- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

High Resolution Melting Temperature Analysis to Identify CRISPR/Cas9 Mutants from Arabidopsis
Published: Vol 8, Iss 14, Jul 20, 2018 DOI: 10.21769/BioProtoc.2944 Views: 9071
Reviewed by: Rainer MelzerRunlai HangMarta Bjornson
Original research article
The authors used this protocol in:

Jul 2017
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
CRISPR/Cas9 made targeted mutagenesis and genome editing possible for many plant species. One of the ways that the endonuclease is used for plant genetics is the creation of loss-of-function mutants, which typically result from erroneous DNA repair through non-homologous end joining (NHEJ) pathway. The majority of erroneous repair events results in single-bp insertion or deletion. While single-bp insertions or deletions (indels) effectively destroy the function of protein-coding genes through frameshift, detection is difficult due to the small size shift. High-resolution melting temperature analysis allows quick detection, and it does not require any additional pipetting steps after the PCR amplification of the region of interest. In this protocol, we will describe the steps required for the analysis of potential homozygous mutants.
Keywords: CRISPR/Cas9Background
CRISPR/Cas9 nuclease is a ribonucleoprotein that is capable of cleaving a DNA double strand at a specific 22 nucleotide sequence. The major advantage of the CRISPR/Cas9 system compared to other nucleases such as zinc-finger nucleases and Transcription Activator-Like Effector Nucleases (TALENs) is that the sequence specificity is conferred by the RNA and does not require separate proteins for each target sequence. This reduces the cost dramatically, and a single construct can target as many as 32 targets. Due to this low cost and efficiency, the CRISPR/Cas9 system is now widely used in many plant species (Baltes and Voytas, 2015; Belhaj et al., 2015).
When a double-stranded DNA break induced by CRISPR/Cas9 is erroneously repaired by the NHEJ pathway, the repaired sequence most frequently results in small indels, among which one bp indels are the most common (Ma et al., 2015; Pan et al., 2016; Ren et al., 2016). One bp indel is too small for a polyacrylamide gel electrophoresis-based method to detect, therefore alternative methods capable of detecting one-bp indels are needed (Zhu et al., 2014).
Currently, the most commonly used method for small indel detection takes advantage of enzymes such as T7 endonuclease (T7E1) and CEL nuclease that cleave mismatches (Yeung et al., 2005; Vouillot et al., 2015). In these methods, the region including the target sequence is amplified by PCR, followed by the melting-annealing cycle to generate heteroduplexes and digestion by the mismatch detecting enzymes. While these methods are very effective in detecting large indels, they are not very effective in detecting a one-bp deletion. Even with T7E1 nuclease, which is better suited for the detection of small indels compared to the CEL nuclease, the efficiency decreases as the indel size decreases (Gohlke et al., 1994).
An alternative method, high-resolution melting (HRM), offers multiple advantages. HRM detects a small shift in the melting temperature (e.g., caused by heteroduplexing) using a dsDNA-binding dye. Firstly, it does not require additional pipetting steps after the PCR amplification step of the target region. Secondly, the method reliably detects single bp indels at a low (5%) concentration. In this protocol, we describe the procedure to analyze the HRM curves of PCR fragments containing CRISPR-generated small indels (Denbow et al., 2017). Four steps required for the detection of CRISPR-induced indels in Arabidopsis include: i) genomic DNA extraction, ii) optimizing the PCR condition, iii) PCR step for analyzing either T1 plants or putative homozygous plants carrying a single bp deletion, and iv) data analysis.
Materials and Reagents
- Tissue disruption
- 2 ml microfuge tubes
- Glass beads, 4 mm diameter (Walter Stern, catalog number: 100E )
- Arabidopsis
- Liquid nitrogen
- 2 ml microfuge tubes
- DNA isolation
- 1.5 ml microfuge tubes
- Chloroform/IAA mixture (24:1)
- Isopropanol (CAS number 67-63-0)
- Ethanol (200 proof) (CAS number 64-17-5)
- 70% ethanol
- Tris-HCl (pH 7.5)
- Tris base (CAS number 77-86-1)
- EDTA free acid (CAS number 60-00-4)
- NaCl (CAS number 7647-14-5)
- CTAB (CAS number 57-09-0)
- RNase A (Sigma-Aldrich, catalog number: R6513 )
- Cesium Chloride (CsCl) (CAS number 7647-17-8)
- 2% CTAB solution (see Recipes)
- TE buffer (see Recipes)
- RNase A stock solution (see Recipes)
- 1 M CsCl solution (see Recipes)
- 1.5 ml microfuge tubes
- PCR
- Optical film (Bio-Rad Laboratories, catalog number: 2239444 )
- 96 well hard-shell black and white plate (Bio-Rad Laboratories, catalog number: hsp9665 )
- LC Green Plus (BioFire Defense, catalog number: BCHM-ASY-0006 )
- Phire Hot Start II DNA polymerase and buffer (Thermo Fisher Scientific, catalog number: F122 )
- dNTP, 100 mM each (Thermo Fisher Scientific, catalog number: 10297018 )
- Mineral oil (Fisher Scientific, catalog number: O121-1 )
- Agarose (CAS number 9012-36-6)
- PCR primers
- Glacial acetic acid (CAS number 64-19-7)
- TAE buffer (see Recipes)
Equipment
- LightScanner system (BioFire Defense, USA)
- PCR machine (Bio-Rad C1000) (Bio-Rad Laboratories, model: C1000 )
- Tissue grinder (Mini-Beadbeater-96, Biospec, OK, USA) (Bio Spec Products, model: Mini-Beadbeater-96 )
- Tabletop centrifuge (Eppendorf, model: 5427 R )
- Vortex mixer (Fisher Scientific, catalog number: 02-215-365 )
Software
- LightScanner software
Procedure
- Genomic DNA isolation
- Harvest 0.1-0.2 g (one mature leaf from Arabidopsis, ~20 mm in length excluding the petiole) of fresh tissue into a 2 ml microfuge tube containing a 4 mm glass bead and immediately freeze in liquid nitrogen. Grind the tissue in liquid nitrogen. Include at least two wild-types (or isogenic background to the mutant) samples. We use the mini-bead beater for grinding the tissues.
