- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Single and Multiplexed Gene Editing in Ustilago maydis Using CRISPR-Cas9

Published: Vol 8, Iss 14, Jul 20, 2018 DOI: 10.21769/BioProtoc.2928 Views: 9949
Reviewed by: Aksiniya AsenovaShahin S. AliAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2017

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
The smut fungus Ustilago maydis is an established model organism for elucidating how biotrophic pathogens colonize plants and how gene families contribute to virulence. Here we describe a step by step protocol for the generation of CRISPR plasmids for single and multiplexed gene editing in U. maydis. Furthermore, we describe the necessary steps required for generating edited clonal populations, losing the Cas9 containing plasmid, and for selecting the desired clones.
Keywords: CRISPR-Cas9Background
The basidiomycete fungus U. maydis is a model organism that has enabled important discoveries in topics like DNA repair and homologous recombination, mating, sexual development, secondary metabolism, filamentous growth, RNA transport and virulence (Holliday, 2004; Steinberg, 2007; Bakkeren et al., 2008; Holloman et al., 2008; Lanver et al., 2017; Niessing et al., 2018). U. maydis is a biotrophic plant pathogen that causes smut disease on maize. As a typical member of the large group of smut fungi its sexual development is intimately coupled with its ability to colonize plants. The popularity of U. maydis as a model organism resides in its short pathogenic life cycle that is completed in less than two weeks, its accessibility for forward and reverse genetics that includes the establishment of self-replicating plasmids (Tsukuda et al., 1988), the development of solopathogenic strains able to cause disease without mating and an available well-annotated genome (Kämper et al., 2006). In U. maydis, homologous recombination-based genome manipulations are very efficient, but become tedious when several genes have to be modified because the number of selectable markers is limited (Schuster et al., 2016). The most recent addition to the technical toolboxes for this organism has been the adaptation of the CRISPR-Cas9 genome editing technology (Schuster et al., 2016) and its development for multiplexed editing (Schuster et al., 2018). The CRISPR-Cas9 genome editing technology, developed from research in bacterial immune systems (Barrangou et al., 2007), has revolutionized molecular biology. The system allows efficient modification of almost any given sequence (Jinek et al., 2012) and has been established for a large number of organisms including filamentous fungi (Deng et al., 2017; Shi et al., 2017).
The CRISPR-Cas9 technology established for U. maydis is based on an all-in-one plasmid approach. A self-replicating plasmid (Tsukuda et al., 1988) is used as a backbone to provide the U. maydis codon-optimized Cas9 gene and the U6 promoter for sgRNA expression. This plasmid is unstable without selection and is rapidly lost (Figure 1 and Schuster et al. [2016]). The system proved very efficient for disruption of single genes reaching on average 70% of edited cells in progeny of one transformant (Schuster et al., 2016). A multiplexing version of the system was generated by using tRNA promoter-based expression cassettes that enabled the expression of several sgRNAs from the same plasmid (Schuster et al., 2018). This modification combined with elevated and longer expression of the Cas9 protein was used for the simultaneous disruption of five effector genes with 70% efficiency (Schuster et al., 2018). The multiplexed version of CRISPR-Cas9 has also been successfully applied for elucidating the contribution of oligopeptide transporters to virulence (Lanver et al., 2018).
Materials and Reagents
- Consumables
- Sterile pipette tip
- Toothpick
- 60 mm x 15 mm round Petri dishes
- Sterile pipette tip
- Competent cells
E. coli competent cells e.g., One Shot TOP10 Chemically Competent E. coli (Thermo Fisher Scientific, catalog number: C404010 ) - Plasmids and double-stranded DNA
- Oligonucleotides 10 pmol/µl
- oMS59: 5’ ATTCGTGATTTACACCAAACACGC 3’
- oMS49: 5’ CCCCTCGTCTCGCGCCTCATTGGTCGAATTG 3’
- oSR224: 5’ CTACACTCAGCACACGATGT 3’
- oMS59: 5’ ATTCGTGATTTACACCAAACACGC 3’
- Enzymes and buffers
- Kits
- Antibiotics
- Reagents
- Tryptone (BD Biosciences)
- Yeast extract (BD Biosciences)
- Sodium chloride (NaCl)
- Peptone (BD Biosciences)
- Sucrose (BD Biosciences)
- Sorbitol (Sigma-Aldrich, catalog number: S1876 )
- Bacto agar (BD Biosciences)
- Potato Dextrose Broth (BD Biosciences)
- Tryptone (BD Biosciences)
- Media (see Recipes)
- Yeast extract tryptone (dYT) liquid medium + 100 µg/ml ampicillin
- YT agar plates + 100 µg/ml ampicillin
- YEPS light liquid medium +/- 2 µg/ml carboxin
- Two layered regeneration agar light (RegAgar) plates (Bottom layer (10 ml) with 4 µg/ml carboxin and top layer (10 ml) without carboxin (Prepare fresh))
- Potato dextrose (PD) agar plates +/- 2 µg/ml carboxin
- Yeast extract tryptone (dYT) liquid medium + 100 µg/ml ampicillin
Equipment
Note: No equipment from specific manufacturers is required. Any equivalent device can be used.
