- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

In vitro Enzymatic Assays of Histone Decrotonylation on Recombinant Histones


Published: Vol 8, Iss 14, Jul 20, 2018 DOI: 10.21769/BioProtoc.2924 Views: 7549
Reviewed by: David CisnerosDamián Lobato-MárquezAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2018
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Class I histone deacetylases (HDACs) are efficient histone decrotonylases, broadening the enzymatic spectrum of these important (epi-)genome regulators and drug targets. Here, we describe an in vitro approach to assaying class I HDACs with different acyl-histone substrates, including crotonylated histones and expand this to examine the effect of inhibitors and estimate kinetic constants.
Keywords: CrotonylationBackground
Posttranslational modifications of histones are an important facet of genome regulation, including gene expression (for example see Pengelly et al., 2013; reviewed in Castillo et al., 2017). Histone modifications alter chromatin structure and/or regulate the binding of proteins, such as nucleosome remodeling factors (reviewed in Bannister and Kouzarides, 2011). Most histone modifications are reversible and can be removed enzymatically. For example, histone acetylation is removed by histone deacetylases (HDACs), of which there exist several classes. In recent years new histone lysine acylations, including succinylation, propionylation, butyrylation, hydroxybutyrylation and crotonylation have emerged as new alternative acylations to the canonical histone acetylation and the functional relevance of many of these newly discovered histone modifications have been demonstrated (reviewed in Sabari et al., 2017). In particular, histone crotonylation is associated with active gene expression and is thought to be influenced by the metabolic state of the cell (Sabari et al., 2015; Fellows et al., 2018). Class I histone deacetylases have recently been shown to also efficiently decrotonylate histones (Wei et al., 2017; Fellows et al., 2018).
Histone deacetylation assays are often performed using fluorescent acetyl substrates, such as BOC-lys(acetyl)-AMC, because this allows high-throughput in vitro approaches, suitable for drug discovery (Wegener et al., 2003a; 2003b and 2003c). However, we found that the analogous fluorescent crotonyl substrate BOC-lys(crotonyl)-AMC was inhibitory to histone deacetylases (Fellows et al., 2018). In this protocol, we describe a method for analysis of the activity of histone deacetylases with in vitro crotonylated histone H3, and we investigate the effect of an inhibitor and estimate kinetic parameters using an in vitro approach. This method does not require the use of radioisotopes. It also does not rely on fluorescent peptide mimics, and thus, the kinetic constants determined may be more representative of those found in vivo. The approach depends on the recognition of histone acylations, such as crotonylation, by specific antibodies. It is a versatile approach, which could be applied to study a variety of other histone modifications, enzymes, and inhibitors.
Materials and Reagents
- 8-strip 0.2 ml non-flex flat cap PCR tubes with individual lids (STARLAB INTERNATIONAL, catalog number: I1402-3700 )
- 250 ml and 1 L cylinders (Corning, catalog number: 3022P-250 , 3022P-1L )
- Pre-cut extra thick blot paper, 7 x 8.4 cm (Bio-Rad Laboratories, catalog number: 1703966 )
- Amersham Protran 0.45 µm nitrocellulose membrane, 300 mm x 4 m (GE Healthcare, catalog number: 10600002 )
- 5, 10 and 25 ml Costar® Stripette® serological pipettes (Corning, catalog numbers: 4487 , 4488 , 4489 )
- 50 ml Falcon® tubes, polypropylene (Corning, catalog number: 352070 )
- Eppendorf® tubes 5.0 ml, Eppendorf QualityTM (Eppendorf, catalog number: 0030119401 )
- X-ray film 18 x 24 cm double sided (Scientific Laboratory Supplies, catalog number: MOL7016 )
- Transparent acetate sheet, e.g., colour copier transparency film (Interaction-Connect, Q-CONNECT®, catalog number: KF00533 )
- Milli-Q® water
- Recombinant human histone H3.1 protein, 1 mg/ml (New England Biolabs, catalog number: M2503S )
Note: Prepare aliquots (e.g., 10 µl aliquots) and store at -80 °C. - Recombinant catalytic domain of human p300 protein (Enzo Life Sciences, catalog number: BML-SE451-0100 ), 100 µg, 15.18 µM (exact concentration may vary by batch, check the tube)
Note: Prepare aliquots (e.g., 10 µl aliquots) and store at -80 °C. - Crotonyl-coenzyme A trilithium salt ~90% pure as per HPLC (Sigma-Aldrich, catalog number: 28007 )
Note: For acetylation we used: Acetyl-coenzyme A (Sigma-Aldrich, catalog number: ACOA-RO ROCHE). Prepare 5 mM stock solution in water, aliquot and store at -80 °C.
