 -size 3000 -minDist 5000 -fragLength 200 - Peak visualization
Display the confident peaks and enriched genome regions by IGV-2.3.31 with the bigwig files generated by bamCoverage. - Peak annotation
Perform peak annotation using homer function annotatePeaks with a default parameter.
>annotatePeaks.pl peaks.txt dm6.bed > annotatedPeaks.txt - Functional analyses
- Generated scatter plot by R package ggplot2.
Create a .csv file containing two columns. The first column contains the log2 values of 3 days’ H3K27me3 levels for protein-coding genes. The second column contains the log2 values of 30 days’ H3K27me3 levels for protein-coding genes.
Generate a scatter plot by R package ggplot2. The results showed that H3K27me3 modification increases with age in head (Figure 2).
>contour = read.csv(file="all_gene_3_30_log2.csv", header=T)
>x=contour[,1]
>y=contour[,2]
>df = data.frame(x,y)
>df <- data.frame(x = x, y = y, d = densCols(x, y, colramp = colorRampPalette(rev(rainbow(10,s=1,v=1,start=1/10, end = 7/10,alpha = 1)))))
>ggplot(df) + geom_point(aes(x, y, col = d), size = 0.01) + geom_density2d(aes(x,y),size=0.3, col="red") + scale_color_identity() + coord_fixed() + geom_abline(slope=1) + scale_y_continuous(limits = c(-4,4)) + scale_x_continuous(limits = c(-4,4))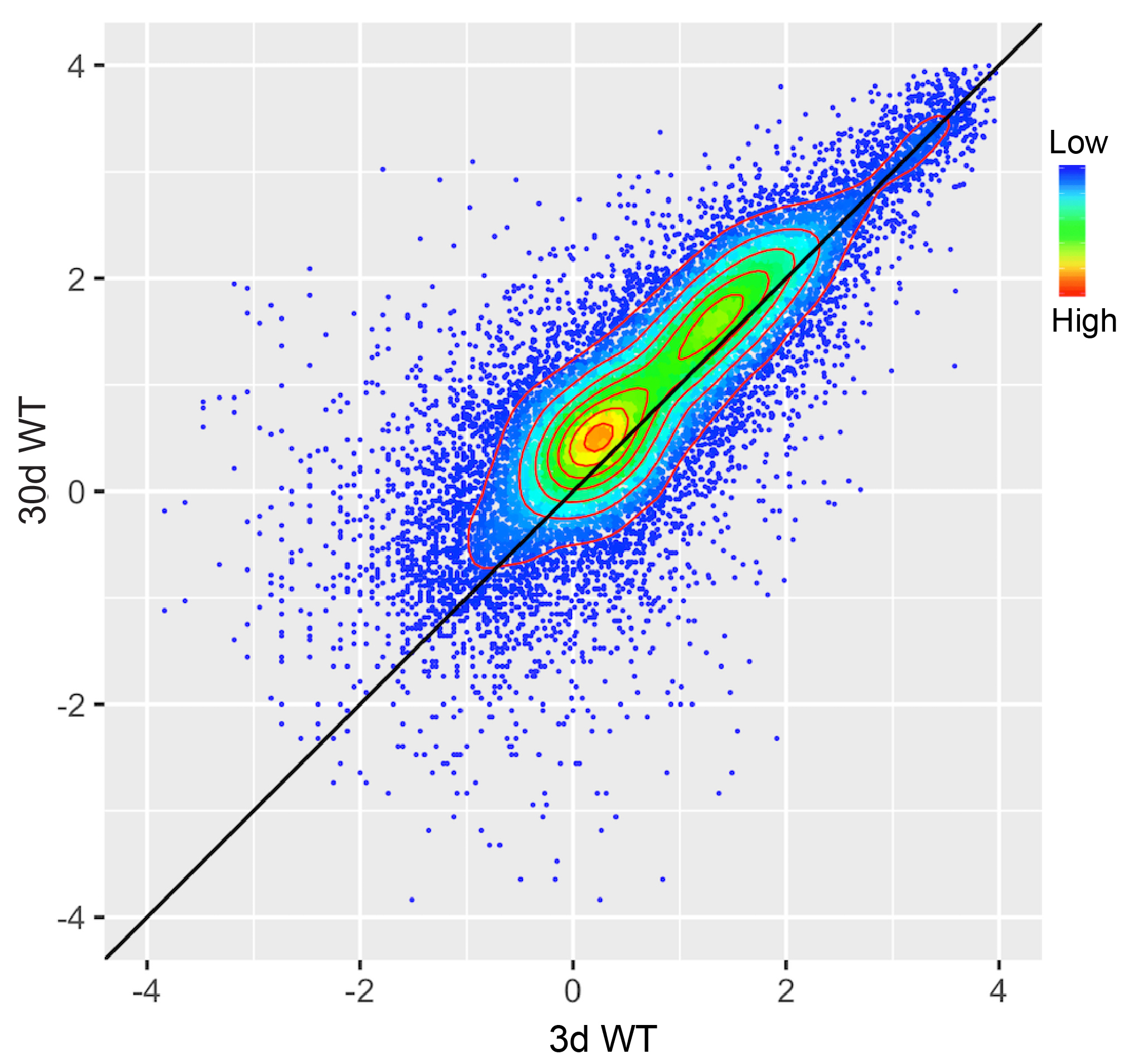
Figure 2. Scatter plot showing the comparison result between the H3K27me3 levels of protein-coding genes in 3 d old flies and 30 d ones. Protein-coding genes: the open reading frames of protein-coding genes annotated in dm6. The dm6 mapped reads were normalized to a scale factor to compare the relative H3K27me3 level quantitatively. Each dot on the plot represents a single gene locus. X- and Y-axis represented log2 mean value of gene’s reference-adjusted reads. Contour lines indicated that H3K27me3 signals were higher in aged flies compared to 3 d old flies (Niu and Liu, unpublished). - Generated Circos plot by J-circos-V1 with the bigwig files. The result showed the distribution of H3K27me3 peak profiles (Figure 3).