- Pipettes
- Computer with internet access
- Plate incubators (28 °C, 37 °C)
- Shakers (28 °C, 37 °C)
- ThermoMixer (e.g., Eppendorf, model: ThermoMixer® C , catalog number: 5382000015)
- Thermocycler (e.g., Analytik, Jena, Biometra, catalog number: 846-2-070-301 )
- Tabletop centrifuge (e.g., Thermo Fisher Scientific, catalog number: 75008801 )
Software
No specific software is required. Any appropriate alignment program like CloneManager 9 Professional Edition (Scientific & Educational Software) or CLC Main Workbench (QIAGEN) can be used.
Procedure
- Select target sequences with E-CRISP (Heigwer et al., 2014)
Note: Although several tools have been developed for sgRNA design, our experience is limited to the E-CRISP tool that proved to be very reliable.- Select U. maydis as the organism in the E-CRISP webpage (http://www.e-crisp.org/E-CRISP/).
- Click on the ‘Enter target sequence’ box and insert target genomic DNA sequence in FASTA format.
Note: Use the coding sequence of the gene you want to inactivate and restrict your search to the most 5´ end of the gene. In case you do not find a suitable sgRNA you can expand the input sequence towards the 3´end. - Click on ‘display advanced options’ on the ‘Start application’ panel and make sure the 3’ PAM is NGG.
Notes:- The E-CRISP tool offers to restrict the search of target sequences to those starting with a G. However, this restriction is not necessary when using our plasmid backbones, because our linearized plasmids retain a G overhang at the 3´ end of the U6 promoter that serves as the first G preferred for initiating transcription at the U6 promoter.
- The E-CRISP tool gives you the option to design truncated target sequences (Fu et al., 2014). We usually use target sequences of 20 nt but have been successful using target sequences as short as 18 nt.
- The E-CRISP tool offers to restrict the search of target sequences to those starting with a G. However, this restriction is not necessary when using our plasmid backbones, because our linearized plasmids retain a G overhang at the 3´ end of the U6 promoter that serves as the first G preferred for initiating transcription at the U6 promoter.
- Click on ‘Start sgRNA search’.
- Select the target sequence with the highest score and most 5’ in the gene. Double check that the target gene(s) correspond to the gene(s) you want to disrupt.
- Select U. maydis as the organism in the E-CRISP webpage (http://www.e-crisp.org/E-CRISP/).
