- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Deoxycholate Fractionation of Fibronectin (FN) and Biotinylation Assay to Measure Recycled FN Fibrils in Epithelial Cells


Published: Vol 8, Iss 16, Aug 20, 2018 DOI: 10.21769/BioProtoc.2972 Views: 7009
Reviewed by: Ralph Thomas BoettcherPiyali SahaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2017

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


Abstract
Fibronectin (FN) is an extracellular matrix protein that is secreted by many cell types and binds predominantly to the cell surface receptor Integrin α5β1. Integrin α5β1 binding initiates the step-wise assembly of FN into fibrils, a process called fibrillogenesis. We and several others have demonstrated critical effects of fibrillogenesis on cell migration and metastasis. While immunostaining and microscopy methods help visualize FN incorporation into fibrils, with each fibril being at least 3 μm in length, the first study that developed a method to biochemically fractionate FN to quantify fibril incorporated FN was published by Jean Schwarzbauer’s group in 1996. Our protocol was adapted from the original publication, and has been tested on multiple cell types including as shown here in MCF10A mammary epithelial and Caki-1 renal cancer epithelial cells. Using two detergent extractions, cellular FN is separated into detergent insoluble or fibril incorporated FN and soluble FN or unincorporated fractions. To determine whether fibrillogenesis utilizes a recycled pool of FN, we have used a Biotin labeled FN (FN-Biotin) recycling assay, that has been modified from a previous study. Using a combination of the recycling assay and deoxycholate fractionation methods, one can quantitatively demonstrate the extent of fibrillogenesis in cells under different experimental conditions and determine the source of FN for fibrillogenesis.
Keywords: Fibronectin (FN)Background
Fibronectin (FN) is a ubiquitously produced extra cellular matrix (ECM) component (Uitto et al., 1989; Mao and Schwarzbauer, 2005). Fibronectin pools are generated transcriptionally that can be increased by several growth factors such as TGF-β1 (Yokoi et al., 2002; Mimura et al., 2004; Tang et al., 2007). The step-wise process of fibrillogenesis involving cell surface receptor Integrin α5β1 engagement with dimeric FN, drives the process of fibrillogenesis in cells (Yang and Hynes, 1996). Integrin α5β1 regulation by receptor activation/inactivation cycles, receptor endocytosis and recycling influences fibrillogenesis (Gao et al., 2000; White et al., 2007; Caswell et al., 2008) with FN contributing to net endocytosis rates of integrins in active conformations (Arjonen et al., 2012). The relevance of FN fibrillogenesis to cellular outcome has been demonstrated by numerous studies investigating fibril-specific functions for the FN protein. For example, a polymeric form of fibronectin or ‘super-fibronectin’ shows anti-metastatic and anti-angiogenic properties against different tumor types (Pasqualini et al., 1996; Yi and Ruoslahti, 2001). In the absence of a FN matrix, as observed in Von Hippel Lindau syndrome, renal cancer characterized by the mutation or loss of the Von Hippel Lindau (VHL) protein (Ohh et al., 1998; Hoffman et al., 2001), introducing a VHL mutant unable to form a fibronectin matrix is insufficient to suppress formation of tumors in SCID mice (Stickle et al., 2004). All disease mutants of the VHL gene in renal cancer also fail to form a FN matrix (Hoffman et al., 2001). Thus, analyses of fibril versus soluble FN maybe of importance to investigations that explore the contribution of the FN matrix to cellular response. Endpoint and real-time assays have been successfully used to study fibrillogenesis (Pankov et al., 2000; Mao and Schwarzbauer, 2005). Both studies rely on microscopy-based approaches to determine fibrillogenesis in cells. Biochemical fractional of FN is a quantitative approach to complement microscopy-based methods to detect levels of fibril incorporated from soluble FN. Using a fractionation assay in combination with a recycling assay, allows us to determine whether FN that is incorporated in the matrix is recycled from an existing pool of FN in the cell or from cell autonomous sources. The FN-Biotin recycling assay is simpler than classical temperature-switching assays that investigate receptor recycling (Roberts et al., 2001). Additionally, the temperature-switching assays quantify protein recycling from a decrease in rate of protein endocytosis; requiring additional lysosomal or proteasomal inhibitors in the assay. Our recycling assay can be adapted to many different proteins that localize intracellularly and at the cell membrane. While performing our FN fractionation protocol we did detect integrin β1 in the soluble and insoluble fractions. It is possible to extend this observation to perform recycling of integrins using a Biotin-conjugated integrin or any protein of interest. Alternatively, it is also possible to perform immunoprecipitation experiments on the recycled protein fraction to determine whether specific proteins interact preferentially with the two FN fractions. This protocol can help answer several questions associated with protein trafficking kinetics, interacting partner proteins and new functional characteristics based on associating proteins in the different fractions.