Manufacturer: Roche Diagnostics, catalog number: 10101893001 . - Trizma®-hydrochloride, ≥ 99.0% (Sigma-Aldrich, catalog number: T3253 )
- Potassium chloride, 99.5-101.0% AnalaR NORMAPUR® (VWR, catalog number: 26764.298 )
- UltraPureTM 0.5 M EDTA, pH 8.0 (Thermo Fisher Scientific, catalog number: 15575020 )
- Tween 20 (Sigma-Aldrich, catalog number: P1379 )
- Glycerol, ≥ 99% (Sigma-Aldrich, catalog number: G5516 )
- Dithiothreitol (DTT) (Thermo Fisher Scientific, catalog number: R0861 )
- Magnesium chloride hexahydrate, ≥ 99.0% (Sigma-Aldrich, catalog number: M2670 )
- Zinc sulfate heptahydrate (Sigma-Aldrich, catalog number: Z0251 )
- Recombinant human HDAC1 protein (Active Motif, catalog number: 31908 ) 50 µg, 0.1 mg/ml, 1.78 µM (exact concentration may vary by batch, check tube)
Note: Prepare aliquots (e.g., 10 µl aliquots) and store at -80 °C. - 2x Laemmli sample buffer (Bio-Rad Laboratories, catalog number: 1610737 )
Note: Add 2-mercaptoethanol to 5% v/v as described by supplier. - 2-mercaptoethanol, ≥ 99.0% (Sigma-Aldrich, catalog number: M6250 )
- Sodium Butyrate, 98% (Sigma-Aldrich, catalog number: 303410 )
Note: Prepare a 1 M solution in water, adjust to pH 7 and aliquot. - Precast RunBlue 4-12% Bis-Tris (Expedeon, catalog number: NBT41227 )
- 20x RunBlue MES run buffer (Expedeon, catalog number: NXB70500 )
- PageRulerTM Prestained protein ladder, 10 to 180 kDa (Thermo Fisher Scientific, catalog number: 26616 )
- 10x Tris/glycine/SDS electrophoresis buffer (Bio-Rad Laboratories, catalog number: 1610732EDU )
- UltraPureTM Tris buffer (Thermo Fisher Scientific, catalog number: 15504020 )
- Glycine, ≥ 99.7% AnalaR NORMAPUR® (VWR, catalog number: 101196X )
- Methanol, ≥ 99.8% AnalaR NORMAPUR® (VWR, catalog number: 20847.307 )
- Sodium chloride, 99.5-100.5% AnalaR NORMAPUR® (VWR, catalog number: 27810.295 )
- Bovine serum albumin, heat shock fraction pH 7 ≥ 98% (Sigma-Aldrich, catalog number: A9647 )
- Anti-crotonyl-histone H3 lys18 (H3K18cr) rabbit polyclonal antibody (PTM Biolabs, catalog number: PTM-517)
- Anti-rabbit IgG HRP linked whole antibody (GE Healthcare, catalog number: NA934-1ML )
- Enhanced chemiluminescence (ECL) Western blotting reagents (GE Healthcare, catalog number: RPN2106 )
- Synthetic crotonylated H3K18cr peptide 95% pure, lyophilized (TGGKAPR-Lys(Crotonyl)-QLATKAA-EDA-Biotin, BioGenes, Peptide 60556.1) (EDA is a spacer amino acid sequence)
- Histone acylation buffer (see Recipe 1)
- HDAC assay buffer (see Recipe 2)
- MES Run buffer (see Recipe 3)
- Tris glycine SDS (TGS) buffer (see Recipe 4)
- Transfer buffer (see Recipe 5)
- Tris-buffered saline, pH 7.5 (TBS) (see Recipe 6)
- TBS with tween 20 (TBS-T) (see Recipe 7)
- TBS-T with 3% (w/v) bovine serum albumin (TBS-T BSA) (see Recipe 8)
Equipment
- Pipettes (Gilson, catalog numbers: F123602 , F123601 , F123600 , F144801 , model: P1000, P200, P20, P2)