- Design sgRNA expression cassettes following examples in Table 1 and insert them into pCas9_sgRNA_0 or pMS73 (Figure 1)
Note: The CRISPR-Cas9 plasmids developed for genome editing in U. maydis confer ampicillin resistance in E. coli and carboxin resistance in U. maydis. These plasmids contain a U. maydis ARS element for autonomous replication (Tsukuda et al., 1988). The plasmid backbones also contain the U. maydis U6 promoter just before an Acc65I site that is used for integration of the sgRNA expression cassette via Gibson Assembly® (Figure 1). Expression of the codon-optimized Cas9 is driven by the constitutive otef promoter in pCas9_sgRNA_0 (Figure 1A) or by the stronger hsp70 promoter in pMS73 (Figure 1B).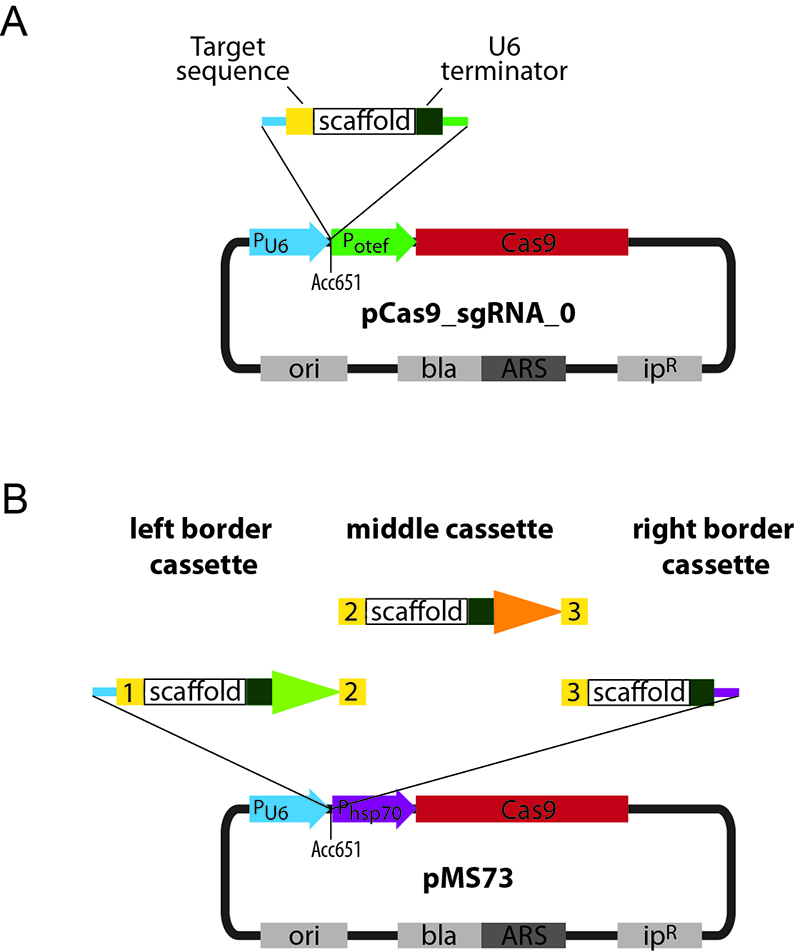
Figure 1. Scheme of the CRISPR-Cas9 plasmids and cloning strategy. A. Schematic representation of pCas9_sgRNA_0 and cloning strategy for the introduction of a single sgRNA. A user-defined targeting fragment can be cloned into the Acc65I-linearized pCas9_sgRNA_0 backbone via Gibson Assembly®. B. Schematic representation of pMS73 and cloning strategy for the introduction of two or more sgRNA expression cassettes. User-defined left and right border cassettes and a variable number of middle cassettes can be cloned into the Acc65I-linearized pMS73 backbone via Gibson Assembly®. The target sequences are used as Gibson overhangs and the number of middle cassettes can be modified depending on the number of target sequences that need to be inserted. ori: Origin of replication, bla: β-lactamase gene that confers resistance to ampicillin in E. coli, ARS: autonomously replicating sequence for replication in U. maydis, ipR: U. maydis ip allele that confers resistance to carboxin. Numbers in yellow boxes indicate different targeting sequences. The scaffold sequence corresponds to the minimal scaffold required for Cas9 binding (Hsu et al., 2013). tRNA promoters used to drive expression of different sgRNAs are indicated by differently colored arrowheads. Cyan line indicates the Gibson overhang to PU6. Green line indicates the Gibson overhang to Potef. Purple line indicates the Gibson overhang to Phsp70.- Introduce your target sequences in the appropriate template (Table 1).
- Purchase the designed sequences as double-stranded DNA fragments.
Note: An alternative to purchasing the sgRNA expression cassettes is their generation by PCR amplification from a template. You can use double-stranded DNA fragments as templates for such a PCR.
Table 1. Templates for generating double-stranded DNA fragments containing the target sequences of interest
*Colors in Table 1 correspond to those in Figure 1.
**The scaffold sequence corresponds to the minimal scaffold required for Cas9 binding (Hsu et al., 2013).
- Introduce your target sequences in the appropriate template (Table 1).
- Cloning of the sgRNA expression cassettes into the CRISPR plasmids via Gibson Assembly® (Gibson et al., 2010)
- Linearize pCas9_sgRNA_0 or pMS73 with Acc65I.
- Clone the sgRNA expression cassettes into the backbone via Gibson Assembly® according to the manufacturer’s protocol.
Note, we have used Gibson Assembly® Master Mix (Instruction Manual, NEB #E2611S/L 10/50 reactions, Version 4.0). - Transform part of the Gibson Assembly® reaction into competent E. coli cells according to the manufacturer’s protocol and select for growth on YT plates containing 100 µg/ml ampicillin. Incubate the selection plates overnight at 37 °C.