- Add 300 μl of Chl/IAA and 300 μl of 2% CTAB solution, vortex for 10 sec.
- Incubate at 65 °C for 30 min.
- Centrifuge at the maximal speed, 4 °C, 10 min.
- Transfer the aqueous phase to a fresh 1.5 ml tube and add 300 μl of Chl/IAA. Vortex for 10 sec, and centrifuge at the maximal speed (18,210 x g), 4 °C, 10 min.
- Transfer the aqueous phase to a fresh 1.5 ml tube and add an equal volume of isopropanol. Vortex for 10 sec.
- Precipitate the genomic DNA by incubating the tube for 10 min at -20 °C.
- Centrifuge at the maximal speed (18,210 x g), 4 °C, 10 min.
- Discard supernatant, wash once with 70% ethanol and dry pellet. Add 50 μl of TE buffer + 0.5 μl RNase stock solution. Mix the contents gently to dissolve the pellet. If it does not dissolve easily, warm the tubes at 65 °C. Incubate for 30 min at 37 °C.
- Add 400 μl of 1 M CsCl solution and mix well.
- Add 800 μl of ethanol, mix and let the genomic DNA precipitate for 20 min at -20 °C.
- Centrifuge at the maximal speed (18,210 x g), 4 °C, 10 min.
- Discard the supernatant and wash the pellet with 700 μl of 70% ethanol. Centrifuge the tubes again to and discard the remaining ethanol.
- Dissolve dried pellet in 50 μl TE buffer. The DNA can be stored at -20 °C.
Note: The method above produces good quality DNA samples, which is important for the reproducibility of the downstream analysis. Other genomic extraction procedures can be used in place of the above method, as long as the DNA quality is comparable.
- Primer design
Primers flanking the predicted editing site should be designed. We typically design a primer set producing an 80-120 bp fragment. Amplicon size > 150 bp might decrease the sensitivity of HRM analysis (Gundry et al., 2013). Tm should be 58 ± 2 °C, and GC content should not exceed 65% (Laurie and George, 2009). - Optimizing the PCR protocol
- Pipet the PCR mixture into a 1.5 ml tube, containing the following components per reaction. We recommend testing temperatures ± 3 °C around the Tm with two technical replicates.
4 μl 5x Phire Reaction Buffer
2 μl dNTP, 2.5 mM each
2 μl forward primer, 5 μM
2 μl reverse primer, 5 μM
2 μl genomic DNA from Step A14, diluted 1/10 in water
0.4 μl Phire Hot Start II DNA Polymerase
5.6 μl water - Run a gradient PCR protocol as follows:

- Run the PCR products on a 4% agarose gel in TAE buffer. Pick the condition that does not produce non-specific secondary bands.
ATTENTION: Additional non-specific bands will complicate the HRM analysis by producing multiple peaks in the decay curve (see the example in Figure 1).
Notes:- For preparing a 4% gel, it is recommended to mix the agarose and TAE and let it hydrate for 30 min prior to microwaving. This step promotes uniform melting.
- Theoretically any PCR enzyme can be used in combination with LC green. In our hands, Phire Hot Start II polymerase produced the most robust results.
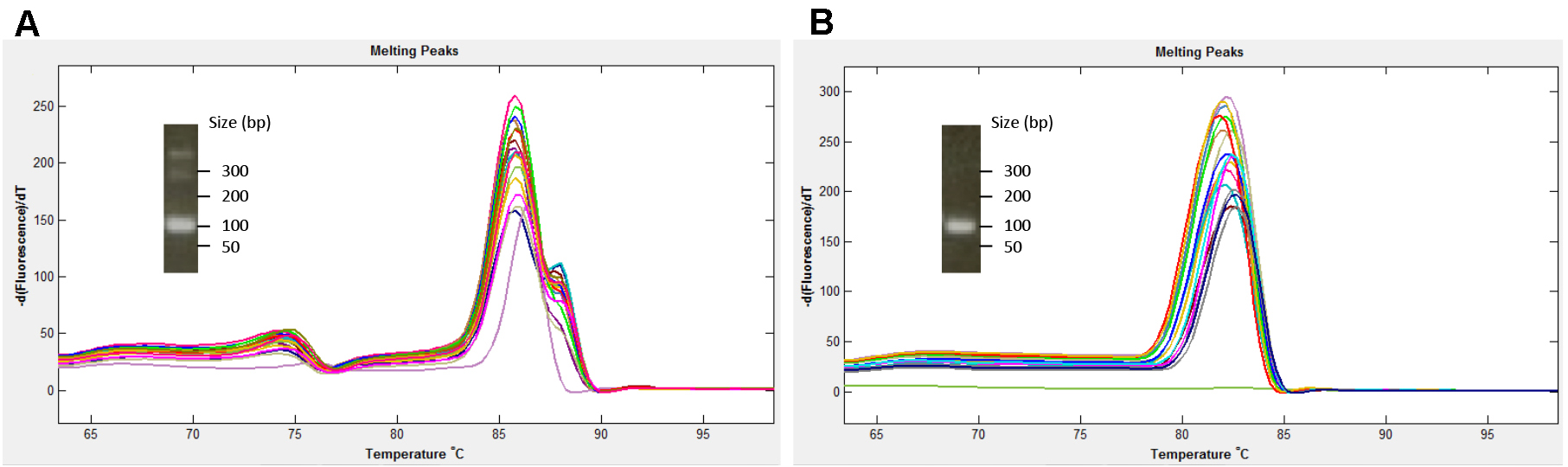
Figure 1. Melt curve influenced by non-specific bands. A. A primer combination that produces non-specific bands. The inset shows a reprehensive PCR product produced using the same combination primers, visualized on 4% agarose gel. B. Melt curves from a primer combination that does not produce non-specific bands. - For preparing a 4% gel, it is recommended to mix the agarose and TAE and let it hydrate for 30 min prior to microwaving. This step promotes uniform melting.
