- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Delivery of the Cas9 or TevCas9 System into Phaeodactylum tricornutum via Conjugation of Plasmids from a Bacterial Donor
Published: Vol 8, Iss 16, Aug 20, 2018 DOI: 10.21769/BioProtoc.2974 Views: 8932
Reviewed by: David CisnerosRan ChenAnonymous reviewer(s)
Original research article
The authors used this protocol in:
Feb 2018
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


Abstract
Diatoms are an ecologically important group of eukaryotic microalgae with properties that make them attractive for biotechnological applications such as biofuels, foods, cosmetics and pharmaceuticals. Phaeodactylum tricornutum is a model diatom with defined culture conditions, but routine genetic manipulations are hindered by a lack of simple and robust genetic tools. One obstacle to efficient engineering of P. tricornutum is that the current selection methods for P. tricornutum transformants depend on the use of a limited number of antibiotic resistance genes. An alternative and more cost-effective selection method would be to generate auxotrophic strains of P. tricornutum by knocking out key genes involved in amino acid biosynthesis, and using plasmid-based copies of the biosynthetic genes as selective markers. Previous work on gene knockouts in P. tricornutum used biolistic transformation to deliver CRISPR-Cas9 system into P. tricornutum. Biolistic transformation of non-replicating plasmids can cause undesired damage to P. tricornutum due to random integration of the transformed DNA into the genome. Subsequent curing of edited cells to prevent long-term overexpression of Cas9 is very difficult as there is currently no method to excise integrated plasmids. This protocol adapts a new method to deliver the Cas9 or TevCas9 system into P. tricornutum via conjugation of plasmids from a bacterial donor cell. The process involves: 1) design and insertion of a guideRNA targeting the P. tricornutum urease gene into a TevCas9 expression plasmid that also encodes a conjugative origin of transfer, 2) installation of this plasmid in Escherichia coli containing a plasmid (pTA-Mob) containing the conjugative machinery, 3) transfer of the TevCas9 expression plasmid into P. tricornutum by conjugation, 4) screening of ex-conjugants for urease knockouts using T7 Endonuclease I and phenotypic screening, and 5) curing of the plasmid from edited cells.
Keywords: CRISPR-Cas9Background
The CRISPR system is a bacterial immune system that recognizes foreign DNA and leads to the activation of a targeted endonuclease, such as Cas9, which will generate a double strand break (DSB) in the invading DNA (Jinek et al., 2012; Wright et al., 2016). This system has been co-opted for genome editing applications whereby a single chimeric guide RNA (gRNA) can program Cas9 to target genes in model organisms, including in P. tricornutum (Daboussi et al., 2014). However, Cas9 generates blunt end DSB and the length of indels generated upon DNA repair is difficult to predict. Thus, in this study we used a dual endonuclease, TevCas9 to target the urease gene. TevCas9 is a dual endonuclease that is generated through the fusion of the I-TevI nuclease domain to Cas9 via a linker region (Wolfs et al., 2016). TevCas9 creates 33-36 bp deletions with non-compatible DNA ends (Wolfs et al., 2016). A previous study used transcription activator-like effector nucleases (TALENs) delivered by biolistic transformation (Weyman et al., 2015) to generate knockouts of the urease gene in P. tricornutum. To create an efficient system for gene editing in P. tricornutum, we adapted and optimized a plasmid-based system to deliver Cas9 or TevCas9 into P. tricornutum via conjugation from a bacterial donor cell (Karas et al., 2015). The new conjugation-based method to deliver Cas9 or TevCas9 described here is simpler, more efficient, and does not require specialized equipment for biolistic transformation (Slattery et al., 2018).
Materials and Reagents
- Pipette tips, 100-1,250 μl (VWR, catalog number: 89079-470 )
- Pipette tips, 1-200 μl (VWR, catalog number: 89079-478 )
- Pipette tips, 0.1-10 μl (VWR, catalog number: 89079-464 )
- 1.5 ml Eppendorf tubes (Corning, Axygen®, catalog number: MCT-150-C )
- 0.2 ml PCR tubes (VWR, catalog number: 20170-012 )
- Parafilm (Bemis, catalog number: PM996 )
- 50 ml centrifuge tubes (Greiner Bio One, catalog number: 227261 )
- NalgeneTM filters (Thermo Fisher Scientific, catalog number: 595-4520 )
- Phaeodactylum tricornutum cells [Culture Collection of Algae and Protozoa (CCAP, UK), catalog number: 1055/1 ] grown in 50 ml Falcon tubes
- TransforMaxTM EPI300TM Chemically competent E. coli cells (Lucigen, catalog number: C300C105 )
- pKS diaCas9_sgRNA plasmid (Addgene, catalog number: 74923 )
- BsaI-HF restriction endonuclease (New England Biolabs, catalog number: R0535S )
- P1 buffer (QIAGEN, catalog number: 19051 )