Note: We have used the heat shock E. coli transformation protocol (Bacterial Transformation Workflow–4 Main Steps) as given at Thermo Fischer website. - Pick some colonies to check for the correct insertion of sgRNA expression cassettes. Use a sterile pipette tip or toothpick to inoculate a single colony into 3 ml of dYT medium with 100 µg/ml ampicillin. Incubate with shaking at 37 °C overnight.
- Isolate the plasmid DNA from bacterial cultures by using e.g., a QIAprep spin miniprep kit according to the manufacturer’s instructions.
- Test for plasmids with inserts by restriction analysis and sequencing.
- For assessing the successful insertion of only one cassette, it is normally sufficient to test plasmids from two transformants or go directly for sequencing. If more than one cassete is to be inserted, it is usually sufficient to analyze plasmids from six transformants by restriction analysis first and follow candidates up by sequencing. When restriction analyses are performed, enzymes should be chosen which cleave outside the cassette(s) and allow to estimate the number of cassettes inserted.
- For plasmid sequencing use the U6-Fwd primer oMS59: ATTCGTGATTTACACCAAACACGC and the reverse primer Potef-Rv oMS49: CCCCTCGTCTCGCGCCTCATTGGTCGAATTG for pCas9_sgRNA_0 derived plasmids or oMS59: ATTCGTGATTTACACCAAACACGC and Phsp70-Rv oSR224: CTACACTCAGCACACGATGT for pMS73-derived plasmids.
- Linearize pCas9_sgRNA_0 or pMS73 with Acc65I.
- Transformation of the CRISPR-Cas9 plasmid in U. maydis protoplasts
- Transform 50 µl of U. maydis protoplasts with no more than 500 ng of the pCas9_sgRNA_0 derived or pMS73 derived plasmids.
- Plate the transformation mixture on a freshly prepared two-layered RegAgar plate containing 4 µg/ml carboxin in the bottom layer and no carboxin in the top layer and incubate four days at 28 °C until transformants are clearly visible.
- Transformation of the CRISPR-Cas9 constructs into U. maydis follows the protocol developed by Schulz et al. (1990).
- Note: See Bösch et al. (2016) for a detailed description of the protocol for the generation of protoplasts and the polyethylene glycol (PEG)-mediated transformation.
- Transform 50 µl of U. maydis protoplasts with no more than 500 ng of the pCas9_sgRNA_0 derived or pMS73 derived plasmids.
- Optional: Transfer single colonies from the transformation plate to 3 ml liquid YEPSL medium containing carboxin (2 µg/ml) to increase CRISPR-Cas9 efficiency.
- Incubate the culture and shake it at 200 rpm and 28 °C overnight.
- Dilute the cultures 1:100 into fresh selection medium and allow them to grow until saturation (approximately 20 h). Repeat three times.
- Incubate the culture and shake it at 200 rpm and 28 °C overnight.
- Isolation of colonies with a defined editing event
- Streak out 10-12 transformants (in case of single gene disruption), or more (in case of multiple gene disruptions) from the original transformation plate (Step 4b) on PD plates without carboxin, or in the case of incubation in liquid medium (Step 5b), spot 1 µl of cells on a PD plate without carboxin and streak them out .
- Incubate the plate for two days at 28 °C.
- Generate a master plate to store colonies to be tested. To do this, pick single colonies with a flat toothpick and transfer to a new PD agar plate. Spread the transferred cells to obtain a larger patch in which cells grow up to form a colony and arrange these using a grid pattern for easier later identification. Incubate overnight at 28 °C (Figure 2).
- This step is of crucial importance because only in the absence of carboxin will the Cas9-containing plasmid be lost. Fungal colonies and cultures grown under selection pressure are heterogeneous populations: a colony (or culture grown with carboxin selection) will contain cells with independent editing events depending on the generation in which editing occurred - except when the CRISPR event took place in the initially transformed cell.
- At this step, you can confirm the loss of the CRISPR-Cas9 plasmid (see Step 8) by replica-plating the single colonies on a PD-carboxin plate in parallel to generate the master plate and selecting those colonies from the master plate which are unable to grow on the carboxin plate.