- Pipet the PCR mixture into a 1.5 ml tube, containing the following components per reaction. We recommend testing temperatures ± 3 °C around the Tm with two technical replicates.
- HRM analysis
- Make a PCR master mix into a 1.5 ml tube, containing the following components per reaction. We strongly recommend at least two technical replicates per sample.
4 μl 5x Phire Reaction Buffer
2 μl dNTP, 2.5 mM each
2 μl forward primer, 5 μM
2 μl reverse primer, 5 μM
2 μl LC Green dye
0.4 μl Phire Hot Start II DNA Polymerase
5.6 μl water - Pipet 18 μl each of the master mix into the 96 well black and white PCR plate. Add 2 μl 1/10 diluted genomic DNA from the unknown samples. Overlay 20 μl mineral oil to each well and seal with an optical film.
Note: Mineral oil is not required if the instrument used for producing HRM is top-heated. - Perform a PCR reaction.

ATTENTION: *The above two steps are critical for forming the heteroduplexes between the PCR products between WT and mutant DNA. - Centrifuge the plate briefly to remove any condensation or air bubble.
- Obtain the melting curve using LightScanner or an equivalent machine capable of performing HRM analysis. In the authors’ lab, the temperature range of 75 °C to 100 °C is used. LightScaner records fluorescence intensity of each well at approximately 0.1 °C interval. The result is automatically saved once the melting is completed as .mat file.
- Make a PCR master mix into a 1.5 ml tube, containing the following components per reaction. We strongly recommend at least two technical replicates per sample.
Data analysis
This step will depend on the software available for the data analysis, typically provided by the manufacturers of machines equipped with HRM analysis capabilities. In this example, we show how to analyze the dataset obtained using LightScanner. Steps described in the following sections can also in Video 1.