- β-mercaptoethanol (VWR, catalog number: 97064-588 )
- Alkaline lysis (P2) buffer (QIAGEN, catalog number: 19052 )
- Neutralization (P3) buffer (QIAGEN, catalog number: 19053 )
- Isopropanol (Sigma-Aldrich, catalog number: 190764 )
- 95% (v/v) EtOH (Commercial Alcohols, catalog number: P016EA95 )
- T4 DNA ligase with buffer (New England Biolabs, catalog number: M0202S )
- 2-Propanol-205 (Caledon Laboratories, catalog number: 8601-7-40 )
- BSA (Sigma-Aldrich, catalog number: A2058 )
- BsaI (New England Biolabs, catalog number: R0535S )
- EZ-10 spin column Miniprep Kit (Bio Basic, catalog number: BS614 )
- EZ-10 spin column PCR product purification kit (Bio Basic, catalog number: BS363 )
- Bacto agar (BD, BactoTM, catalog number: 214030 )
- Agar A (Bio Basic, catalog number: FB0010 )
- Yeast extract (BioShop, catalog number: YEX401 )
- Bacteriological tryptone (BioShop, catalog number: TRP402 )
- Sodium chloride (NaCl) (Fisher Scientific, Fisher Chemical, catalog number: S271-500 )
- AmpliTaq 360 DNA polymerase, 10x buffer, and 25 mM Magnesium Chloride (Thermo Fisher Scientific, Applied BiosystemsTM, catalog number: 4398828 )
- ZeocinTM (InvivoGen, catalog number: ant-zn-5p )
- Ampicillin (BioShop, catalog number: AMP201.100 )
- Gentamicin sulfate (Bio Basic, catalog number: GB0217 )
- D-Glucose (BioShop, catalog number: GLU501.1 )
- Zymolyase 20 T (BioShop, catalog number: ZYM001.1 )
- Tris (Fisher Scientific, Fisher BioReagents, catalog number: BP152-5 )
- Glycerol (Fisher Scientific, Fisher Chemical, catalog number: G33-1 )
- Sodium sulfate (Na2SO4) (Fisher Scientific, Fisher Chemical, catalog number: S421-300LB )
- Potassium chloride (KCl) (Merck, EMD Millipore, catalog number: PX1405-1 )
- Sodium bicarbonate (NaHCO3) (Merck, EMD Millipore, catalog number: SX0320-1 )
- Potassium bromide (KBr) (The Science Company, catalog number: NC-2136 )
- Boric acid (H3BO3) (BioShop, catalog number: BOR001.1 )
- Sodium fluoride (NaF) (Sigma-Aldrich, catalog number: S7920 )
- Magnesium chloride hexahydrate (MgCl2•6H2O) (BioShop, catalog number: MAG510.1 )
- Calcium chloride dihydrate (CaCl2•2H2O) (Fisher Scientific, catalog number: C79-3 )
- Strontium chloride hexahydrate (SrCl2•6H2O) (Sigma-Aldrich, catalog number: 255521 )
- Sodium nitrate (NaNO3) (Fisher Scientific, catalog number: S343-500 )
- Sodium phosphate monobasic monohydrate (NaH2PO4•H2O) (Sigma-Aldrich, catalog number: S9638 )
- Iron(III) chloride hexahydrate (FeCl3•6H2O) (Sigma-Aldrich, catalog number: 236489 )
- Na2EDTA•2H2O (Bio Basic, catalog number: EB0185 )
- Copper (II) sulfate pentahydrate (CuSO4•5H2O) (Bio Basic, catalog number: CDB0063 )
- Sodium molybdate dihydrate (Na2MoO4•2H2O) (Avantor Performance Materials, catalog number: 3764-01 )
- Zinc sulfate heptahydrate (ZnSO4•7H2O) (Alfa Aesar, catalog number: A12915-36 )
- Cobalt (II) chloride hexahydrate (CoCl2•6H2O) (Sigma-Aldrich, catalog number: C3169 )
- Manganese(II) chloride tetrahydrate (MnCl2•4H2O) (BioShop, catalog number: MAN222 )
- Selenous acid (H2SeO3) (Sigma-Aldrich, catalog number: 229857 )
- Nickel(II) sulfate (NiSO4) (Sigma-Aldrich, catalog number: 656895 )
- Sodium orthovanadate (Na3VO4) (BioShop, catalog number: SPP310 )
- Potassium chromate (K2CrO4) (Sigma-Aldrich, catalog number: 216615 )
- Thiamine-HCl (Sigma-Aldrich, catalog number: 47858 )
- Biotin (Sigma-Aldrich, catalog number: B4639 )
- Cyanocobalamin (Sigma-Aldrich, catalog number: C3607 )
- Zymogen solution (see Recipes)
- Lysis buffer for P. tricornutum cells (see Recipes)
- LB medium (see Recipes)
- SOC media (see Recipes)
- LB agar plates (see Recipes) containing 100 μg ml-1 ampicillin or 100 μg ml-1 ampicillin with 40 μg ml-1 gentamicin
- 50% L1, 5% LB, 1% agar plate (1 L) (see Recipes)
- L1 media (see Recipes)
- L1 agar plates (see Recipes) containing 50 μg ml-1 ZeocinTM
- 2x Aquil salt (see Recipes)
- NP stock (see Recipes)
- 1,000x L1 trace metals (see Recipes)
- Vitamin solution (see Recipes)
Equipment
- Qubit® 2.0 Fluorometer (Thermo Fisher Scientific, InvitrogenTM, model: Qubit® 2.0 , catalog number: Q32866)
- Heat block (VWR, catalog number: 13259-030 )
- Water bath (Fisher Scientific, model: IC 2100 )
- Micropipettes (PIPETMAN, variable volume)
- Binocular Microscope (Leitz Wetzlar, model: Labolux12 )
- Hemocytometer
- Table top centrifuge (Hettich Lab Technology, catalog number: 2004-01 )
- Centrifuge (Beckman Coulter, model: Avanti® J-26 XPI , catalog number: 393127)
- PCR thermal cycler (Bio-Rad Laboratories, model: T100TM, catalog number: 1861096 )
- UV-visible spectrophotometer (Biochrom, model: ULTROSPEC 2100® , catalog number: 80-2112-21)
- Incubator shaker (Thermo Fisher Scientific, model: Large Incubated and Refrigerated, catalog number: SHKE5000-7 )
- Vortex
- Autoclave
- Refrigerator
Procedure
- Designing Cas9-sgRNA targeting the urease gene
- Obtain the pKSconj diaCas9 or diaTevCas9 (pPtGE34 or pPtGE35) plasmid (Figure 1) from Addgene (107932 or 107999).