Materials and Reagents
- 15 ml conical tube (Thermo Fisher Scientific, catalog number: 339650 )
- Pipette tip
1,250 μl (USA Scientific, TipOne, catalog number: 1112-1720 )
200 μl (USA Scientific, TipOne, catalog number: 1110-1700 )
20 μl (USA Scientific, TipOne, catalog number: 1123-1710 )
10 μl (USA Scientific, TipOne, catalog number: 1111-3700 ) - Aluminum foil (Walmart, 551605957)
- Beaker (100 ml, WWR, catalog number: 890000-200 )
- Filter paper (GE Healthcare, catalog number: 1001-929 )
- T75 flask (Corning, catalog number: 353824 )
- Six-well tissue culture plates (Corning, catalog number: 3506 )
- Cell lifter (Fisher Scientific, FisherbrandTM, catalog number: 08-100-240 )
- 23 G needle (BD, Precision Glide, catalog number: 305193 )
- Microcentrifuge tubes (Eppendorf, catalog number: 022600028 )
- MCF10A breast cell lines (ATCC, catalog number: CRL-10317 )
- Caki-1 renal cancer cell lines (ATCC, catalog number: HTB-46 )
- Protein Ladder (Bio-Rad Laboratories, catalog number: 1610375 )
- PVDF protein transfer membrane (Merck, catalog number: IPVH08100 )
- Fibronectin antibody (Santa Cruz Biotechnology, catalog number: sc-59826 )
- Actin antibody (Abcam, catalog number: ab8227 )
- GAPDH antibody (Abcam, catalog number: ab9484 )
- Biotin labeled fibronectin (CYTOSKELETON, catalog number: FNR03 )
- Streptavidin conjugated IRDye® (LI-COR, catalog number: 926-32230 )
- IRDye 800CW Secondary Antibody (LI-COR, catalog number: 925-32210 )
- DMEM/F-12 HEPES (Thermo Fisher Scientific, GibcoTM, catalog number: 11330057 )
- Insulin (Thermo Fisher Scientific, GibcoTM, catalog number: 12585014 )
- EGF (Thermo Fisher Scientific, GibcoTM, catalog number: PHG0313 )
- Hydrocortisone (Corning, catalog number: 354203 )
- Horse Serum (Thermo Fisher Scientific, GibcoTM, catalog number: 16050122 )
- Penicillin-Streptomycin (Thermo Fisher Scientific, GibcoTM, catalog number: 15140122 )
- Cholera toxin (Sigma-Aldrich, catalog number: C8052-2MG )
- Skimmed Milk powder (Thermo Fisher Scientific, catalog number: LP0031B )
- BSA (Fisher Scientific, Fisher BioReagentsTM, catalog number: BP9703100 )
- McCoy's 5A (ATCC, catalog number: 30-2007 )
- FBS (Corning, catalog number: 35-011-CV )
- 0.05% Trypsin-EDTA (Thermo Fisher Scientific, GibcoTM, catalog number: 25300062 )
- SDS (Fisher Scientific, Fisher BioReagentsTM, catalog number: BP166-100 )
- Sodium deoxycholate (Fisher Scientific, Fisher BioReagentsTM, catalog number: BP349-100 )
- Tris Base (Fisher Scientific, Fisher BioReagentsTM, catalog number: BP152-1 )
- Tris-HCl (Fisher Scientific, Fisher BioReagentsTM, catalog number: BP153-1 )
- Glycine (Fisher Scientific, Fisher ChemicalTM, catalog number: G48-212 )
- Tween 20 (Fisher Scientific, Fisher BioReagentsTM, catalog number: BP337-500 )
- Sodium Hydroxide pellets (Fisher Scientific, Fisher ChemicalTM, catalog number: S318-1 )
- Ethanol, absolute (200 proof) (Fisher Scientific, Fisher BioReagentsTM, catalog number: BP2818500 )
- N-ethylmaleimide (Thermo Fisher Scientific, catalog number: 23030 )
- Iodoacetic acid (Thermo Fisher Scientific, catalog number: 35603 )
- DTT (Fisher Scientific, Fisher BioReagentsTM, catalog number: BP172-25 )
- Bromophenol blue (Fisher Scientific, Fisher BioReagentsTM, catalog number: BP115-25 )
- Nitrocellulose (GE Healthcare, catalog number: 10600012 )
- NaCl (Fisher Scientific, Fisher BioReagentsTM, catalog number: BP358-1 )
- KCl (Fisher Scientific, Fisher ChemicalTM, catalog number: P217-3 )
- Na2HPO4 (MP Biomedicals, catalog number: 02191440.1 )
- KH2PO4 (MP Biomedicals, catalog number: 02195453.1 )
- 100% Methanol (Fisher Scientific, Fisher ChemicalTM, catalog number: A935-4 )
- Glacial acetic acid (Fisher Scientific, catalog number: A38-212 )
- Phenylmethylsulphonyl fluoride (PMSF) (ACROS ORGANICS, catalog number: 215740050 )
- Ethylenediaminetetraacetic acid (EDTA) (Fisher Scientific, catalog number: 5312-500 )
- Growth media for MCF10A cells (see Recipes)
- Growth media for Caki-1 cells (see Recipes)
- 10x PBS (see Recipes)
- 1x PBS (see Recipes)
- 10x TBS (see Recipes)
- 1x TBS (see Recipes)
- 1x TBS 0.2% Tween (see Recipes)
- 1x SDS Running Buffer (see Recipes)
- 1x Transfer Buffer (see Recipes)
- 2 M DTT (see Recipes)
- 5x Loading Dye (see Recipes)
- Blocking Buffer (see Recipes)
- Primary antibody buffer (see Recipes)
- Secondary antibody buffer (see Recipes)
- Acid wash (see Recipes)