Overview: The analysis consists basically of four steps: 1) Designating experimental groups; 2) Determining the range of the melt curves to be analyzed; 3) Selecting the reference curve (typically the curve generated from WT; and 4) Calling mutations based on the difference from the reference curve.
- Open LightScanner software, and click on Small amplicon. In the pop-up window, select the saved experiment file (.mat file) to be analyzed.
- Define the subsets
- Click on Subsets tab at the top, and click New subset button. In the 96 well format grid at the top, click on the wells that contained the samples. Each primer combination should be one subset. Once done, click Add Marked to Subset. Define all the subsets (Figure 2).
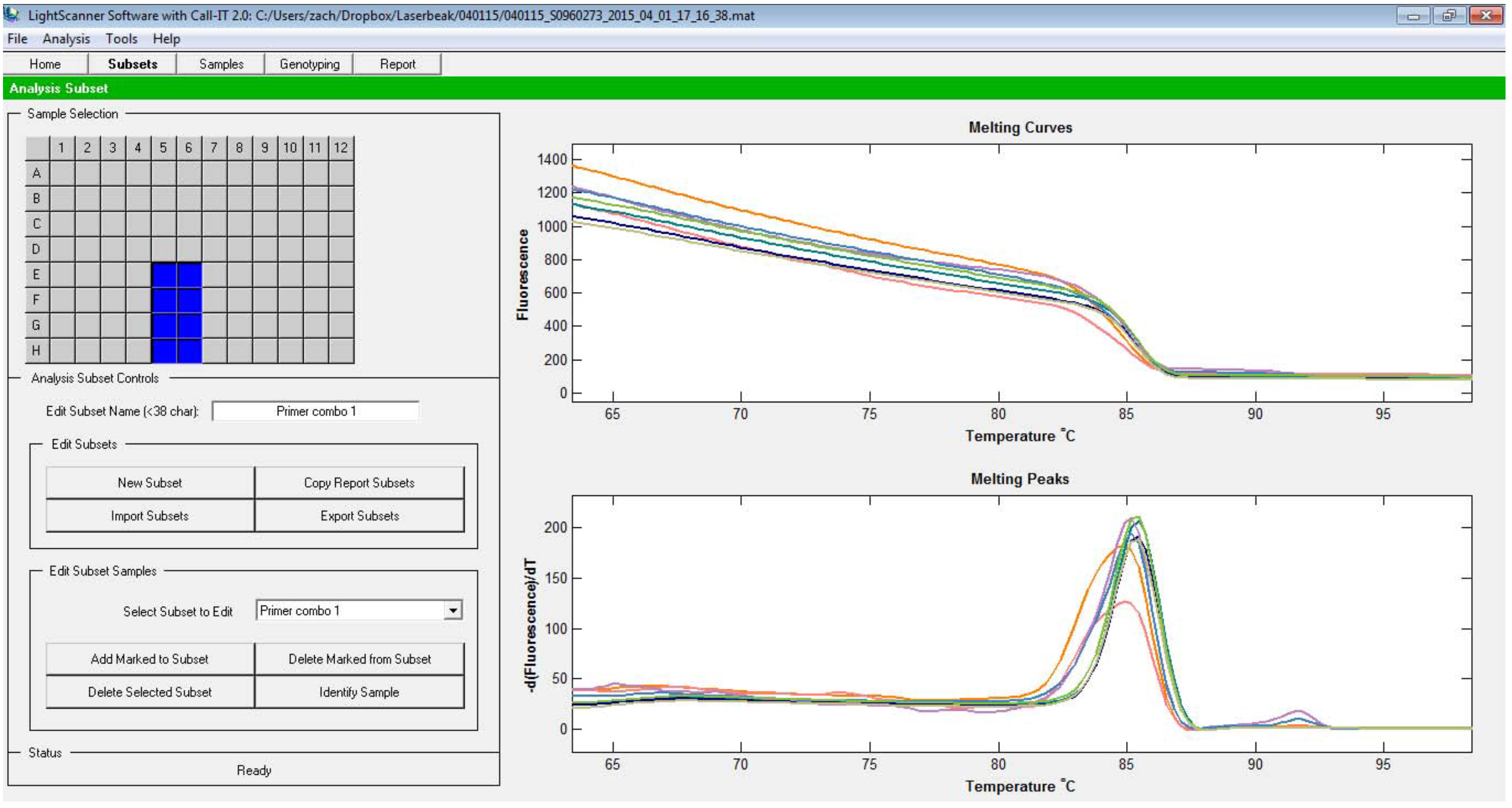
Figure 2. Subsets selection screen. Typically, all reactions with one primer combination is designated as one subset. - Once subsets are defined, you can view a specific subset by clicking on Select Subset to Edit button.
- Click on Subsets tab at the top, and click New subset button. In the 96 well format grid at the top, click on the wells that contained the samples. Each primer combination should be one subset. Once done, click Add Marked to Subset. Define all the subsets (Figure 2).
- Add sample names
Open Samples tab at the top, and type in all the sample names (Figure 3).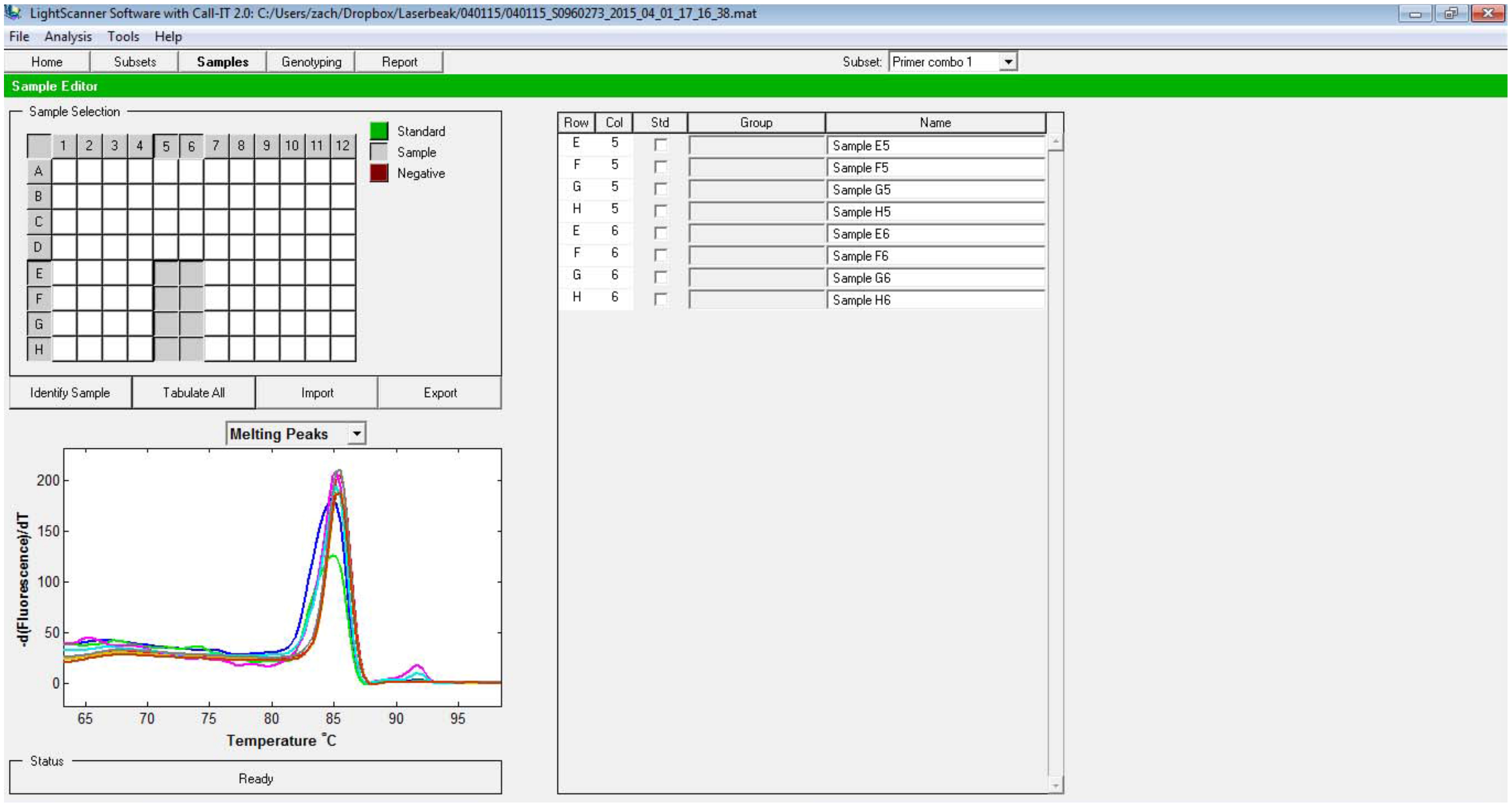
Figure 3. Sample names can be designated under Samples tab - Call the differences between the samples
- Click on Genotyping tab at the top, then click on Normalize tab to select the range that encompasses the range to be analyzed. Include the plateau before and after the melting (Figure 4).