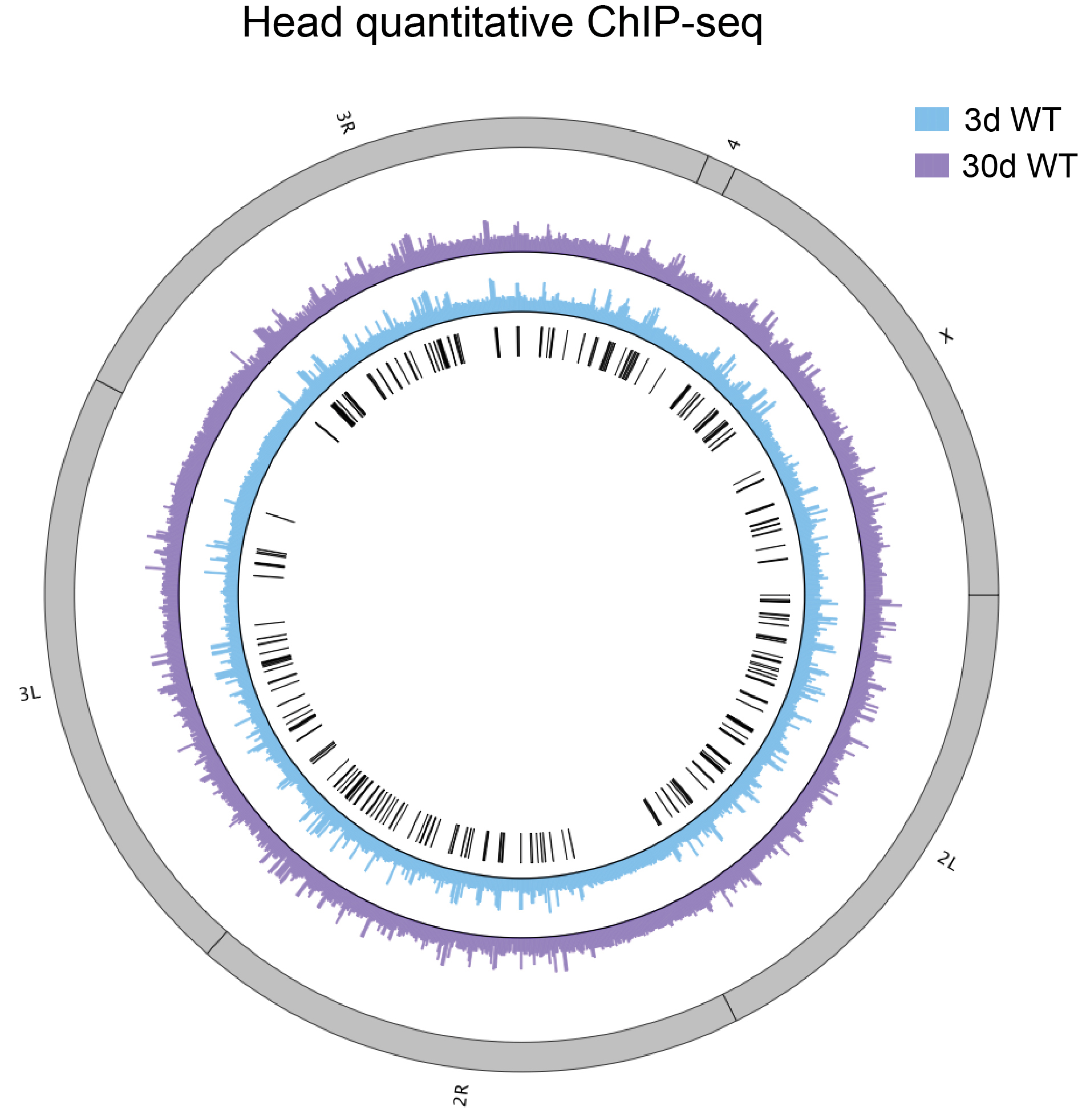
Figure 3. Circos plot showing peak profiles of H3K27me3 in 3d and 30d old flies. Black boxes and lines (innermost circle) represented common peak regions, corresponding to their chromosomal locations. Chromosome ideogram was in grey (outermost ring). ChIP-seq was from head tis sues of 3 d and 30 d old male flies. Circos plot of the H3K27me3 epigenome illustrates peak profiles with age in head (Niu and Liu, unpublished).