- Pipette controller PIPETBOY acu2 (VWR, INTEGRA Biosciences, catalog number: 612-2964 )
- Fume hood (e.g., Protector Xstream Laboratory Hood, Labconco)
- Milli-Q Water Purification System (Merck, catalog number: ZRXQ003WW )
- GE Healthcare AmershamTM HypercasetteTM Autoradiography Cassette (GE Healthcare, catalog number: RPN11643 )
- Vortex-genie 2 (Scientific Industries, model: Vortex-Genie 2 , catalog number: SI-0236)
- Microcentrifuge (STARLAB INTERNATIONAL, model: Mini Fuge, catalog number: N2631-0007 )
- Two T100TM Thermal Cyclers (Bio-Rad Laboratories, catalog number: 1861096 )
- Amersham electrophoresis power supply EPS 301 (GE Healthcare, catalog number: 18113001 )
- XCell SureLockTM Mini-Cell Electrophoresis System (Thermo Fisher Scientific, catalog number: EI0001 )
- Transblot® SD semi-dry electrophoretic transfer cell (Bio-Rad Laboratories, catalog number: 1703940 )
- MI-5 x-ray film processor (Jet X-Ray, model: Mi5 )
- Epson expression 1680 scanner (Seiko Epson, model: Epson Expression 1680 )
Note: Equivalent models of equipment specified here may also be used.
Software
- ImageJ version 1.50 b
- Windows Excel 2016
- Adobe Illustrator CC 2015.3
- Graphpad Prism version 7.0
- Epson Scan Software in professional mode
Note: Other versions of this software or similar software may be used but the instructions specified in the data analysis section may then differ.
Procedure
We found that decrotonylation by HDACs did not occur on synthetic, modified histone tail peptides, necessitating the use of the full-length recombinant histone. In our assay, we first crotonylate histones in vitro using recombinant histone H3.1 and the catalytic domain of p300 (‘p300’). We then follow decrotonylation by class I HDACs using specific antibodies, here against histone H3 crotonylated at lysine 18 (H3K18cr). H3K18cr is the dominant histone crotonylation mediated by p300 (Sabari et al., 2015) and appears to be the most abundant histone crotonylation in histones isolated from intestinal cells (Fellows et al., 2018). Importantly, highly specific antibodies have been generated that recognize H3K18cr (Sabari et al., 2015). We describe an assay for decrotonylation to address three questions: can HDAC1 remove the crotonyl group and how much of it is required, how effective is the HDAC inhibitor butyrate at preventing decrotonylation by HDAC1 and what are the kinetic parameters of HDAC1 regarding the decrotonylation of histone H3? The same steps can be performed for other acylations, such as acetylation, butyrylation or propionylation using the relevant acyl-CoA and antibody. This protocol can also be used to test other HDAC enzymes, using the relevant recombinant protein, and other HDAC inhibitors such as Trichostatin A (TSA) and Vorinostat (suberoylanilide hydroxamic acid, SAHA).