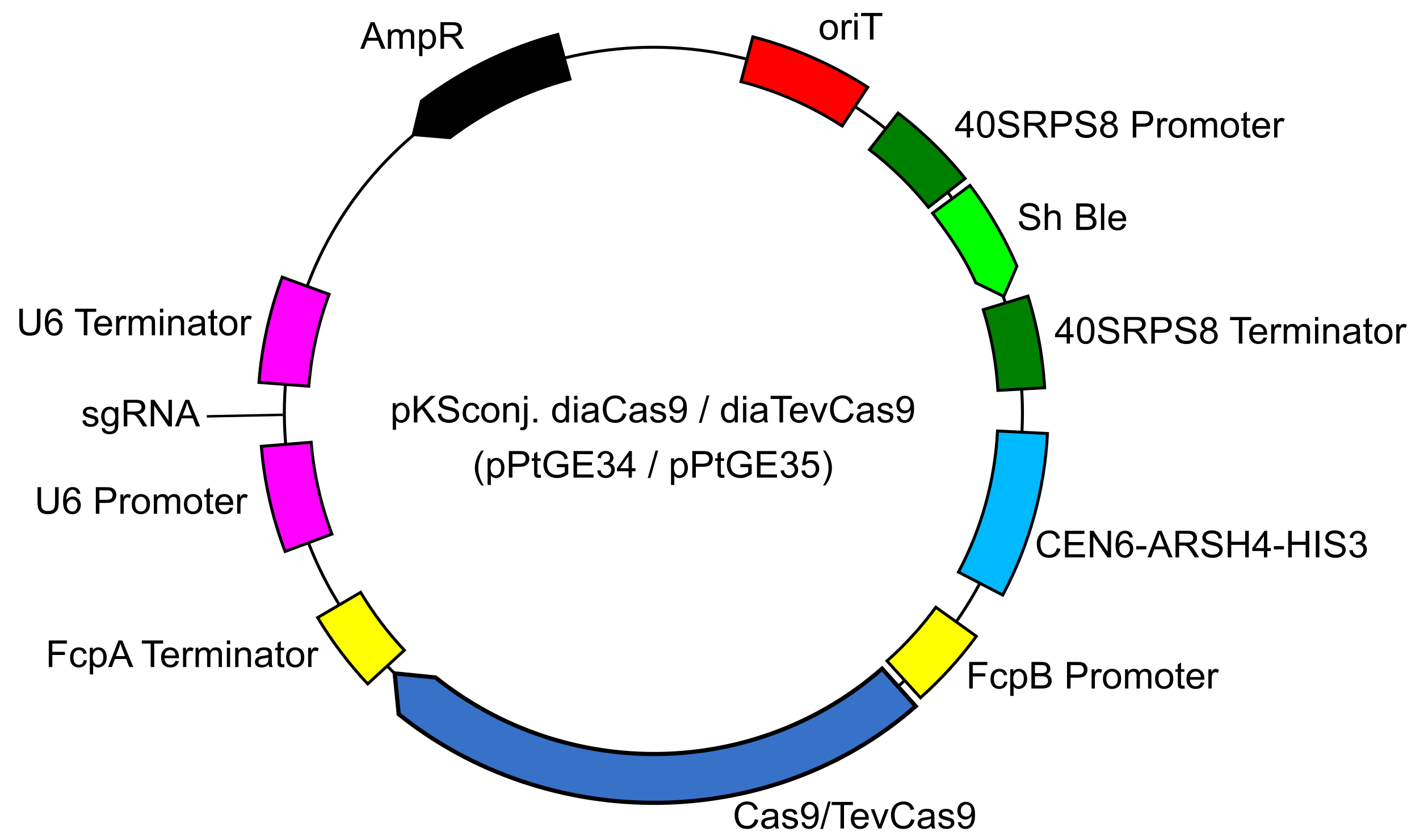
Figure 1. Plasmid map for pKSconj. diaCas9/diaTevCas9 (pPtGE34/pPtGE35) - Download the P. tricornutum urease gene sequence. NCBI Gene ID: 7194881 (PHATRDRAFT_29702).
- Format the sequence in FASTA format with a sequence identifier on line 1 preceded by a > followed by the nucleotide sequence.
- Use a custom Perl script to search for Cas9 and TevCas9 gRNA target sites in the urease gene, ~15 bp downstream of a 5’-CNNNG-3’ I-TevI cleavage site (see Supplemental files). A typical TevCas9 target site is illustrated in Figure 2.