- 1 M Tris-HCl pH 8.0 (see Recipes)
- 1 M Tris-HCl pH 8.8 (see Recipes)
- 100 mM Phenylmethylsulphonyl fluoride (PMSF) (see Recipes)
- 100 mM Iodoacetic acid (see Recipes)
- 100 mM N-Ethylmaleimide (NEM) (see Recipes)
- 0.5 M Ethylenediaminetetraacetic acid (EDTA) pH 8.0 (see Recipes)
- Deoxycholate lysis buffer (see Recipes)
- SDS lysis buffer (see Recipes)
Equipment
- Magnetic stirrer (Corning, catalog number: 440936 )
- Rotor wheel (Thermo Fisher Scientific, catalog number: 88881001 )
- Heating block (Thermo Fisher Scientific, catalog number: 88870001 )
- Orbital Shaker (Corning, catalog number: 6780-FP )
- Microcentrifuge (Eppendorf, model: 5424 R , catalog number: 5404000138)
- Centrifuge (Thermo Fisher Scientific, model: SorvallTM Legend T plus , catalog number: 75004367)
- Incubator (PHC, Panasonic, model: MCO-170AICUVL-PA )
- Pipettes (5 ml; 10 ml, Fisher Scientific, FisherBrandTM, catalog numbers: 13-676-10C ; 13-676-10F )
- Tissue culture hood (Thermo Fisher Scientific, catalog number: 1333 )
- Autoclave
- Odyssey Fc Imaging System (LI-COR, model number: 2800 )
- Light microscope (Zeiss, model: ID03 )
- Ice bucket
- -20 °C freezer (Fisher Scientific, catalog number: 13-986-148 )
- SDS PAGE apparatus and Transfer tank (Bio-Rad Laboratories, catalog number: 1658029fc )
- Tray to set up transfer (12 ½" x 17 ½")
Software
- LI-COR Lite (https://www.licor.com/bio/products/software/image_studio_lite/download.html)
- ImageJ (https://imagej.nih.gov/ij/download.html)
- Microsoft excel
Procedure
- Deoxycholate fractionation of FN in MCF10A and Caki-1 cells
- MCF10A breast cell lines or Caki-1 renal cancer cells should be maintained to reach no more than 80-90% confluence in a T75 tissue culture flask in a 5% CO2 buffered incubator. Refer Figure 1 for the quick start protocol.
- Once the cells are approximately 80% confluent in the flask, aspirate the cell culture medium, rinse the cells once in 15 ml 1x PBS and aspirate before adding 2-3 ml 0.05% Trypsin-EDTA. Place the flask in a 5% CO2 buffered incubator for 5-7 min.
- Once the cells are successfully dislodged from the flask which you can check using a light microscope, add 3 ml of growth medium to the flask to neutralize the action of trypsin.
- Transfer the 6 ml cell suspension into a 15 ml conical tube and centrifuge at 300 x g for 5 min at room temperature.
- After centrifugation, discard the supernatant and resuspend the pellet in growth medium.
- For plating in 6-well dishes, plate cells at a confluence that will reach 80% on the day of the experiment. For Caki-1 cells the plating number is 100,000 cells/well and MCF10A cells 70,000 cells/well. Cells in one well of the six-well plate are sufficient to visualize the detergent soluble and insoluble FN fractions for each experimental condition.
Note: Cell density of 80% provides sufficient soluble and insoluble FN protein for quantification. If cell density is a variable in your experimental condition, the 80% density is ideal since cells are not too confluent to trigger cellular pathways that may interfere with your experimental read-out. - Incubate the cells at 37 °C at 5% CO2.
- The following day, check to see whether optimum cell density has been reached. If yes, the cells can be processed for deoxycholate fractionation of FN.
Note: Since the entire fractionation procedure is performed on ice, it is important at this stage that you have cold 1x PBS, cold deoxycholate lysis buffer (inhibitors freshly added) and cold SDS lysis buffer (inhibitors freshly added). Refer ‘Recipes’ section. - Aspirate medium from wells and place the plates on ice in an ice bucket. Add 10 ml 1x cold PBS to remove any trace of growth medium in the cells and aspirate completely. Rinse again with 1x PBS if you see traces of media in the well. It is important at this point to make sure all PBS is aspirated so that subsequent addition of lysis buffer is not diluted.
- To each well, add 300 μl of cold (4 °C) deoxycholate lysis buffer containing inhibitors and immediately scrape the cells using a cell lifter.
- Transfer all the cells in the lysis buffer into an Eppendorf tube and lyse using a 23 G syringe needle at least 20 times.
- Place the tubes on a rotor wheel in the cold room and allow to rotate for 30 min. Rotor speed is not critical for lysis. Lysis time can be extended to 1 h at this point without affecting results (optional).