Part I: In vitro decrotonylation assay
- In vitro crotonylation of recombinant histone H3.1 using the catalytic domain of p300
- Defrost all reagents on ice.
- Dilute the 5 mM crotonyl-CoA stock solution 1:50 in histone acylation buffer.
- Prepare the reaction mix in a 0.2 ml PCR tube as described in Table 1.
Table 1. Histone H3 crotonylation reaction set-up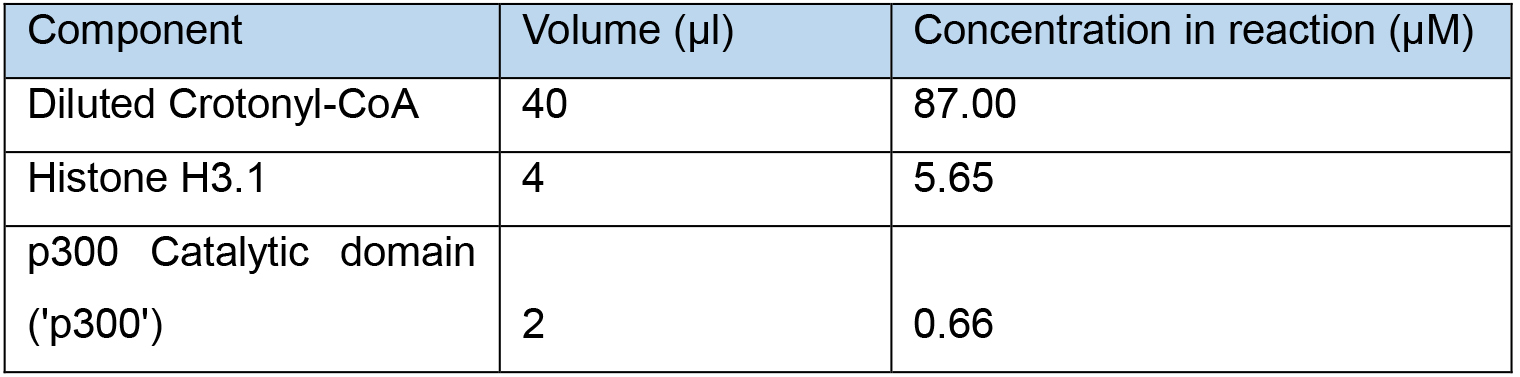
- Mix well by pipetting or brief vortexing.
- Incubate for 2 h at 30 °C in a thermocycler.
- Transfer to a thermocycler at 65 °C and incubate for 5 min to stop the reaction.
Note: Histone tails are unstructured. Therefore heat inactivation is unlikely to affect the modification. - Place the tube on ice.
- Defrost all reagents on ice.
- HDAC1 mediated histone decrotonylation assay
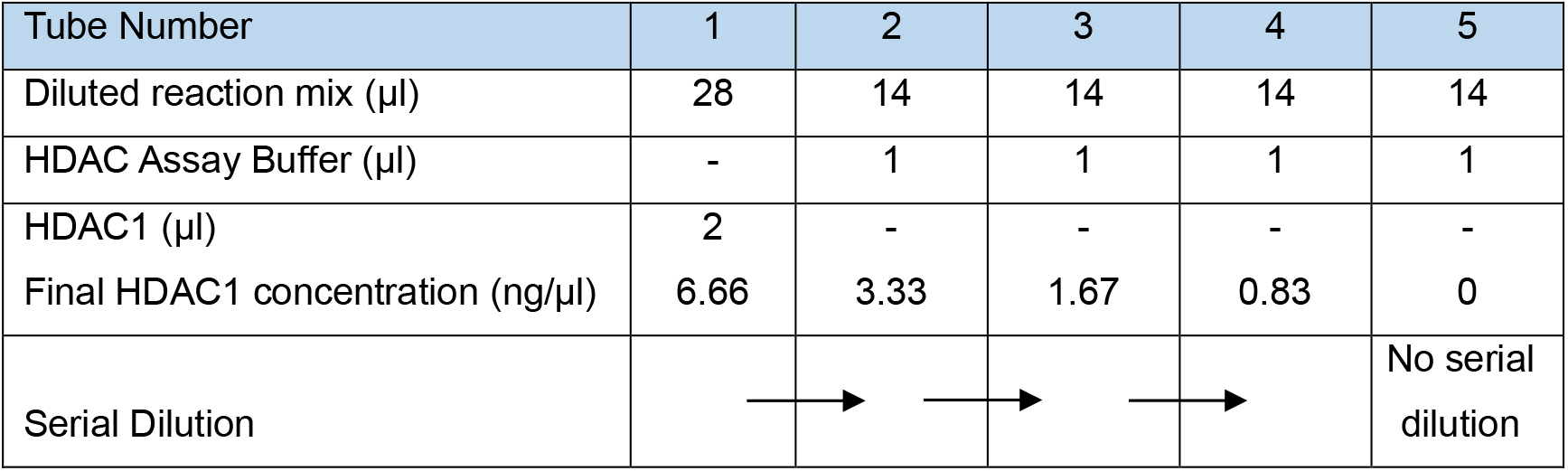
- Decrotonylation assay with different concentrations of HDAC1
We verify the activity of the HDAC1 enzyme towards the substrate and demonstrate the minimum amount required to observe the decrotonylation reaction.- Dilute the reaction from Part I Procedure A 1:3 in HDAC assay buffer, i.e., 46 μl of reaction mix and 92 μl of HDAC assay buffer.
- Cut 3 tubes away from an 8-tube strip leaving a 5-tube strip.
- Label these PCR tubes as 1-5.
- On ice, add diluted reaction mix, HDAC assay buffer and HDAC1 as described below and shown in Table 2.
Note: This sets up a two-fold serial dilution of HDAC1 in tubes 1-4 whilst keeping the other reagents constant. Tube 5 is the negative control without any HDAC1. As soon as HDAC1 has been added to tube 1, perform all steps as quickly as possible as the reaction will start from this point. Whilst it is impossible to capture the first few seconds of the reaction, as it takes a few seconds to set it up, it is important to be quick so that the initial phase of the reaction is recorded.