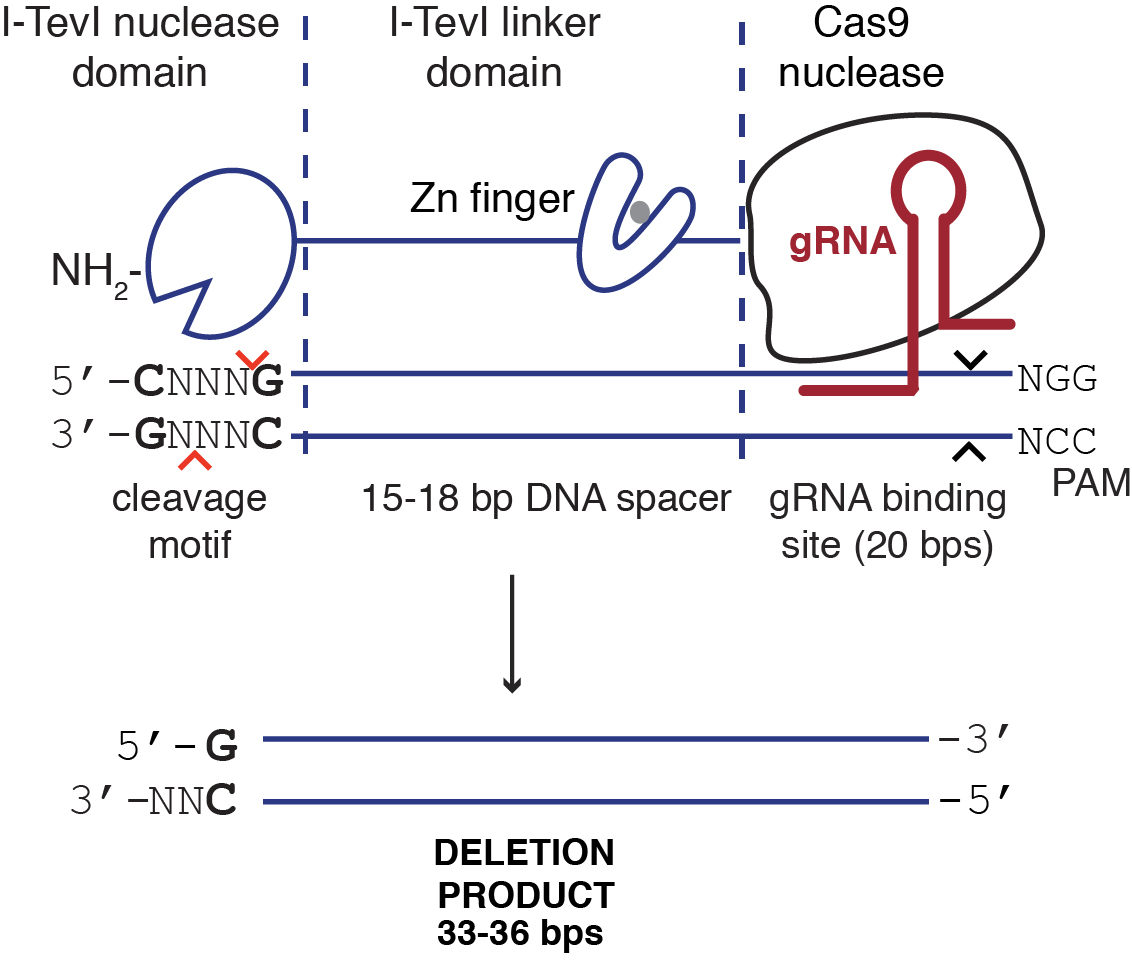
Figure 2. Illustration of TevCas9 target site with predicted deletion size - Select four target sites from the list of predicted sites for the P. trincornutum urease gene. Order an oligonucleotide corresponding to the predicted gRNA binding site (underlined sequence in Table 1), and the complementary oligonucleotide. Design the oligonucleotides with top strand 5’-TCGA-3’ and bottom strand 3’-CAAA-5’ BsaI restriction cut site overhangs to facilitate cloning into the Cas9 and TevCas9 vectors (Table 1).
Table 1. Sequences of gRNAs targeting the urease gene in P. tricornutum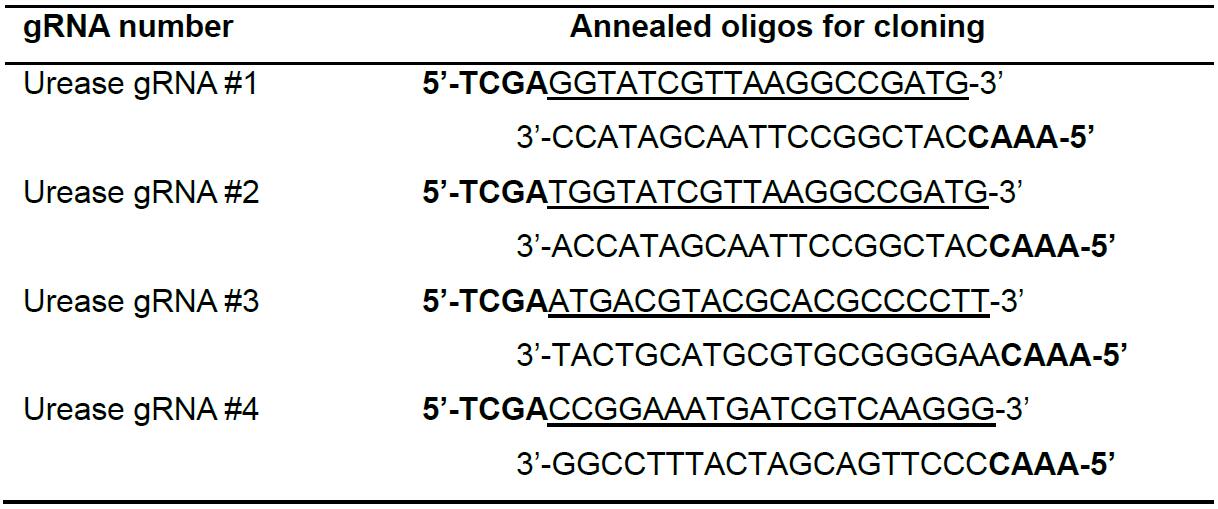
- Obtain the pKSconj diaCas9 or diaTevCas9 (pPtGE34 or pPtGE35) plasmid (Figure 1) from Addgene (107932 or 107999).
- Cloning gRNAs into pKSconj diaCas9 and pKSconj diaTevCas9 plasmids
- Phosphorylate and anneal the ordered oligos to generate gRNA cloning inserts. Prepare a 50 μl gRNA phosphorylation reaction (Table 2) in a 0.2 ml PCR tube. Place the reaction in a thermocycler and incubate at 37 °C for 30 min and then denature at 95 °C for 5 min. Next, slowly cool to 50 °C at a rate of -1 °C min-1 to allow annealing of gRNA top and bottom strands.
Table 2. Reagents for gRNA phosphorylation
- Clone gRNAs into the pKSconj diaCas9 and pKSconj diaTevCas9 plasmids using Golden Gate assembly (Table 3). Place the reaction in a thermocycler and run the protocol as described in Table 4.