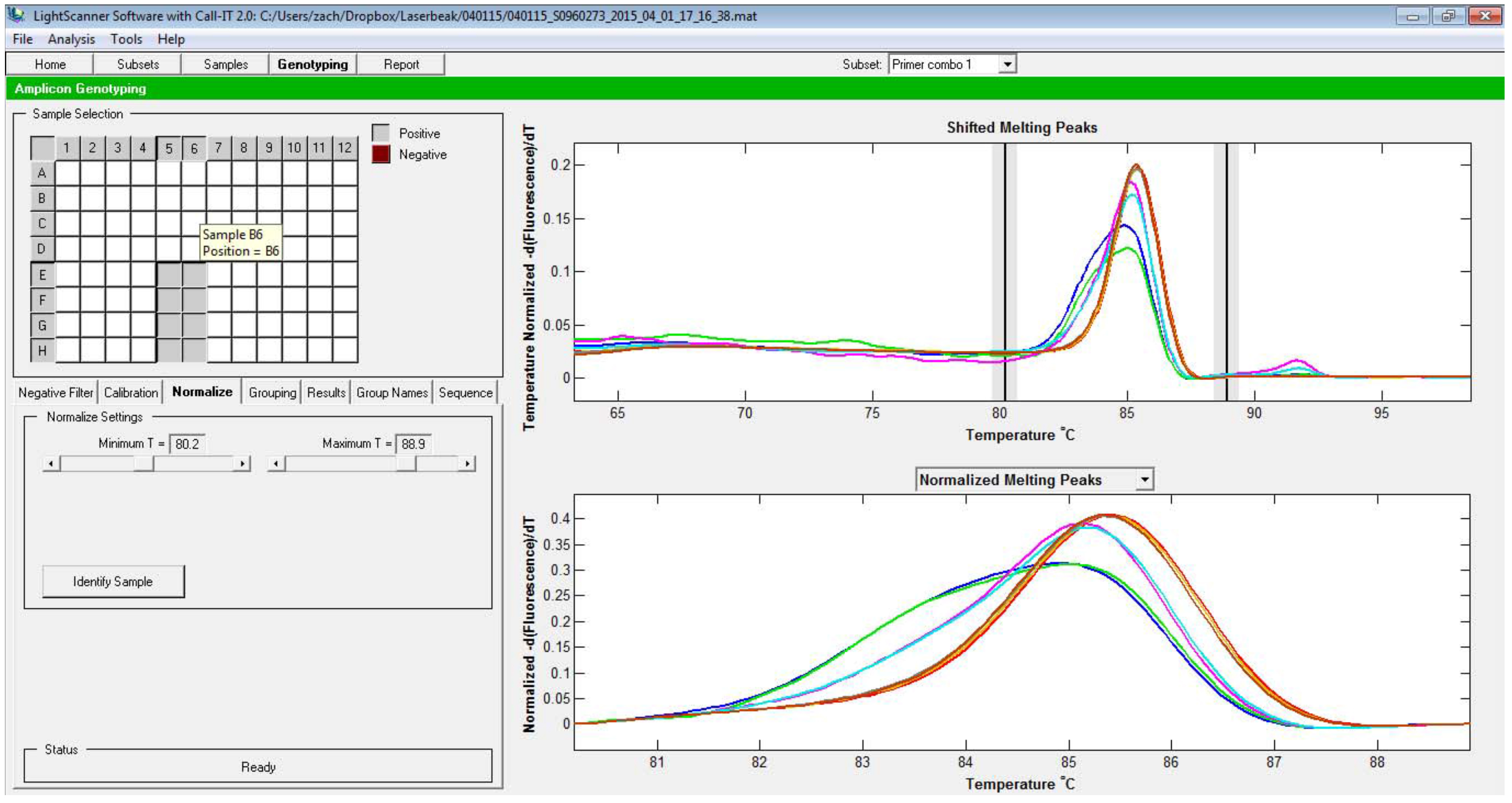
Figure 4. Selection of the melt curve window to be analyzed. The two vertical lines represent the beginning and the end of the regions used for normalization. - In case you are trying to call the difference between the mutants and the WT, the melt curve from the WT should be set as the baseline. To do this, click Select baseline, click on the well that contained the sample to be used as the baseline, then Finish selection. After this, the normalized melt curve (the bottom graph) is recalculated to set WT curve as the baseline (Figure 5).