- To tube 1, add 28 μl of diluted reaction mix.
- To tubes 2-5, add 14 μl of diluted reaction mix.
- Add 1 μl HDAC assay buffer to tubes 2-5 to allow for the HDAC1 volume.
- Add 2 μl of HDAC1 to tube 1 and mix well (0.1 mg/ml, 1.78 µM stock. 0.12 µM final concentration).
- Remove 15 μl from tube 1, add to tube 2 and mix well.
- Remove 15 μl from tube 2, add to tube 3 and mix well.
- Repeat with tubes 3 and 4 but not tube 5.
- To tube 1, add 28 μl of diluted reaction mix.
- Vortex all tubes briefly by holding both sides of the strip.
- Spin tubes briefly to bring all of the liquid to the bottom of the tube.
- Incubate in a thermocycler at 30 °C for 2 h.
- As soon as the time is up, remove from the thermocycler.
- If the sample is to be run on a gel, add 15 μl (an equal volume) of 2x Laemmli with 5% (v/v) 2-mercaptoethanol and incubate at 95 °C for 1 min to stop the reaction.
- If the sample is to be dot-blotted, just incubate at 95 °C for 1 min to stop the reaction.
- If the sample is to be run on a gel, add 15 μl (an equal volume) of 2x Laemmli with 5% (v/v) 2-mercaptoethanol and incubate at 95 °C for 1 min to stop the reaction.
- At this point the tubes can be frozen at -20 °C for at least a few days or continued with further analysis as explained in Part II.
- Dilute the reaction from Part I Procedure A 1:3 in HDAC assay buffer, i.e., 46 μl of reaction mix and 92 μl of HDAC assay buffer.