Table 3. Reagents for Golden Gate assembly
Table 4. Thermocycler setting for Golden Gate assembly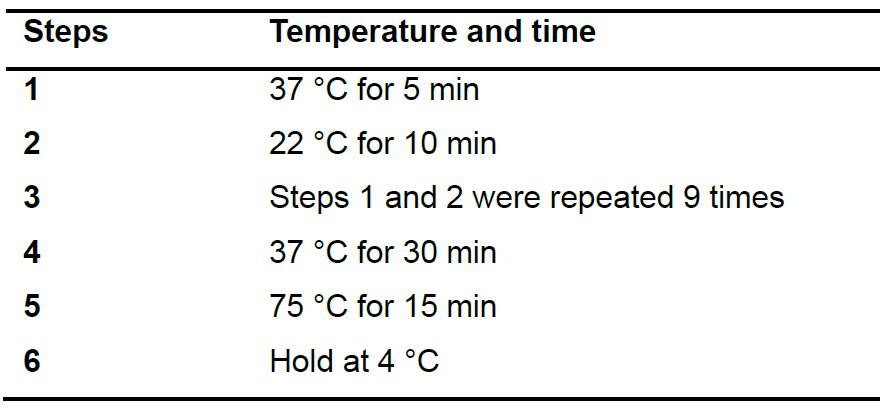
- Transform the product from the Golden Gate reaction into Epi300 E. coli cells using heat shock as follows: For one reaction, mix 3 μl of Golden Gate assembly product with 30 μl of Epi300 E. coli cells. Incubate the reaction on ice for 30 min and heat shock at 42 °C for 45 sec, then incubate on ice for 2 min. Add 400 μl of SOC to the reaction and recover cells at 37 °C with shaking at 225 RPM for 90 min. Plate 150 μl of recovered cells onto LB + Ampicillin media and incubate at 37 °C for 16 h/overnight.
- The following day, inoculate 1 to 5 colonies into separate glass test tubes containing 3 ml of LB + Ampicillin and incubate at 37 °C with shaking at 225 RPM for 16 h/overnight.
- After overnight growth, take 3 ml of overnight culture to isolate plasmid DNA (Bio Basic Inc. plasmid miniprep kit). Send isolated plasmid DNA for Sanger sequencing at the London Regional Genomics Centre using a sequencing primer upstream of the gRNA insertion site to identify a plasmid containing a properly cloned gRNA.
- Transform the properly cloned gRNA plasmid into Epi300 E. coli containing the pTA-Mob (Strand et al., 2014) plasmid, obtained from Dr. Rahmi Lale, using the heat shock protocol described in Step 3 with 1 μl of plasmid and 30 μl of E. coli cells.
- Phosphorylate and anneal the ordered oligos to generate gRNA cloning inserts. Prepare a 50 μl gRNA phosphorylation reaction (Table 2) in a 0.2 ml PCR tube. Place the reaction in a thermocycler and incubate at 37 °C for 30 min and then denature at 95 °C for 5 min. Next, slowly cool to 50 °C at a rate of -1 °C min-1 to allow annealing of gRNA top and bottom strands.
- Transfer of plasmid into P. tricornutum via conjugation from E. coli
Note: This protocol was adapted from a published protocol (Karas et al., 2015).- P. tricornutum growth conditions: 18 °C under cool white fluorescent light (75 μE m-2 sec-1) with a photoperiod of 16 h light and 8 h dark in 50 ml Falcon tubes at the Biotron Experimental Climate Change Research Centre at Western University.
- Briefly, adjust 250 μl of P. tricornutum liquid culture to 1.0 x 108 cells ml-1, plate onto a 50% L1, 1% agar plate, and grow for 4 days.
- Add 2 ml of L1 media to the plate, scrape and collect cells into a 1.5 ml Eppendorf tube.
- Count the cells using a hemocytometer and adjust the concentration to 5 x 108 cells ml-1.
- Grow 3 ml E. coli cultures containing the diaCas9 or diaTevCas9 plasmids with gRNAs at 37 °C overnight. Dilute 1 ml of overnight culture to 50 ml of LB with 100 μg ml-1 ampicillin and 40 μg ml-1 gentamicin and grow the culture to an OD600 of 0.8-1.0.
- Centrifuge the culture for 10 min at 3,000 x g in a 50 ml Falcon tube.
- Discard the supernatant and resuspend the pellet in 500 μl SOC media.
- Mix 200 μl of P. tricornutum from Step 4 with 200 μl of E. coli from Step 7 and plate them onto a 50% L1, 5% LB, 1% agar plate.
- Incubate the plate for 90 min at 30 °C, then for 2 days at 18 °C with light.
- After 2 days, add 1.5 ml of L1 media to the plate, scrape and collect cells in a 1.5 ml Eppendorf tube.
- Plate 500 μl of the scraped cells onto a 50% L1, 1% agar plate containing 50 μg ml-1 ZeocinTM.
- Parafilm the plate and incubate the plate for 14 days at 18 °C with light. A typical conjugation experiment should yield 1.0 x 102 - 1.3 x 104 total exconjugants.