- Transfer the Eppendorf tubes to a microfuge and centrifuge at 21,130 x g for 30 min at 4 °C. If the microfuge has a maximum speed of 15,871 x g, you can increase the centrifugation time to 45 min.
- Check to see if you spot a pellet in the size of a pinhead. The pellets are usually very difficult to see and sometimes require a second round of centrifugation.
Note: It is also a good idea to place the Eppendorf tubes in the microfuge and mark the cap of the tube where the pellet will settle. This makes it easier to know where to look for the pellet. - Transfer the supernatant to a new Eppendorf tube and label it ‘soluble FN’. This protein fraction contains detergent soluble FN. To the soluble FN fraction, add 5x SDS loading dye and store in a -20 °C freezer until ready to resolve proteins on a gel.
- To the pellet, add 20 μl cold SDS lysis buffer containing inhibitors and mix the pellet with the buffer using a pipette tip.
- Heat the pellet to 95 °C for 1 min for the pellet to mix completely with the SDS lysis buffer. This protein fraction contains the deoxycholate ‘insoluble FN’.
Note: Heating the pellet for 1 min with SDS lysis buffer is a necessary step to completely resuspend the pellet. - Add 5x SDS loading dye, mix and store in the -20 °C freezer.
- Before loading ‘soluble FN and ‘insoluble FN’ samples on an SDS-PAGE gel, heat samples to 95 °C for 5 min and centrifuge at 21,130 x g at RT for 1 min.

Figure 1. Flowchart for extraction and separation of the two FN fractions
- Biotinylation assay to measure recycled FN in MCF10A cells
- The MCF10A cells are seeded under the same conditions and incubated in a 5% CO2 buffered incubator as described in Procedure A.
- Reconstitute one vial of lyophilized FN-Biotin with 10 μl sterile distilled water and allow the tube to sit for a few minutes at room temperature before tapping the tube a few times to mix. The concentration of the stock will now be 2 mg/ml.
- Add 20 μl of the stock solution to 2 ml serum free media to make a working concentration of 20 μg/ml FN-Biotin medium. To make 2 ml serum free media containing a working concentration of 20 μg/ml FN-Biotin, you will need a total of two vials of lyophilized FN-Biotin. In our hands, reducing the working concentration to 10 μg/ml proved insufficient to determine FN uptake by MCF10A cells using immunoblotting.
- Before beginning the biotinylation assay, serum starve the cells for 2 h. Serum starvation is performed by aspirating growth media from the cells, washing cells two times each with 2 ml 1x PBS and adding serum-free medium to the cells.
- After the 2 h incubation, aspirate the serum-free medium from cells, wash cells in 6-well plates twice each with 2 ml 1x PBS, aspirate PBS completely and add 500 μl of FN-Biotin media to the cells.
- Incubate for 30 min in the 37 °C incubator.
- After incubation, aspirate the FN-Biotin medium, wash with 2 ml 1x PBS, aspirate and add 1 ml acid wash (see Recipes) for 30 sec. The acid wash step is critical to remove cell surface bound FN-Biotin. It is important not to exceed acid wash to more than 1 min as it begins to affect cell morphology.
- Remove the acid wash and rinse the well four times each with 2 ml 1x PBS.
- Aspirate the PBS and add growth medium to the cells without FN-Biotin and with or without test compounds depending on your experimental question. Since we were interested to investigate whether TGFβ1 and TGFβ2 increases FN fibrillogenesis via recycling, we had a control condition with only growth medium, growth medium containing 10 ng/ml TGFβ1 and growth medium containing 10 ng/ml TGFβ2 (Varadaraj et al., 2017).
- To determine whether FN is recycled to form fibrils, perform a deoxycholate fractionation as described in Procedure A, Steps 8-17, using 300 μl of deoxycholate lysis buffer per well.
- Resolving the FN protein on a 5% gel and immunoblotting
- Run the soluble and pellet lysates on a 5% SDS-PAGE gel.
- Load 1/10th of the total volume of soluble fraction and the entire volume of the pellet fraction. If you load 1/10th, keep the volume consistent for all experimental conditions.
- Let the gel run till only the top two ladders of the protein ladder are remaining within the gel -250 kDa and 150 kDa. The gel run usually takes 3-4 h at 80 V.
- After the run is complete, transfer the gel using either Nitrocellulose or PVDF membranes at 25 V overnight (12-16 h) in a cold room. Alternatively, the transfer tank can be placed in an ice bucket during the transfer procedure.
- After the transfer is complete, place the membrane in an immunoblotting box and add 5 ml of blocking buffer (sufficient volume to cover the membrane) for 1 h at room temperature and place on an orbital shaker at 55 rpm.
- After blocking, remove the blocking solution and wash the membrane 2-3 times with 1x TBS using gentle agitation. Make sure all traces of milk has left the membrane. Rinse until the TBS washes are clear.
- Add FN antibody at a dilution of 1:1,000 in primary antibody buffer and allow the membrane to incubate overnight on an orbital shaker set at a speed of 55 rpm at 4 °C.
- After primary antibody incubation, rinse three times by placing the membrane on an orbital shaker set at a speed of 55 rpm for 5 min in 10 ml 1x TBS 0.2% Tween.