Figure 2. Example of a master plate containing colonies to be tested. Generation of a master plate is advisable to facilitate the screening process. Single colonies derived from individual U. maydis transformants were transferred to a new PD plate following a grid pattern and labeled with numbers for identification. Transfers were made with flat toothpicks, cells were placed in the center of a grid square and slightly spread to increase the area in which cells can grow up and form a colony. A. Master plate before incubation; B. Master plate after incubation overnight at 28 °C.
- Streak out 10-12 transformants (in case of single gene disruption), or more (in case of multiple gene disruptions) from the original transformation plate (Step 4b) on PD plates without carboxin, or in the case of incubation in liquid medium (Step 5b), spot 1 µl of cells on a PD plate without carboxin and streak them out .
- Colony PCR-based screen for CRISPR-Cas9 induced mutations
- Design primers enabling the amplification of a PCR product of about 500 bp containing the target site.
- With a sterile pipet tip or a toothpick, pick a small amount of a colony from the master plate (Step 6c) and place it in a well of a 96-well plate containing 20 µl of 20 mM NaOH.
- Shake the plate for 30 min at room temperature at 800 rpm using e.g., a ThermoMixer.
- Set up a 20 µl PCR reaction using a high fidelity polymerase e.g., Phusion DNA Polymerase or BioMix Red as suggested by the manufacturer. Use 1 µl of the lysed cells as DNA template.
Note: In case of multiplexing, a separate PCR for each target site needs to be performed. If you have many colonies and/or sites to test, we recommend using kits like the Plate Seq Kit PCR. In this case, perform the PCR reaction in this step directly in the 96-well-PCR-plate provided in the kit. - Amplify the PCR products in a Thermocycler following the polymerase manufacturer’s instructions.
- Sequence the PCR fragment either after cleanup with e.g., the Wizard SV Gel and PCR cleanup System or e.g., in the Plate Seq Kit from Eurofins.
- Design primers enabling the amplification of a PCR product of about 500 bp containing the target site.
- Loss of the CRISPR-Cas9 plasmid
- If you have not screened for the loss of the plasmid before (see note after Step 6), pick some cells with a toothpick from the master plate and streak out on a PD plate containing carboxin. Incubate for two days at 28 °C. Go back to the master plate and use colonies that were unable to grow on the carboxin plate, these have lost the plasmid.
Note: We observe retention of the plasmid in colonies on the master plate in about 5% of the cases. - Growth on the carboxin plate means that some cells still contain the plasmid. If this is the case and it is this colony that contains the desired mutation, inoculate cells from this strain from the master plate in 3 ml YESPSL medium without carboxin and grow overnight at 28 °C.
- Take cells from this overnight culture and spread on a PD plate without carboxin to obtain single colonies. Incubate for two days at 28 °C.
- Replica-plate single colonies on both, a PD plate with carboxin and a PD plate without carboxin, and incubate overnight at 28 °C. Select colonies that have lost the plasmid i.e., are unable to grow on the plate containing carboxin.
- If necessary, repeat the loss of plasmid procedure until there is no growth on selection plates.
- If you have not screened for the loss of the plasmid before (see note after Step 6), pick some cells with a toothpick from the master plate and streak out on a PD plate containing carboxin. Incubate for two days at 28 °C. Go back to the master plate and use colonies that were unable to grow on the carboxin plate, these have lost the plasmid.