- Screening for urease knockouts in P. tricornutum induced by Cas9 and TevCas9 (see Figure 3 for an overview of screening procedure) - T7 Endonuclease I assay
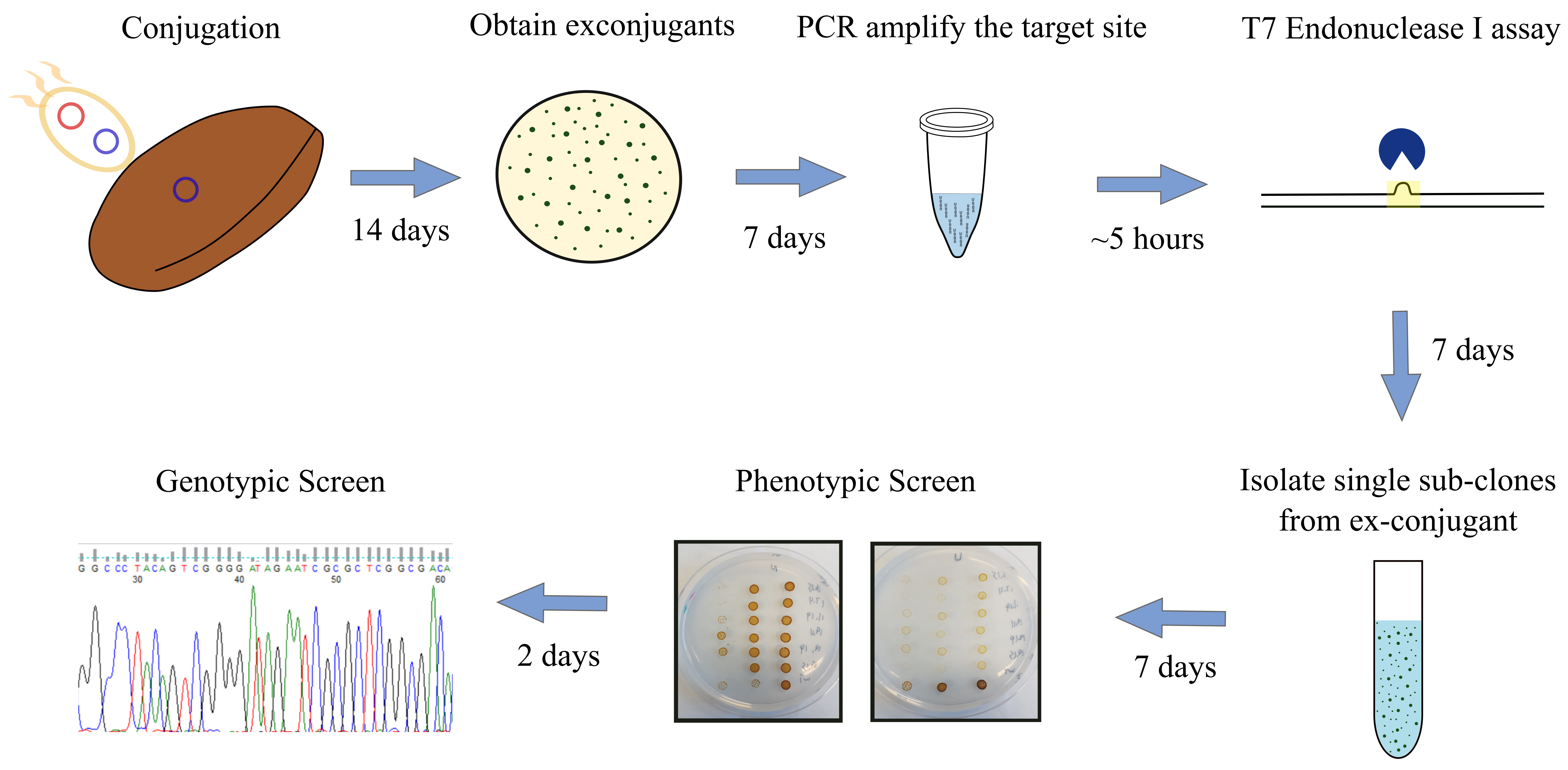
Figure 3. Overview of screening procedure for P. tricornutum urease knockouts. pKSconj diaCas9 or pKSconj diaTevCas9 plasmid is delivered to P. tricornutum by E. coli. Screen the exconjugants with T7EI assay. Isolate sub-clones from exconjugants and perform phenotypic screen by plating sub-clones onto urea and nitrate plates. Verify the deletion length by Sanger Sequencing.- After 14 days, randomly select ten P. tricornutum exconjugants and streak them onto an L1 + ZeocinTM plate and grow for 7 days.
- Scrape 50% of the cells from one streak and place it into 3 ml of L1 media with 50 μg ml-1 ZeocinTM. Suspend the other 50% of cells in 100 μl TE (pH 8.0) buffer, flash freeze at -80 °C for 15 min, and heat lyse at 95 °C for 5 min.
- Use the lysed P. tricornutum exconjugant cells as the template for PCR amplification of the gRNA target site using AmpliTaq DNA polymerase. Design the PCR so the product will be ~500 bp using primers located ~250 bp upstream and downstream of the gRNA target site. Analyze the PCR products on a 1% agarose gel to confirm the size and presence of the PCR product.
- Denature the PCR product at 95 °C for 5 min and slowly cool to 50 °C and then freeze the sample at -20 °C for 2 min to generate heteroduplexes of edited and unedited target sites.
- Use the PCR product as the substrate for a T7EI assay (Table 5). Incubate the reaction at 37 °C for 15 min and analyze the digested product on a 1% agarose gel. PCR products from exconjugant that yield two bands on the agarose gel (500 bp and 250 bp) contains editing at the target site by Cas9 or TevCas9. An example of the T7EI assay gel is shown in Figure 4.