- Add IR Dye 800CW secondary antibody at a dilution of 1 μl in 15 ml secondary antibody buffer for 1 h on an orbital shaker set at a speed of 55 rpm. It is important to protect the membrane from light. If the immunoblot boxes are transparent, cover the box with aluminum foil throughout the incubation and subsequent wash steps.
- To visualize FN-Biotin, perform Steps C1-C6 following which add Streptavidin conjugated IR Dye secondary antibody at a dilution of 1 μl in 15 ml secondary antibody buffer for 1 h on an orbital shaker set at a speed of 55 rpm. It is important to protect the membrane from light. If the immunoblot boxes are transparent, cover the box with aluminum foil throughout the incubation and subsequent wash steps.
- After antibody incubation, rinse three times by placing the membrane on an orbital shaker set at a speed of 55 rpm for 5 min in 10 ml 1x TBS 0.2% Tween.
- Remove 1x TBS 0.2% Tween and replace with 10 ml 1x TBS before imaging on the LI-COR scanner.
- An image scan of the membrane will appear as shown in Figure 2A.
- To probe for a loading control protein, run 1/10th of the soluble FN fractions on a 10% gel and immunoblot for GAPDH (Figure 2B) or Actin. Actin and GAPDH primary antibodies are used at a dilution of 1 μl in 5 ml (1 μg/5 ml) and can be reused several times if the diluted antibody is stored at 4 °C.
Note: HRP conjugated secondary antibodies can also be used and processed using an ECL detection method. In that case, you will need an HRP conjugated Streptavidin secondary antibody.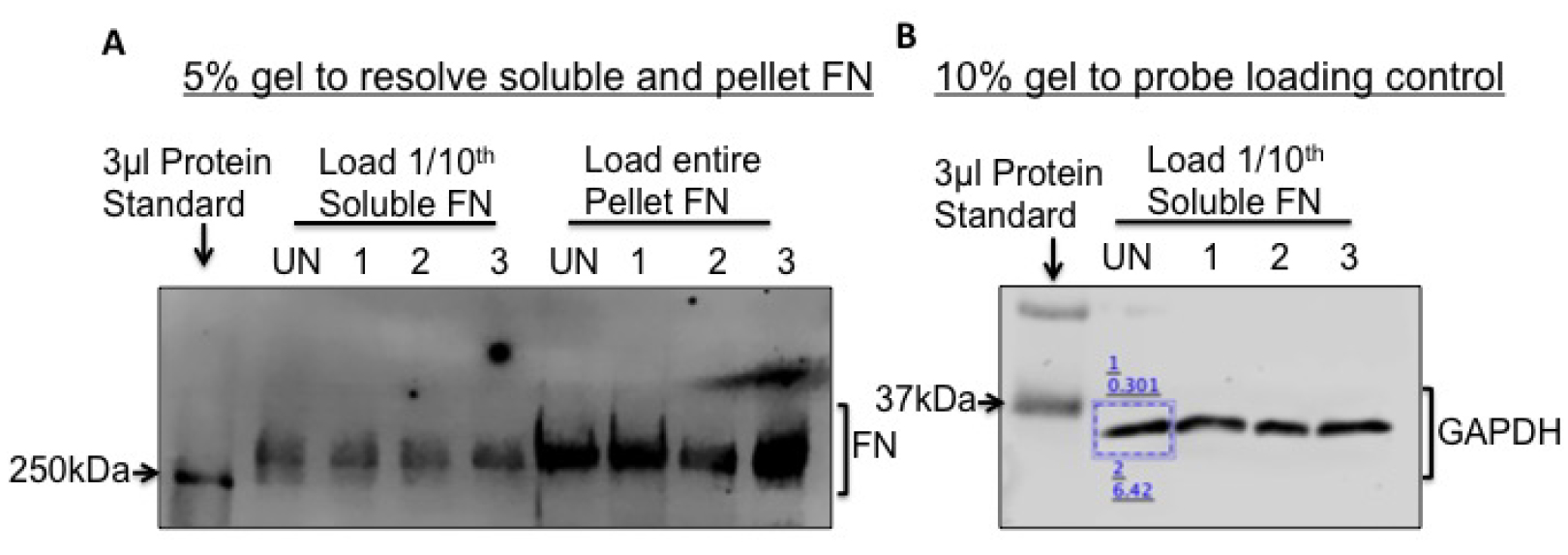
Figure 2. Fibronectin fractionation and analysis by immunoblotting. To visualize and quantify soluble and pellet FN, load 1/10th of the soluble and the entire pellet fractions from Caki-1 cells, on a 5% SDS-PAGE gel as shown in (A). A band above the 250 kDa MW ladder is FN. UN, 1, 2 and 3 refer to ‘untreated’ and three different treatment conditions (B). To normalize for protein loading, load 1/10th of the soluble FN on a 10% SDS-PAGE gel and immunoblot for GAPDH. Pixel intensities of the protein bands are quantified using the LI-COR Lite software by drawing a rectangle (blue box) around the band. The lower value denotes the background-corrected band intensity and the upper value denotes the background.