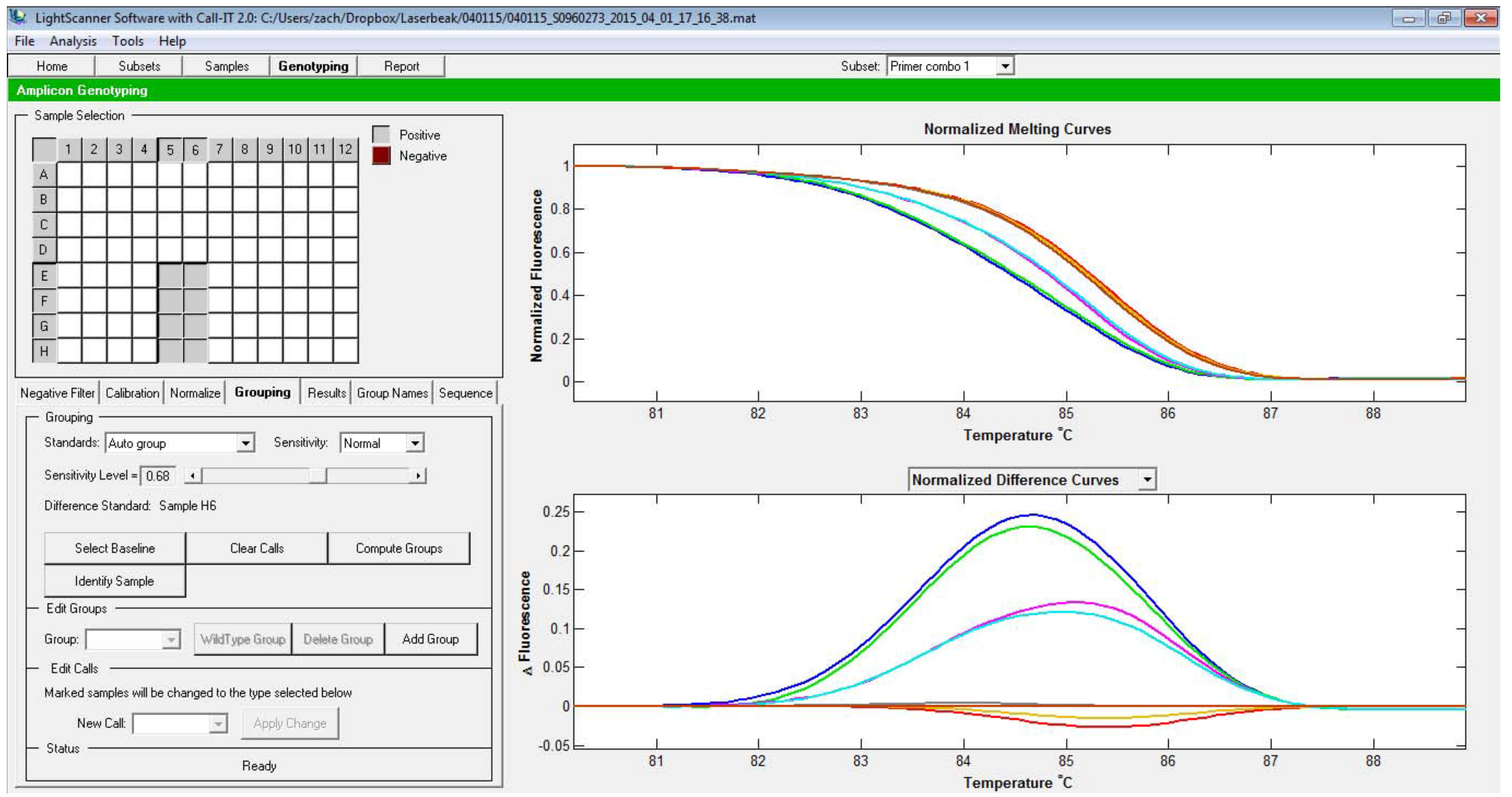
Figure 5. Selection of the baseline curve. Note the difference with Figure 3, in which the baseline is not designated. Other curves are recalculated based on the baseline curve. - Now, the groups can be called either automatically or manually. To call the different groups automatically, select Auto group option, then change the sensitivity level (Figure 6).
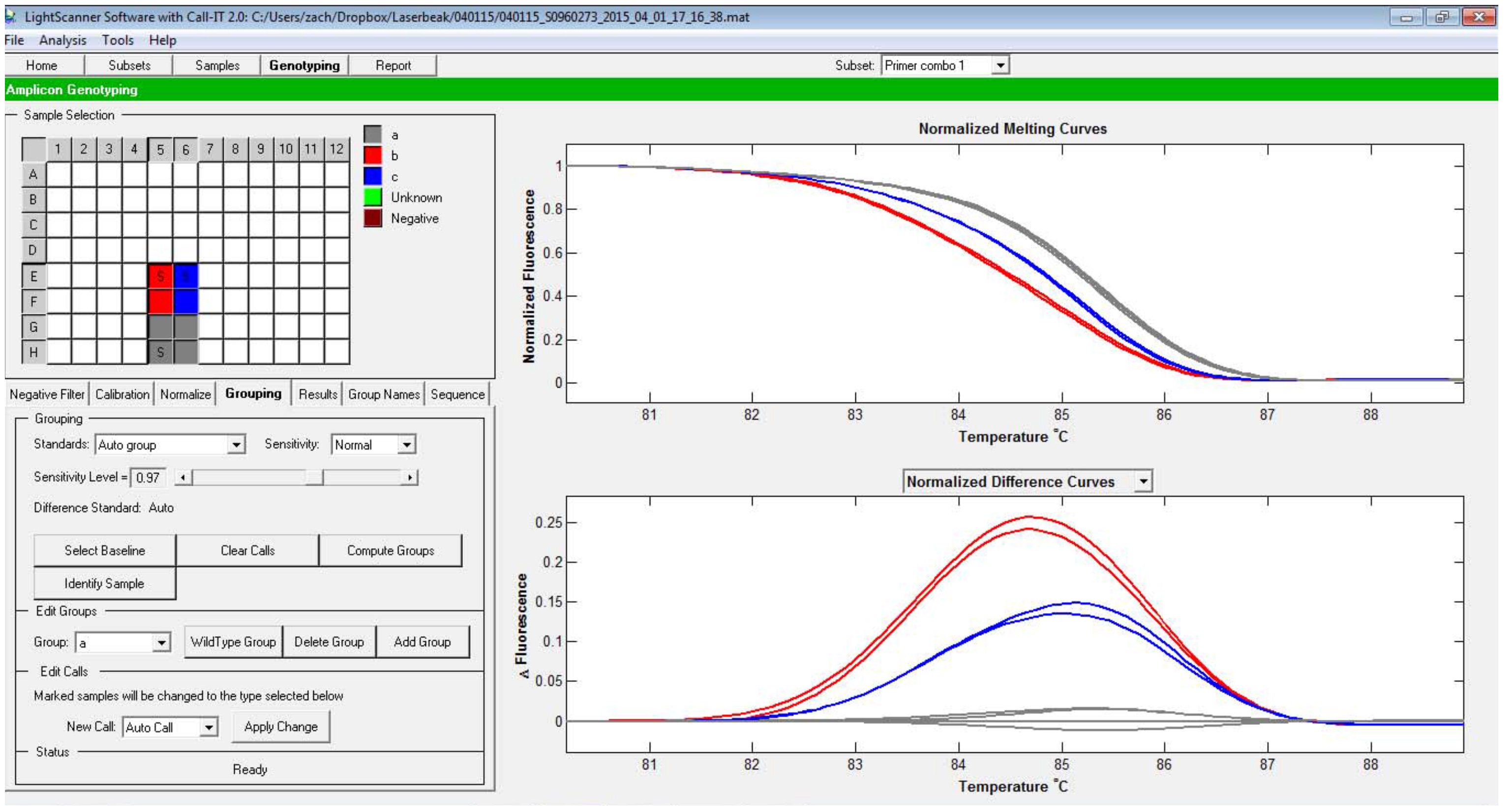
Figure 6. Automatic group calling. In this case, samples shown in red and blue are called as separate groups from the