Table 5. Reagents for T7EI assay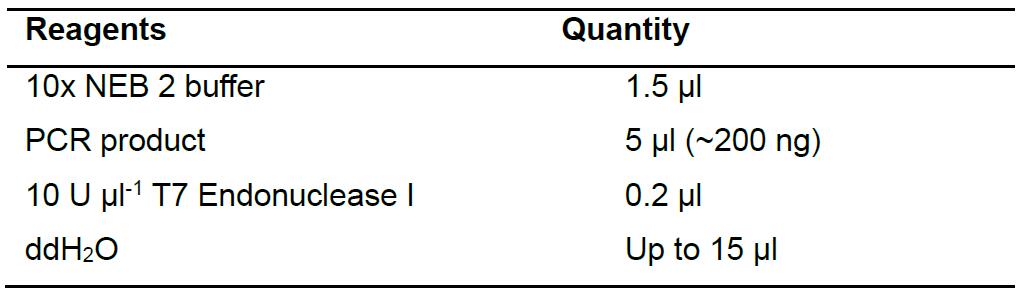
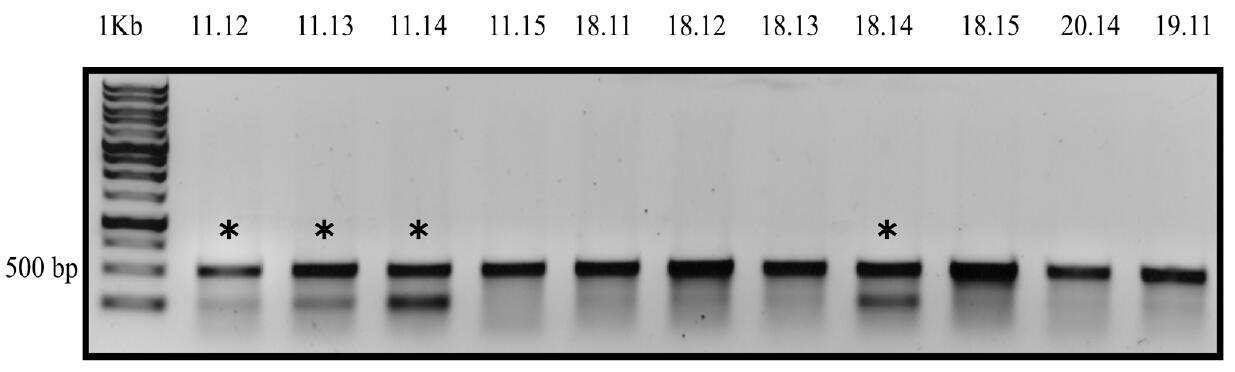
Figure 4. Example analysis of Cas9 activity by T7EI assay. P. tricornutum colonies were resuspended in TE (pH 8.0) buffer followed by flash freezing at -80 °C for 15 min followed by cell lysis at 95 °C for 12 min. The urease gene target site was PCR amplified. PCR product was denatured at 95 °C and cooled down to 50 °C to allow random reannealing. T7E1 assay was performed using the randomly annealed PCR product as template and incubated at 37 °C for 15 min with T7EI and NEB 2 buffer. Products were loaded onto a 1% agarose gel at 100 V for 40 min. Asterisks (*) denote colonies positive for editing. The numbers (11.12, 11.13….) stand for different colonies. - Dilute the liquid cultures from Step D2 of P. tricornutum exconjugants that were identified to have editing to 10-4 and plate onto L1+ ZeocinTM plates and grow for 10-14 days to obtain sub-clones.
- Phenotypic screening of urease knockouts (see Figure 5 for an example of the phenotypic screen)
- Streak sub-clones first onto L1 + ZeocinTM plates containing 2.1 mM urea with a pipette tip, use the same tip to streak the sub-clone onto an L1 + ZeocinTM plate containing 4.2 mM nitrate and grow for 5 days. Sub-clones that grow on L1 + ZeocinTM with nitrate but not on L1 + ZeocinTM with urea are urease knockouts.

Figure 5. Example phenotypic screen of P. tricornutum sub-clones on urea and nitrate plates. Dilutions of Cas9 and TevCas9 sub-clones were spot plated onto L1 with nitrate and L1 with urea plates. - PCR amplify the target site, as in Steps D2-D3, from sub-clones identified as urease knockouts and send the PCR product for Sanger sequencing.
- Streak sub-clones first onto L1 + ZeocinTM plates containing 2.1 mM urea with a pipette tip, use the same tip to streak the sub-clone onto an L1 + ZeocinTM plate containing 4.2 mM nitrate and grow for 5 days. Sub-clones that grow on L1 + ZeocinTM with nitrate but not on L1 + ZeocinTM with urea are urease knockouts.
- Curing of the pKSconj diaCas9 and diaTevCas9 plasmids from urease knockout sub-clones
- Inoculate identified urease knockout sub-clones in 3 ml of L1 media without ZeocinTM and grow for 7 days.
- After 7 days, dilute (10-4) and plate the liquid cultures onto L1 plates and grow for 7 days to obtain single colonies.
- Randomly select 10 colonies and streak them onto L1 and L1 + ZeocinTM plates and grow for 5 days. Growth on L1 plates but not on L1 + ZeocinTM plates indicates colonies that are successfully cured of the plasmid.
Data analysis
- Ten exconjugants of P. tricornutum containing the pKSconj diaCas9 or diaTevCas9 plasmid with different gRNAs were randomly selected for T7EI analysis.
- From those, 5 sub-clones were randomly selected for phenotypic screening.
- To determine the removal of the plasmid, 10 colonies were randomly selected for Zeocin sensitivity screening.
Notes
- Multiple gRNAs may need to be designed as the activity of Cas9 and TevCas9 varies when using different gRNAs.
- The number of edited colonies may vary, more exconjugants may have to be selected to identify edited clones and obtain more accurate editing efficiencies.