Data analysis
Typically, deoxycholate extractions and biotinylation assays are tested independently at least three times to get statistically significant results. To quantify FN that is in fibrils (pellet fraction) compared to the non-fibril (soluble FN) fraction, immunoblots of resolved FN and GAPDH are required. Follow the below step-by-step bullet points to perform data analysis of the samples.
- Using the LI-COR Lite software, quantify all the protein bands individually and export to an excel sheet.
- If the experimental question is to test whether FN fibril increases with treatment, calculate the pixel intensities of soluble FN to corresponding GAPDH or Actin levels.
- After normalizing soluble FN levels, calculate the ratio of pellet FN pixel intensities to the normalized soluble FN levels. In Figure 2A, soluble FN in UN (untreated) and treatment conditions 1, 2 and 3 are normalized to GAPDH in UN, 1, 2 and 3 respectively.
- Calculate pixel intensities for pellet FN in UN, 1, 2 and 3.
- Finally calculate ratios for UN pellet FN versus normalized UN soluble FN, sample 1 pellet FN versus normalized sample 1 soluble FN and so on.
- To represent the analyzed data as a figure in a publication, convert the ratios to fold difference/relative difference, create a bar graph of the fold difference between UN and treated samples, the UN sample being ‘1’, calculate SEM and perform appropriate statistics (Refer Figure 4E in Varadaraj et al., 2017).
If the data clearly demonstrates that pellet FN increases with treatment X, it is likely that both soluble and pellet FN increase, in which case you may wrongly infer that only the pellet FN is increasing. To check if this were the case, it is essential to perform the same experiment but extract total protein instead of deoxycholate fractions. If the total FN levels remain the same between treatments, then you can conclude that only the fibril fractions are changing with treatments. Alternatively, if the total FN protein increases with treatment, then the increase in pellet FN may be due to an increase in the total protein and not specifically a difference in pellet FN fraction between treatments. Our study (refer Figure 1C in Varadaraj et al., 2017) showed that TGFβ1 and TGFβ2 increases FN fibril fraction, we measured total FN levels in parallel experiments to fractionation experiments to confirm that FN fibril fraction was increasing with treatments.
Recipes
- Growth media for MCF10A cells
- To 500 ml of DMEM/F12 media:
Add 25 ml of horse serum, 1.25 ml of Insulin from a 4 mg/ml Insulin stock solution, 350 μl of Cholera toxin from 150 μg/ml stock, 215 μl of EGF from 50 μg/ml stock and 500 μl Hydrocortisone from 500 μg/ml stock - To make stock solution of Cholera toxin:
Dissolve 2 mg Cholera toxin in 13.3 ml DMEM/F12 (without additives) and store as 350 μl aliquots in the -20 °C freezer. We have used aliquots of Cholera toxin that has been stored for no longer than 6 months - To make EGF stock solution:
Dissolve 1 mg lyophilized EGF in 20 ml sterile dH2O and store as 215 μl aliquots in the -20 °C freezer - To make Hydrocortisone stock solution:
Dissolve 50 mg Hydrocortisone in 100 ml 200 proof ethanol and store as 10 ml aliquots in the -20 °C freezer
- To 500 ml of DMEM/F12 media:
- Growth media for Caki-1 cells
To 500 ml McCoy's 5A medium add 50 ml FBS and 5 ml Penicillin-Streptomycin (optional)
Note: *These buffers can be stored at room temperature.
- 10x PBS (1 L) *
Weigh 80 g NaCl, 2 g KCl, 14.4 g Na2HPO4, 2.4 g KH2PO4 and add 500 ml sterile dH2O and adjust pH using HCl to pH 7.2 to 7.4
Make up the final volume to 1 L using sterile dH2O - 1x PBS (1 L) *
Dilute 100 ml of 10x PBS with 900 ml sterile dH2O
10x TBS (1L) *- Weigh 87.66 g NaCl, 12.11 g Tris base and add 500 ml sterile dH2O
- Adjust pH using HCl to pH 8.0 and make up final volume to 1 L using sterile dH2O
- 1x TBS (1 L) *
Dilute 1 part of 10x TBS with 9 parts sterile dH2O - 1x TBS 0.2% Tween (1 L) *
Add 2 ml Tween 20 to 1x TBS - 1x SDS Running Buffer (10 L) *
- Add 303 g Tris Base, 1,440 g Glycine and 100 g SDS
- Make up to a total volume of 10 L using sterile dH2O
- 1x Transfer Buffer (1 L)
Add 1.86 g Glycine, 3.02 g Tris Base, 150 ml 100% Methanol and bring upto a total volume of 1 L with dH2O
Note: The transfer buffer can be stored at 4 °C. - 2 M DTT
Weigh 3.08 g of DTT (FW 154.25) in 10 ml sterile dH2O