Data analysis
The sequencing data should be analyzed by alignment against the wild-type sequence using software like CloneManager or CLC Main Workbench. Frameshifts mutations are generated when the number of nucleotides deleted or inserted cannot be divided by three. Alternatively, indels that generate a premature stop codon can be used. It is mandatory to inspect the sequencing chromatograms carefully and check for the presence of overlapping peaks. Overlapping peaks are a sign of genetic heterogeneity in the sequenced sample (Figure 3).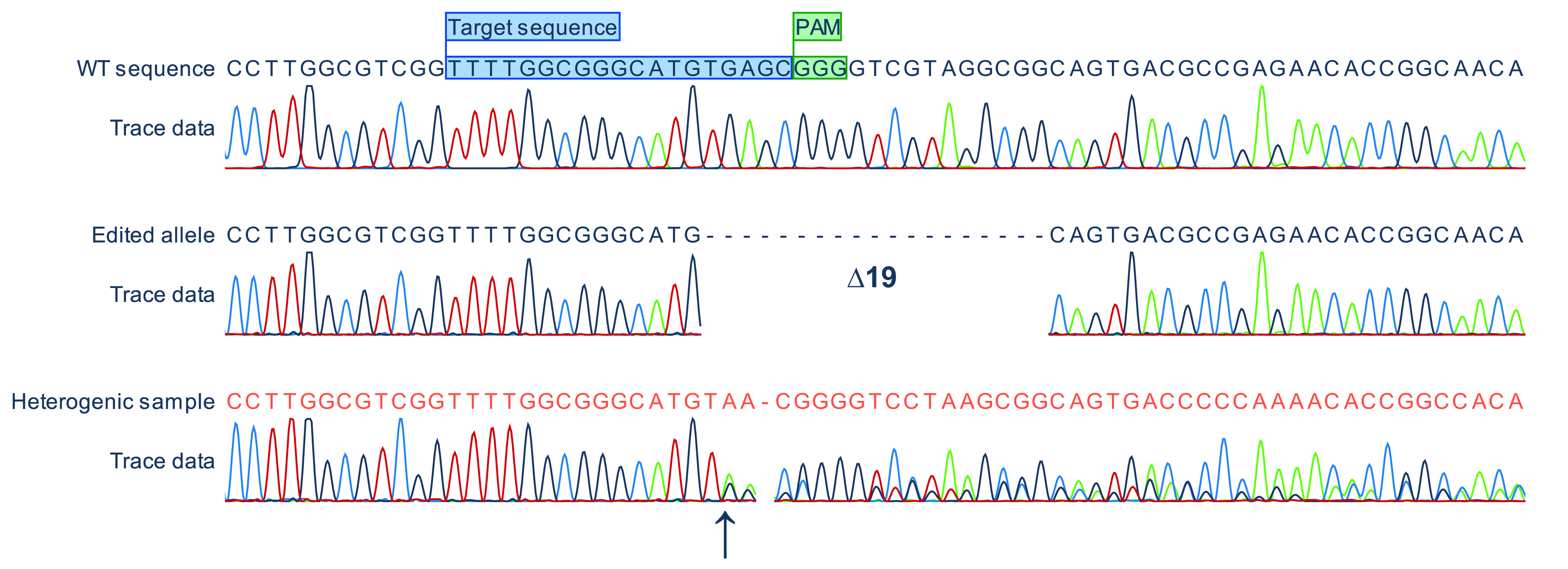
Figure 3. Representative sequencing results. Alignment of nucleotide sequences and chromatograms of a wild-type (WT) allele, a corresponding edited allele lacking 19 bp and a heterogenic sample from the same experiment. Note that a sequence is provided (dark orange) by the sequencing company for the heterogenic sample taking into account the highest peaks and incorrectly suggesting sequence homogeneity. Heterogeneity can only be recognized by careful analysis of the chromatogram which in this case shows that the sequence up to the arrow (predicted Cas9 cleavage site) is homogeneous and becomes heterogeneous from this point onwards. Target sequence and PAM sequence are depicted as colored boxes in the wild-type sequence. The alignment was generated with CLC Main Workbench.
Notes
- The CRISPR-Cas9 system in U. maydis has proven to be highly efficient for the disruption of single genes where up to 100% of the tested transformants contained edited cells (Schuster et al., 2016). For sgRNA with lower editing efficiency, the efficiency could be improved by higher expression of the Cas9 protein using a stronger promoter (i.e., Phsp70 instead of Potef) and by extending exposure to Cas9 by maintaining the transformed cells in liquid culture under conditions selecting for the Cas9 expressing plasmid (Schuster et al., 2018). Using longer Cas9 exposure times, we have been able to improve editing using numerous sgRNAs. We have rarely been confronted with target sequences that either do not work at all or that give very low gene editing efficiency. In such cases, changing the target sequence provides an efficient solution.
- Two independently edited strains have been analyzed for off-target mutations and no off-targets where found via whole genome sequencing (Schuster et al., 2016). Since off-targets will be specific for each sgRNA, we recommend generating three independent mutants in a given gene or group of genes. If these mutants all show the same phenotypic alteration and this phenotype can be complemented by introducing the wild-type gene or genes, you have convincing evidence that the phenotype is caused by the generated mutation(s).
- The genomic location of genes to be edited should be taken into account when designing a multiplexing experiment. Two or more simultaneous CRISPR events in the same chromosome could lead to undesired chromosomal deletions or rearrangements. In such cases, sequential editing should be performed.