Recipes
- Zymolyase solution
200 mg zymolyase 20 T (USB)
9 ml dH2O
1 ml 1 M Tris pH 7.5
10 ml 50% glycerol - Lysis buffer for P. tricornutum cells (1 L)
10 ml 1 M Tris pH 8.0
2 ml 0.5 M EDTA
988 ml dH2O - LB medium (1 L)
10 g tryptone
5 g yeast extract
10 g NaCl
1 L dH2O
Sterilize by autoclave at 121 °C with 1 atmosphere pressure for 15 min - SOC media (1 L)
20 g tryptone
5 g yeast extract
0.5 g NaCl
10 ml 250 mM KCl
5 ml 2 M MgCl
20 ml 1 M glucose
900 ml dH2O - LB agar plates (containing 100 μg ml-1 ampicillin or 100 μg ml-1 ampicillin with 40 μg ml-1 gentamicin) (1 L)
10 g tryptone
5 g yeast extract
10 g NaCl
15 g Agar
0.1 g ampicillin or 0.1 g ampicillin with 0.4 g gentamicin
1 L dH2O - 50% L1, 5% LB, 1% agar plate (1 L)
500 ml L1
50 ml LB
10 g bacto agar - L1 media (1 L)
500 ml 2x Aquil salt
2 ml NP stock
500 μl vitamin solution
1 ml 1,000x L1 trace metals
497.5 ml dH2O
Sterilize by vacuum filtration with NalgeneTM filters - L1 agar containing 50 μg ml-1 ZeocinTM (1 L)
500 ml L1 media
10 g bacto agar
0.5 g ZeocinTM
500 ml dH2O - 2x Aquil salt (1 L)
24.5 g NaCl
4.09 g Na2SO4
0.7 g KCl
0.2 g NaHCO3
0.1 KBr
0.03 g H3BO3
0.003 g NaF
11.1 g MgCl2•6H2O
1.54 g CaCl2•2H2O
0.017 g SrCl2•6H2O - NP stock (100 ml)
37.5 g NaNO3
2.5 g NaH2PO4•H2O - 1,000x L1 trace metals (1 L)
3.15 g FeCl3•6H2O
4.36 g Na2EDTA•2H2O
0.25 ml CuSO4•5H2O (9.8 g L-1 dH2O)
3.0 ml Na2MoO4•2H2O (6.3 g L-1 dH2O)
1.0 ml ZnSO4•7H2O (22.0 g L-1 dH2O)
1.0 ml CoCl2•6H2O (10.0 g L-1 dH2O)
1.0 ml MnCl2•4H2O (180.0 g L-1 dH2O)
1.0 ml H2SeO3 (1.3 g L-1 dH2O)
1.0 ml NiSO4•6H2O (2.7 g L-1 dH2O)
1.0 ml Na3VO4 (1.84 g L-1 dH2O)
1.0 ml K2CrO4 (1.94 g L-1 dH2O) - Vitamin solution (1 L)
200 mg Thiamine-HCl
10 ml of 0.1 g L-1 biotin
1 ml of 1 g L-1 cyanocobalamin
Acknowledgments
This work was supported by Designer Microbes Inc., an NSERC ENGAGE Grant to D.R.E. and Designer Microbes Inc. (EGP/486420-2015), an Ontario Centres of Excellence VIP-1 Grant to Designer Microbes Inc. (OCE-VIP-1/23879), and NSERC Discovery Grant to D.R.E. (RGPIN-2015-04800).
This protocol was developed from the following published paper: Slattery et al. (2018).
Competing interests
The authors declare no conflicts of interests.
References
- Daboussi, F., Leduc, S., Marechal, A., Dubois, G., Guyot, V., Perez-Michaut, C., Amato, A., Falciatore, A., Juillerat, A., Beurdeley, M., Voytas, D. F., Cavarec, L. and Duchateau, P. (2014). Genome engineering empowers the diatom Phaeodactylum tricornutum for biotechnology. Nat Commun 5: 3831.
- Gibson, D. G. (2009). Synthesis of DNA fragments in yeast by one-step assembly of overlapping oligonucleotides. Nucleic Acids Res 37: 6984-6990.
- Gibson, D.G. (2011). Gene and genome construction in yeast. Curr Protoc Mol Biol Chapter 3, Unit 3.22.
- Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A. and Charpentier, E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337(6096): 816-821.
- Karas, B. J., Diner, R. E., Lefebvre, S. C., McQuaid, J., Phillips, A. P., Noddings, C. M., Brunson, J. K., Valas, R. E., Deerinck, T. J., Jablanovic, J., Gillard, J. T., Beeri, K., Ellisman, M. H., Glass, J. I., Hutchison, C. A., 3rd, Smith, H. O., Venter, J. C., Allen, A. E., Dupont, C. L. and Weyman, P. D. (2015). Designer diatom episomes delivered by bacterial conjugation. Nat Commun 6: 6925.
- Noskov, V. N., Karas, B. J., Young, L., Chuang, R. Y., Gibson, D. G., Lin, Y. C., Stam, J., Yonemoto, I. T., Suzuki, Y., Andrews-Pfannkoch, C., Glass, J. I., Smith, H. O., Hutchison, C. A., 3rd, Venter, J. C. and Weyman, P. D. (2012). Assembly of large, high G+C bacterial DNA fragments in yeast. ACS Synth Biol 1(7): 267-273.
- Slattery, S.S., Diamond, A., Wang, H., Therrien, J.A., Lant, J.T., Jazey, T., Lee, K., Klassen, Z., Desgagné-Penix, I., Karas, B.J., et al. (2018). An expanded plasmid-based genetic toolbox enables Cas9 genome editing and stable maintenance of synthetic pathways in Phaeodactylum tricornutum. ACS Synth Biol 7: 328–338.
- Strand, T. A., Lale, R., Degnes, K. F., Lando, M. and Valla, S. (2014). A new and improved host-independent plasmid system for RK2-based conjugal transfer. PLoS One 9(3): e90372.
- Weyman, P. D., Beeri, K., Lefebvre, S. C., Rivera, J., McCarthy, J. K., Heuberger, A. L., Peers, G., Allen, A. E. and Dupont, C. L. (2015). Inactivation of Phaeodactylum tricornutum urease gene using transcription activator-like effector nuclease-based targeted mutagenesis. Plant Biotechnol J 13(4): 460-470.
- Wolfs, J. M., Hamilton, T. A., Lant, J. T., Laforet, M., Zhang, J., Salemi, L. M., Gloor, G. B., Schild-Poulter, C. and Edgell, D. R. (2016). Biasing genome-editing events toward precise length deletions with an RNA-guided TevCas9 dual nuclease. Proc Natl Acad Sci U S A 113(52): 14988-14993.
- Wright, A. V., Nuñez, J. K., Doudna, J. A. (2016). Biology and applications of CRISPR systems: Harnessing nature's toolbox for genome engineering. Cell 164(1-2): 29-44.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Wang, H., Slattery, S. S., Karas, B. J. and Edgell, D. R. (2018). Delivery of the Cas9 or TevCas9 System into Phaeodactylum tricornutum via Conjugation of Plasmids from a Bacterial Donor. Bio-protocol 8(16): e2974. DOI: 10.21769/BioProtoc.2974.
Category
Microbiology > Microbial genetics > Mutagenesis
Molecular Biology > DNA > Mutagenesis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.