Aliquot and store in the -20 °C freezer - 5x Loading Dye
- To make a total volume of 100 ml loading dye:
- Add 50 ml glycerol to a beaker, 10 g SDS, 33 ml 1 M Tris pH 6.8 and 17 ml water
- Allow the solution to mix by placing on a magnetic stirrer set at 37 °C for the SDS to dissolve completely
- Pick a small amount of Bromophenol blue powder using a pipette tip and mix into the solution
- To 90 ml of this solution add 10 ml of 2 M DTT
- Store as 2 ml aliquots in the -20 °C freezer
- To make a total volume of 100 ml loading dye:
- Blocking Buffer
Weigh 0.25 g skimmed milk powder and dissolve in 1x TBS
Prepare 10 ml fresh for each use - Primary antibody buffer
Weigh 0.05 g BSA and dissolve in 1x TBS
Prepare 10 ml fresh for each use - Secondary antibody buffer
Weigh 0.05 g BSA and dissolve in 1x TBS 0.2% Tween
Prepare 10 ml fresh for each use - Acid wash
Weigh 14.6 g NaCl and add 2.5 ml glacial acetic acid
Dilute with sterile dH2O up to 500 ml and store at room temperature - 1 M Tris-HCl pH 8.0 (1 L) *
- Weigh 157.6 g of Tris-HCl salt and dissolve in 900 ml sterile dH2O
- After the salt has dissolved, adjust pH using NaOH to pH 8.0 and add water up to a total of 1 L
- 1 M Tris-HCl pH 8.8 (1 L) *
- Weigh 157.6 g of Tris-HCl salt and dissolve in 900 ml sterile dH2O
- After the salt has dissolved, adjust pH using NaOH to pH 8.8 and add water up to a total of 1 L
- 100 mM Phenylmethylsulphonyl fluoride (PMSF)
Weigh 0.174 g in 10 ml ethanol to obtain a 100 mM stock solution (FW 174.2)
Aliquot and store in the -20 °C freezer - 100 mM Iodoacetic acid
Weigh 0.185 g in 10 ml sterile dH2O to obtain a 100 mM stock solution (FW 185.95)
Aliquot and store in the -20 °C freezer - 100 mM N-Ethylmaleimide (NEM)
Weigh 0.125 g in 10 ml ethanol to obtain a 100 mM stock solution (FW 125.13)
Aliquot and store in the -20 °C freezer - 0.5 M Ethylenediaminetetraacetic acid (EDTA) pH 8.0 (FW 292.24) (0.5 L) *
- Weigh 73.06 g and add 400 ml sterile dH2O
- Allow the salt to dissolve and adjust pH to 8.0
- Add water to make up to a final volume of 500 ml
- Deoxycholate lysis buffer (0.1 L)
2% Sodium Deoxycholate
0.02 M Tris-HCl pH 8.8
2 mM PMSF
2 mM EDTA
2 mM Iodoacetic Acid
2 mM N-ethylmalemide- To make 100 ml buffer:
Add 2 g Sodium deoxycholate salt, 2 ml 1 M Tris-HCl pH 8.8 and remaining sterile dH2O upto a total volume of 100 ml
Note: This buffer can be stored in the refrigerator for no more than 3 months. - To make 10 ml of this buffer with inhibitors:
Add 20 μl of 100 mM PMSF stock, 40 μl of 0.5 M EDTA stock, 20 μl Iodoacetic acid from 100 mM stock and 20 μl NEM from 100 mM stock
Note: Inhibitors should be added freshly before use at final concentrations of 2 mM PMSF, 2 mM EDTA pH 8.0, 2 mM Iodoacetic Acid and 2 mM NEM.
- To make 100 ml buffer:
- SDS lysis buffer (0.1 L) *
- To make 100 ml buffer:
Add 1 g SDS and 2.5 ml 1 M Tris-HCl pH 8.0 and water up to 100 ml - For 10 ml of the SDS lysis buffer:
Add 20 μl of 100 mM PMSF stock, 40 μl of 0.5 M EDTA stock, 20 μl Iodoacetic acid from 100 mM stock and 20 μl NEM from 100 mM stock
Note: Inhibitors should be added freshly before use at final concentrations of 2 mM PMSF, 2 mM EDTA pH 8.0, 2 mM Iodoacetic Acid and 2 mM NEM.
- To make 100 ml buffer:
Acknowledgments
Research reported in this publication was funded in part by National Institute On Minority Health and Health Disparities of the National Institutes of Health under Award Number U54MD012388 and National Cancer Institute of the National Institutes of Health under the awards for the Partnership of Native American Cancer Prevention U54CA143924 (UACC) and U54CA143925 (NAU) to AV and National Institutes of Health grant P20 GM109091 to K.M. We would also like to acknowledge the work from Jean Schwarzbauer (Princeton University) and Donaldson, J.G (National Institutes of Health) that helped develop protocols for our studies.
Competing interests
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Authors declare no conflicts of interest or competing interests.
References
- Arjonen, A., Alanko, J., Veltel, S. and Ivaska, J. (2012). Distinct recycling of active and inactive β1 integrins. Traffic 13(4): 610-625.
- Caswell, P. T., Chan, M., Lindsay, A. J., McCaffrey, M. W., Boettiger, D. and Norman, J. C. (2008). Rab-coupling protein coordinates recycling of α5β1 integrin and EGFR1 to promote cell migration in 3D microenvironments. J Cell Biol 183(1): 143-155.