ones that group with the baseline. - Alternatively, groups can be set manually. To do this, under Edit groups, press Add group. Then click on the wells that contained the samples belonging to the same group (e.g., replicates from one genotype), then under Edit Call, press Apply changes. Now the group is displayed in a different color. The process can be repeated to add additional groups (Figure 7).
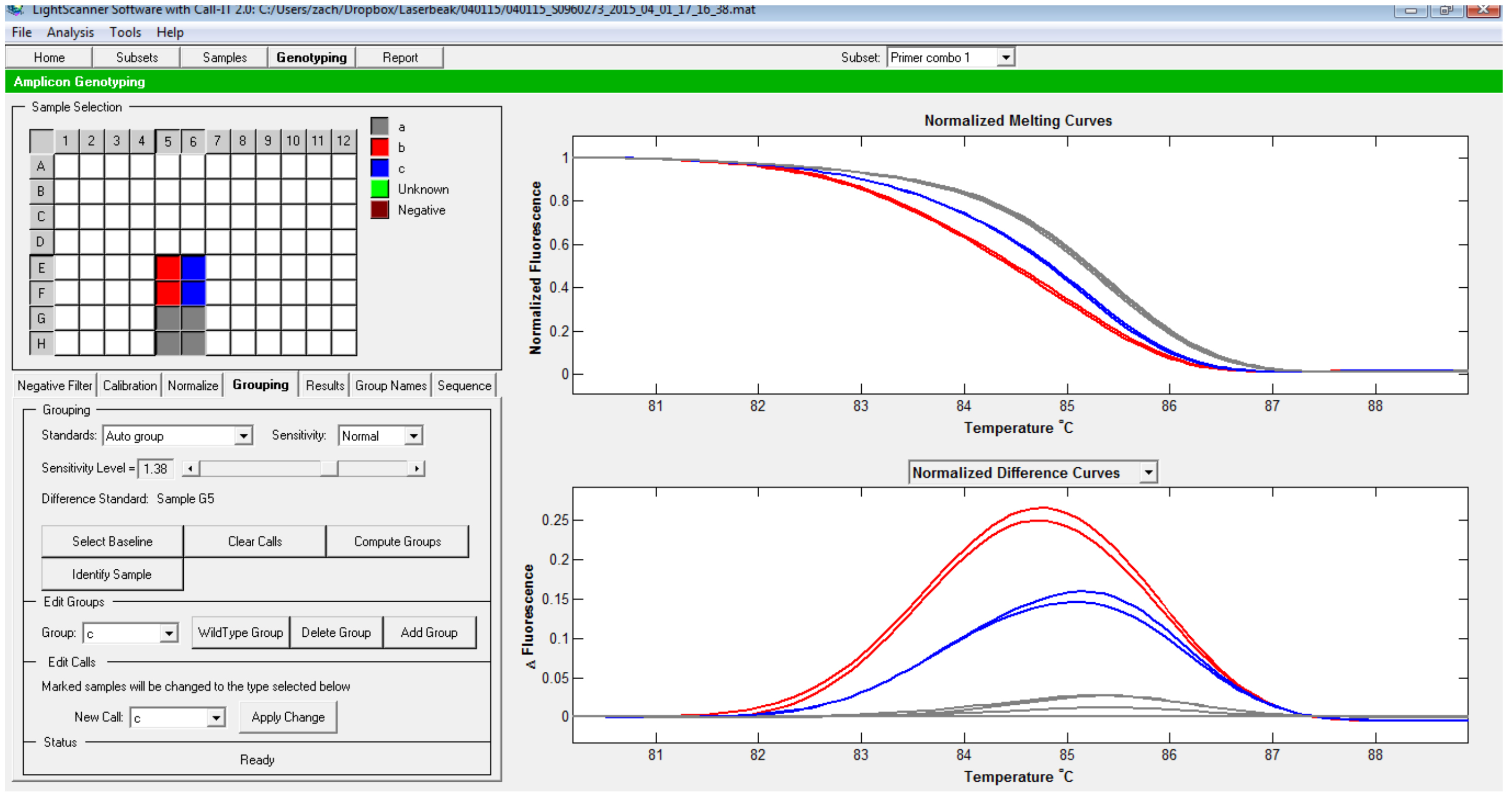
Figure 7. Manual selection of groups. Groups have been manually designated using the procedure detailed in the protocol.