Recipes
- Yeast extract tryptone (dYT) medium
This medium consists of:
1.6% (w/v) tryptone (BD Biosciences)
1% (w/v) yeast extract (BD Biosciences)
0.5% (w/v) NaCl in double distilled water
Note: Autoclave and store at room temperature before use. - Yeast extract tryptone (YT) agar medium
This medium consists of:
0.8% (w/v) tryptone (BD Biosciences)
0.5% (w/v) yeast extract (BD Biosciences)
0.5% (w/v) NaCl
1.3% Bacto agar (BD Biosciences) in double distilled water
Note: Autoclave and immediately pour into Petri dishes (20 ml per Petri dish). - YEPSL liquid medium
This medium consists of:
0.4% (w/v) yeast extract (BD Biosciences)
0.4% (w/v) peptone (BD Biosciences)
2% (w/v) sucrose (BD Biosciences) in double distilled water
Note: Autoclave and store at room temperature before use. - Regeneration agar light (RegAgar)
This medium consists of:
1% (w/v) yeast extract (BD Biosciences)
0.4% (w/v) peptone (BD Biosciences)
0.4% (w/v) sucrose (Roth)
18.22% (w/v) sorbitol (Sigma)
1.5% Bacto agar (BD Biosciences) in double distilled water
Notes:- Autoclave and store at room temperature before use.
- During U. maydis transformation, the agar is melted in a microwave oven, mixed well and cooled down to 50-60 °C. Carboxin (4 µg/ml final concentration) is added and 10 ml of this supplemented RegAgar is poured into a Petri dish. Once this is solidified, an additional 10 ml of molten RegAgar without carboxin is poured as a top layer into the same Petri dish. This second layer should be poured about 25-30 min before plating the transformation mixture (see transformation protocol by Bösch et al., 2016).
- Autoclave and store at room temperature before use.
- Potato dextrose (PD) agar medium
This medium consists of:
2.4% Potato Dextrose Broth (BD Biosciences)
2% Bacto agar (BD Biosciences) in double distilled water
Note: Autoclave and immediately pour into Petri dishes (20 ml per Petri dish).
Acknowledgments
This work was funded by the Max Planck Society. The protocol is adapted from Schuster et al. (2016 and 2018). We would like to acknowledge present and past members of the Kahmann Lab who use CRISPR-Cas9 mediated genome editing regularly and provided feedback on the protocol. The authors declare that there are no conflicts of interest nor competing interests related to this article.
References
- Bakkeren, G., Kamper, J. and Schirawski, J. (2008). Sex in smut fungi: Structure, function and evolution of mating-type complexes. Fungal Genet Biol 45 Suppl 1: S15-21.
- Barrangou, R., Fremaux, C., Deveau, H., Richards, M., Boyaval, P., Moineau, S., Romero, D. A. and Horvath, P. (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315(5819): 1709-1712.
- Bösch, K., Frantzeskakis, L., Vraneš, M., Kämper, J., Schipper, K. and Göhre, V. (2016). Genetic manipulation of the plant pathogen Ustilago maydis to study fungal biology and plant microbe interactions. J Vis Exp (115): e54522.
- Deng, H., Gao, R., Liao, X. and Cai, Y. (2017). CRISPR system in filamentous fungi: Current achievements and future directions. Gene 627: 212-221.
- Fu, Y., Sander, J. D., Reyon, D., Cascio, V. M. and Joung, J. K. (2014). Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol 32(3): 279-284.
- Gibson, D. G., Smith, H. O., Hutchison, C. A., 3rd, Venter, J. C. and Merryman, C. (2010). Chemical synthesis of the mouse mitochondrial genome. Nat Methods 7(11): 901-903.
- Heigwer, F., Kerr, G. and Boutros, M. (2014). E-CRISP: fast CRISPR target site identification. Nat Methods 11(2): 122-123.
- Holliday, R. (2004). Early studies on recombination and DNA repair in Ustilago maydis. DNA Repair (Amst) 3(6): 671-682.
- Holloman, W. K., Schirawski, J. and Holliday, R. (2008). The homologous recombination system of Ustilago maydis. Fungal Genet Biol 45 Suppl 1: S31-39.
- Hsu, P. D., Scott, D. A., Weinstein, J. A., Ran, F. A., Konermann, S., Agarwala, V., Li, Y., Fine, E. J., Wu, X., Shalem, O., Cradick, T. J., Marraffini, L. A., Bao, G. and Zhang, F. (2013). DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31(9): 827-832.
- Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A. and Charpentier, E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337(6096): 816-821.