- Exploring the effect of an HDAC inhibitor on histone decrotonylation
Here, butyrate is introduced in serial dilution which allows testing of the ability of the compound to inhibit the decrotonylation. This can be used to estimate the concentration of inhibitor required to halve the amount of created product (IC50).- Dilute the reaction mix from Part I A 1:3 in HDAC assay buffer i.e., 46 μl of reaction mix and 92 μl of HDAC assay buffer.
- Label an 8-strip of PCR tubes as 1-8.
- On ice, add diluted reaction mix, HDAC assay buffer, butyrate and HDAC1 as described below and shown in Table 3.
Note: A double reaction is prepared in tube 1 and single reactions in tubes 2-6, allowing a two-fold serial dilution of butyrate to be performed in tubes 1-6 whilst keeping the other reagents constant. Tube 7 and 8 are negative controls, tube 7 is without butyrate and tube 8 is without butyrate and HDAC1. As soon as HDAC1 has been added to tube 1, perform all steps as quickly as possible as the reaction will start from this point. - To tube 1, add 28 μl of diluted reaction mix.
- To tubes 2-8, add 14 μl of diluted reaction mix.
- Add 1 μl HDAC assay buffer to tubes 2-7 to allow for the volume of butyrate added to tube 1.
- Add 2 μl HDAC assay buffer to tube 8 to allow for the volume of butyrate and HDAC1.
- Add 2 μl of 160 mM butyrate to tube 1 to give a final concentration of 10 mM once all reagents have been added.
- Add 2 μl of HDAC1 to tube 1 and mix well (0.1 mg/ml, 1.78 µM stock. 0.11 µM final concentration).
- Add 1 μl of HDAC1 to tubes 2-7.
- Remove 16 μl from tube 1, add to tube 2 and mix well.
- Remove 16 μl from tube 2, add to tube 3 and mix well.
- Repeat with tubes 3, 4, 5 and 6 but not tubes 7 or 8.
- Vortex all tubes by holding both sides of the strip.
- Incubate at 30 °C for 2 h.
- As soon as the time is up, remove from the thermocycler.
- If the sample is to be run on a gel, add 16 μl (an equal volume) of 2x Laemmli with 5% (v/v) 2-mercaptoethanol and incubate at 95 °C for 1 min.
- If the sample is to be dot-blotted, just incubate at 95 °C for 1 min.
- If the sample is to be run on a gel, add 16 μl (an equal volume) of 2x Laemmli with 5% (v/v) 2-mercaptoethanol and incubate at 95 °C for 1 min.