Note: For an accurate prediction, the variation in ΔFluorescence among the WT samples should be < 0.05. The manual mode helps to identify different samples and examine if there is any apparent difference between them. The authors use ΔFluorescence > 0.1 compared to the WT as a rule-of-thumb for potential mutants and analyze the PCR fragments through Sanger sequencing later.
- Click on Genotyping tab at the top, then click on Normalize tab to select the range that encompasses the range to be analyzed. Include the plateau before and after the melting (Figure 4).
Recipes
- 2% CTAB solution
100 mM Tris-HCl (pH 8.0)
20 mM EDTA (pH 8.0)
1.4 M NaCl
2% CTAB
Autoclave - TE buffer
10 mM Tris-HCl (pH 8.0)
1 mM EDTA (pH 8.0)
Autoclave - 100x RNase A stock solution
10 mM Tris-HCl (pH 7.5)
15 mM NaCl
10 mg/ml RNase A
Boil at 100 °C for 5 min, cool and store at -20 °C - 1 M CsCl
Dissolved in distilled water, autoclaved - 50x TAE buffer
2 M Tris base
50 mM EDTA (pH 8.0)
5.71% (v/v) glacial acetic acid
Acknowledgments
This work was supported by The National Science Foundation MCB 1052048 (S.O.), IOS 1403593, and Texas A&M University AgriLife research. The protocol has been partially adapted from our previous publication (Denbow et al., 2017).The authors declare no conflicts of interest.
References
- Baltes, N. J. and Voytas, D. F. (2015). Enabling plant synthetic biology through genome engineering. Trends Biotechnol 33(2): 120-131.
- Belhaj, K., Chaparro-Garcia, A., Kamoun, S., Patron, N. J. and Nekrasov, V. (2015). Editing plant genomes with CRISPR/Cas9. Curr Opin Biotechnol 32: 76-84.
- Denbow, C. J., Lapins, S., Dietz, N., Scherer, R., Nimchuk, Z. L. and Okumoto, S. (2017). Gateway-compatible CRISPR-Cas9 vectors and a rapid detection by high-resolution melting curve analysis. Front Plant Sci 8: 1171.
- Gohlke, C., Murchie, A. I., Lilley, D. M. and Clegg, R. M. (1994). Kinking of DNA and RNA helices by bulged nucleotides observed by fluorescence resonance energy transfer. Proc Natl Acad Sci U S A 91(24): 11660-11664.
- Gundry, C. N., Vandersteen, J. G., Reed, G. H., Pryor, R. J., Chen, J. and Wittwer, C. T. (2003). Amplicon melting analysis with labeled primers: a closed-tube method for differentiating homozygotes and heterozygotes. Clin Chem 49: 396-406.
- Laurie, A. D. and George, P. M. (2009). Evaluation of high-resolution melting analysis for screening the LDL receptor gene. Clin Biochem 42: 528-35.
- Ma, X., Zhang, Q., Zhu, Q., Liu, W., Chen, Y., Qiu, R., Wang, B., Yang, Z., Li, H., Lin, Y., Xie, Y., Shen, R., Chen, S., Wang, Z., Chen, Y., Guo, J., Chen, L., Zhao, X., Dong, Z. and Liu, Y. G. (2015). A robust CRISPR/Cas9 system for convenient, high-efficiency multiplex genome editing in monocot and dicot plants. Mol Plant 8(8): 1274-1284.
- Pan, C., Ye, L., Qin, L., Liu, X., He, Y., Wang, J., Chen, L. and Lu, G. (2016). CRISPR/Cas9-mediated efficient and heritable targeted mutagenesis in tomato plants in the first and later generations. Sci Rep 6: 24765.
- Ren, C., Liu, X., Zhang, Z., Wang, Y., Duan, W., Li, S. and Liang, Z. (2016). CRISPR/Cas9-mediated efficient targeted mutagenesis in Chardonnay (Vitis vinifera L.). Sci Rep 6: 32289.
- Vouillot, L., Thelie, A. and Pollet, N. (2015). Comparison of T7E1 and surveyor mismatch cleavage assays to detect mutations triggered by engineered nucleases. G3 (Bethesda) 5(3): 407-415.
- Yeung, A. T., Hattangadi, D., Blakesley, L. and Nicolas, E. (2005). Enzymatic mutation detection technologies. Biotechniques 38(5): 749-758.
- Zhu, X., Xu, Y., Yu, S., Lu, L., Ding, M., Cheng, J., Song, G., Gao, X., Yao, L., Fan, D., Meng, S., Zhang, X., Hu, S. and Tian, Y. (2014). An efficient genotyping method for genome-modified animals and human cells generated with CRISPR/Cas9 system. Sci Rep 4: 6420.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Denbow, C., Ehivet, S. C. and Okumoto, S. (2018). High Resolution Melting Temperature Analysis to Identify CRISPR/Cas9 Mutants from Arabidopsis. Bio-protocol 8(14): e2944. DOI: 10.21769/BioProtoc.2944.
Category
Plant Science > Plant molecular biology > DNA > Mutagenesis
Molecular Biology > DNA > Mutagenesis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.