- Kämper, J., Kahmann, R., Bolker, M., Ma, L. J., Brefort, T., Saville, B. J., Banuett, F., Kronstad, J. W., Gold, S. E., Muller, O., Perlin, M. H., Wosten, H. A., de Vries, R., Ruiz-Herrera, J., Reynaga-Pena, C. G., Snetselaar, K., McCann, M., Perez-Martin, J., Feldbrugge, M., Basse, C. W., Steinberg, G., Ibeas, J. I., Holloman, W., Guzman, P., Farman, M., Stajich, J. E., Sentandreu, R., Gonzalez-Prieto, J. M., Kennell, J. C., Molina, L., Schirawski, J., Mendoza-Mendoza, A., Greilinger, D., Munch, K., Rossel, N., Scherer, M., Vranes, M., Ladendorf, O., Vincon, V., Fuchs, U., Sandrock, B., Meng, S., Ho, E. C., Cahill, M. J., Boyce, K. J., Klose, J., Klosterman, S. J., Deelstra, H. J., Ortiz-Castellanos, L., Li, W., Sanchez-Alonso, P., Schreier, P. H., Hauser-Hahn, I., Vaupel, M., Koopmann, E., Friedrich, G., Voss, H., Schluter, T., Margolis, J., Platt, D., Swimmer, C., Gnirke, A., Chen, F., Vysotskaia, V., Mannhaupt, G., Guldener, U., Munsterkotter, M., Haase, D., Oesterheld, M., Mewes, H. W., Mauceli, E. W., DeCaprio, D., Wade, C. M., Butler, J., Young, S., Jaffe, D. B., Calvo, S., Nusbaum, C., Galagan, J. and Birren, B. W. (2006). Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature 444(7115): 97-101.
- Lanver, D., Muller, A. N., Happel, P., Schweizer, G., Haas, F. B., Franitza, M., Pellegrin, C., Reissmann, S., Altmuller, J., Rensing, S. A. and Kahmann, R. (2018). The biotrophic development of Ustilago maydis studied by RNA-Seq analysis. Plant Cell 30(2): 300-323.
- Lanver, D., Tollot, M., Schweizer, G., Lo Presti, L., Reissmann, S., Ma, L. S., Schuster, M., Tanaka, S., Liang, L., Ludwig, N. and Kahmann, R. (2017). Ustilago maydis effectors and their impact on virulence. Nat Rev Microbiol 15(7): 409-421.
- Niessing, D., Jansen, R. P., Pohlmann, T. and Feldbrugge, M. (2018). mRNA transport in fungal top models. Wiley Interdiscip Rev RNA 9(1).
- Schulz, B., Banuett, F., Dahl, M., Schlesinger, R., Schafer, W., Martin, T., Herskowitz, I. and Kahmann, R. (1990). The b alleles of U. maydis, whose combinations program pathogenic development, code for polypeptides containing a homeodomain-related motif. Cell 60(2): 295-306.
- Schuster, M., Schweizer, G. and Kahmann, R. (2018). Comparative analyses of secreted proteins in plant pathogenic smut fungi and related basidiomycetes. Fungal Genet Biol 112: 21-30.
- Schuster, M., Schweizer, G., Reissmann, S. and Kahmann, R. (2016). Genome editing in Ustilago maydis using the CRISPR-Cas system. Fungal Genet Biol 89: 3-9.
- Shi, T. Q., Liu, G. N., Ji, R. Y., Shi, K., Song, P., Ren, L. J., Huang, H. and Ji, X. J. (2017). CRISPR/Cas9-based genome editing of the filamentous fungi: the state of the art. Appl Microbiol Biotechnol 101(20): 7435-7443.
- Steinberg, G. (2007). Tracks for traffic: microtubules in the plant pathogen Ustilago maydis. New Phytol 174(4): 721-733.
- Tsukuda, T., Carleton, S., Fotheringham, S. and Holloman, W. K. (1988). Isolation and characterization of an autonomously replicating sequence from Ustilago maydis. Mol Cell Biol 8(9): 3703-3709.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Schuster, M., Trippel, C., Happel, P., Lanver, D., Reißmann, S. and Kahmann, R. (2018). Single and Multiplexed Gene Editing in Ustilago maydis Using CRISPR-Cas9. Bio-protocol 8(14): e2928. DOI: 10.21769/BioProtoc.2928.
Category
Microbiology > Microbial genetics > Genome editing
Molecular Biology > DNA > DNA modification
Microbiology > Microbial genetics > CRISPR-Cas9
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.