- Gao, B., Curtis, T. M., Blumenstock, F. A., Minnear, F. L. and Saba, T. M. (2000). Increased recycling of α5β1 integrins by lung endothelial cells in response to tumor necrosis factor. J Cell Sci 113 Pt 2: 247-257.
- Hoffman, M. A., Ohh, M., Yang, H., Klco, J. M., Ivan, M. and Kaelin, W. G., Jr. (2001). von Hippel-Lindau protein mutants linked to type 2C VHL disease preserve the ability to downregulate HIF. Hum Mol Genet 10(10): 1019-1027.
- Mao, Y. and Schwarzbauer, J. E. (2005). Fibronectin fibrillogenesis, a cell-mediated matrix assembly process. Matrix Biol 24(6): 389-399.
- Mimura, Y., Ihn, H., Jinnin, M., Asano, Y., Yamane, K. and Tamaki, K. (2004). Epidermal growth factor induces fibronectin expression in human dermal fibroblasts via protein kinase C delta signaling pathway. J Invest Dermatol 122(6): 1390-1398.
- Ohh, M., Yauch, R. L., Lonergan, K. M., Whaley, J. M., Stemmer-Rachamimov, A. O., Louis, D. N., Gavin, B. J., Kley, N., Kaelin, W. G., Jr. and Iliopoulos, O. (1998). The von Hippel-Lindau tumor suppressor protein is required for proper assembly of an extracellular fibronectin matrix. Mol Cell 1(7): 959-968.
- Pankov, R., Cukierman, E., Katz, B. Z., Matsumoto, K., Lin, D. C., Lin, S., Hahn, C. and Yamada, K. M. (2000). Integrin dynamics and matrix assembly: tensin-dependent translocation of α5β1 integrins promotes early fibronectin fibrillogenesis. J Cell Biol 148(5): 1075-1090.
- Pasqualini, R., Bourdoulous, S., Koivunen, E., Woods, V. L., Jr. and Ruoslahti, E. (1996). A polymeric form of fibronectin has antimetastatic effects against multiple tumor types. Nat Med 2(11): 1197-1203.
- Roberts, M., Barry, S., Woods, A., van der Sluijs, P. and Norman, J. (2001). PDGF-regulated rab4-dependent recycling of αvβ3 integrin from early endosomes is necessary for cell adhesion and spreading. Curr Biol 11(18): 1392-1402.
- Stickle, N. H., Chung, J., Klco, J. M., Hill, R. P., Kaelin, W. G., Jr. and Ohh, M. (2004). pVHL modification by NEDD8 is required for fibronectin matrix assembly and suppression of tumor development. Mol Cell Biol 24(8): 3251-3261.
- Tang, C. H., Yang, R. S., Chen, Y. F. and Fu, W. M. (2007). Basic fibroblast growth factor stimulates fibronectin expression through phospholipase C γ, protein kinase C α, c-Src, NF-κB, and p300 pathway in osteoblasts. J Cell Physiol 211(1): 45-55.
- Uitto, J., Olsen, D. R. and Fazio, M. J. (1989). Extracellular matrix of the skin: 50 years of progress. J Invest Dermatol 92(4 Suppl): 61S-77S.
- Varadaraj, A., Jenkins, L. M., Singh, P., Chanda, A., Snider, J., Lee, N. Y., Amsalem-Zafran, A. R., Ehrlich, M., Henis, Y. I. and Mythreye, K. (2017). TGF-β triggers rapid fibrillogenesis via a novel TbetaRII-dependent fibronectin-trafficking mechanism. Mol Biol Cell 28(9): 1195-1207.
- White, D. P., Caswell, P. T. and Norman, J. C. (2007). αvβ3 and α5β1 integrin recycling pathways dictate downstream Rho kinase signaling to regulate persistent cell migration. J Cell Biol 177(3): 515-525.
- Yang, J. T. and Hynes, R. O. (1996). Fibronectin receptor functions in embryonic cells deficient in alpha 5 beta 1 integrin can be replaced by alpha V integrins. Mol Biol Cell 7(11): 1737-1748.
- Yi, M. and Ruoslahti, E. (2001). A fibronectin fragment inhibits tumor growth, angiogenesis, and metastasis. Proc Natl Acad Sci U S A 98(2): 620-624.
- Yokoi, H., Mukoyama, M., Sugawara, A., Mori, K., Nagae, T., Makino, H., Suganami, T., Yahata, K., Fujinaga, Y., Tanaka, I. and Nakao, K. (2002). Role of connective tissue growth factor in fibronectin expression and tubulointerstitial fibrosis. Am J Physiol Renal Physiol 282(5): F933-942.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Varadaraj, A., Magdaleno, C. and Mythreye, K. (2018). Deoxycholate Fractionation of Fibronectin (FN) and Biotinylation Assay to Measure Recycled FN Fibrils in Epithelial Cells. Bio-protocol 8(16): e2972. DOI: 10.21769/BioProtoc.2972.
Category
Biochemistry > Protein > Self-assembly
Biochemistry > Protein > Isolation and purification
Molecular Biology > Protein > Isolation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.