- Generated scatter plot by R package ggplot2.
Recipes
- Standard Drosophila media
ddH2O to make up 1 L of the foodSucrose 36 g/L Maltose 38 g/L Yeast 22.5 g/L Agar 5.4 g/L Maizena 60 g/L Soybean flour 8.25 g/L Sodium benzoate 0.9 g/L Methyl-p-hydroxybenzoate 0.225 g/L Methyl-p-hydroxybenzoate 6.18 ml/L - Glycine, 2.5 M
- Weigh 187.6 mg glycine and add 1 ml Milli-Q water
- Heat the mixture at 37 °C and mix it until glycine dissolves
- Store at room temperature (20-25 °C) for no more than 1 month
- ChIP Wash Buffer
- Mix 150 mM NaCl, 2 mM EDTA (pH 8.0), 20 mM Tris-HCl (pH 8.0), 1% Triton X-100 (volume/volume), and 0.1% SDS (volume/volume, use 10% SDS solution)
- Dilute the solution with Milli-Q water
- Store at 4 °C for no more than 6 months
- ChIP Final Wash Buffer
Mix 500 mM NaCl, 2 mM EDTA (pH 8.0), 20 mM Tris-HCl (pH 8.0), 1% Triton X-100 (volume/volume), and 0.1% SDS (volume/volume, use 10% SDS solution).
Note: Prepare the solution with Milli-Q water. Store at 4 °C for no more than 6 months. - ChIP Elution Buffer
Mix 100 mM NaHCO3 and 1% SDS (volume/volume, use 10% SDS solution).
Note: Freshly prepare the solution with Milli-Q water before elution step. Always put the solution at room temperature (20-25 °C) and ensure that there are no SDS precipitates before use.
Acknowledgments
This article is a part of works from our published article (Ma et al., 2018). This work was supported by grants from the National Program on Key Basic Research Project of China to N.L. (2016YFA0501900) and the National Natural Science Foundation of China to N.L. (31371326).
Competing interests
The authors declare that no competing interests exist.
References
- An, J., Lai, J., Sajjanhar, A., Batra, J., Wang, C. and Nelson, C. C. (2015). J-Circos: an interactive Circos plotter. Bioinformatics 31(9): 1463-1465.
- Heinz, S., Benner, C., Spann, N., Bertolino, E., Lin, Y. C., Laslo, P., Cheng, J. X., Murre, C., Singh, H. and Glass, C. K. (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38(4): 576-589.
- Langmead, B. and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9(4): 357-359.
- Ma, Z. J., Wang, H., Cai, Y. P., Wang, H., Niu, K. Y., Wu, X. F., MA, H. H., Yang, Y., Tong, W. H., Liu, F., Liu, Z. D., Zhang, Y. Y., Liu, R., Zhu, Z. J. and Liu, N. (2018). Epigenetic drift of H3K27me3 in aging links glycolysis to healthy longevity in Drosophila. eLife 7: e35368.
- Orlando, D. A., Chen, M. W., Brown, V. E., Solanki, S., Choi, Y. J., Olson, E. R., Fritz, C. C., Bradner, J. E. and Guenther, M. G. (2014). Quantitative ChIP-Seq normalization reveals global modulation of the epigenome. Cell Rep 9(3): 1163-1170.
- Pohl, A. and Beato, M. (2014). bwtool: a tool for bigWig files. Bioinformatics 30(11): 1618-1619.
- Ramirez, F., Dundar, F., Diehl, S., Gruning, B. A. and Manke, T. (2014). deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 42(Web Server issue): W187-191.
- Robinson, J. T., Thorvaldsdottir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G. and Mesirov, J. P. (2011). Integrative genomics viewer. Nat Biotechnol 29(1): 24-26.
Article Information
Copyright
![]() Niu et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Niu et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Niu, K., Liu, R. and Liu, N. (2018). Quantitative ChIP-seq by Adding Spike-in from Another Species. Bio-protocol 8(16): e2981. DOI: 10.21769/BioProtoc.2981.
- Ma, Z. J., Wang, H., Cai, Y. P., Wang, H., Niu, K. Y., Wu, X. F., MA, H. H., Yang, Y., Tong, W. H., Liu, F., Liu, Z. D., Zhang, Y. Y., Liu, R., Zhu, Z. J. and Liu, N. (2018). Epigenetic drift of H3K27me3 in aging links glycolysis to healthy longevity in Drosophila. eLife 7: e35368.
Category
Systems Biology > Epigenomics > Sequencing
Molecular Biology > DNA > DNA sequencing
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.