- At this point the tubes can be frozen at -20 °C for at least a few days or continued with further analysis as explained in Part II.
Table 3. Reaction set-up with a serial dilution of butyrate and constant HDAC1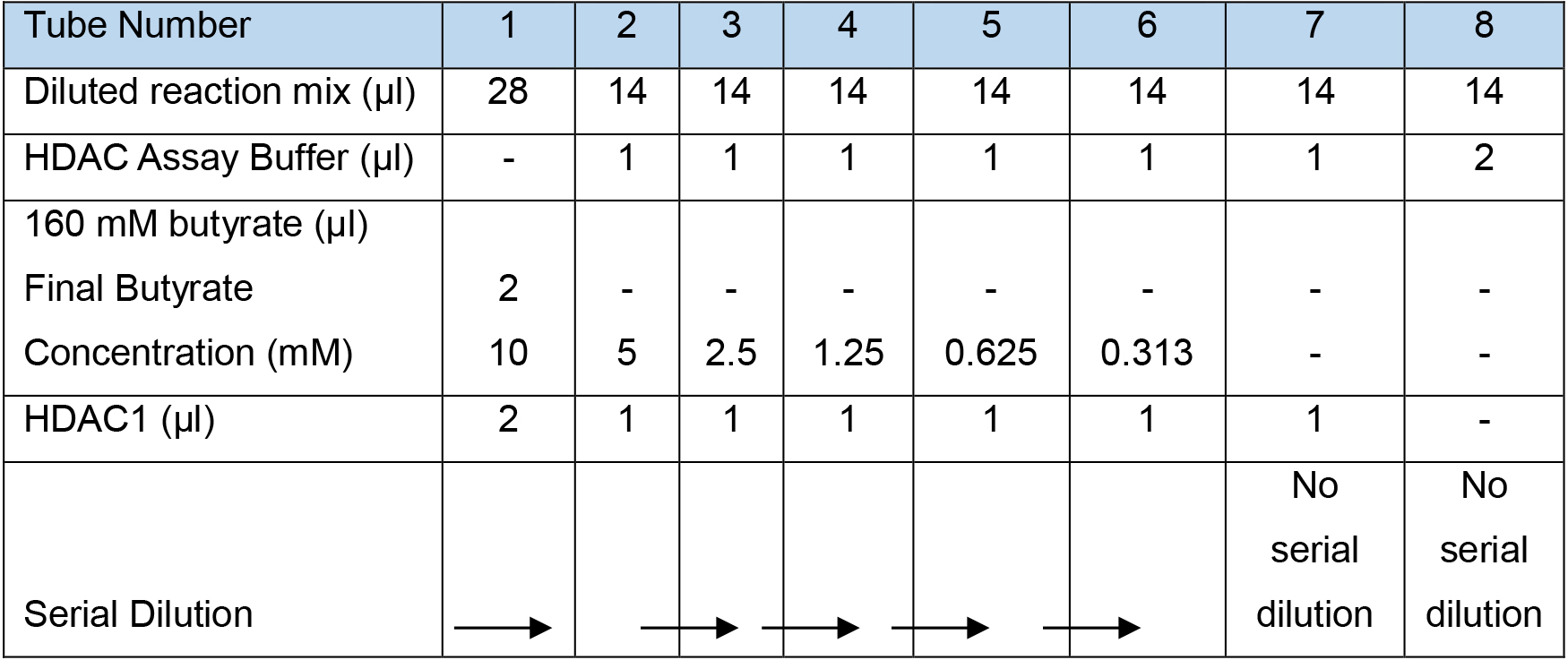
- Dilute the reaction mix from Part I A 1:3 in HDAC assay buffer i.e., 46 μl of reaction mix and 92 μl of HDAC assay buffer.
- Determination of kinetic parameters of histone decrotonylation
Determination of kinetic parameters such as the maximum initial rate allows assessment of whether the enzyme follows Michaelis-Menten kinetics and is a useful way to find out important kinetic parameters such as Kcat, Vmax and Km. It allows comparison of an enzyme’s activity with different substrates or comparison of the activity of different enzymes with the same substrate. This requires the change in substrate or product concentration to be plotted over time so that the initial rate can be determined (see Figure 1 for illustration). The concentration of the enzyme must be optimized so that the initial rate is within a measurable timeframe. Initial rates are then plotted against the substrate concentration at the start of the reaction, to determine the maximum rate (Vmax) and substrate concentration at half Vmax (Km) as shown in Figure 1. As the reactions performed here are analyzed by antibody-based approaches, they cannot be measured in real time. Therefore, it is necessary to set up a reaction for each time point. Here, six identical reactions are set up and stopped at 0, 0.5, 1, 2, 4 and 10 min. The shortest time point that is practical with the method we present is 30 sec. To reliably determine kinetic parameters, it is also necessary to perform the reaction three times for each substrate concentration, and five different substrate concentrations are recommended. This means that 90 reactions are required (5 substrate concentrations x 6 time points x 3 replicates). These have been grouped into three replicate sets named A, B, and C. In our hands, and as described below, the initial reaction occurs in the first minute. Reducing the amount of enzyme may allow longer reaction times and thus more time points during the initial phase. However, this must be balanced against keeping the change in substrate concentration for all reactions distinguishable from background noise.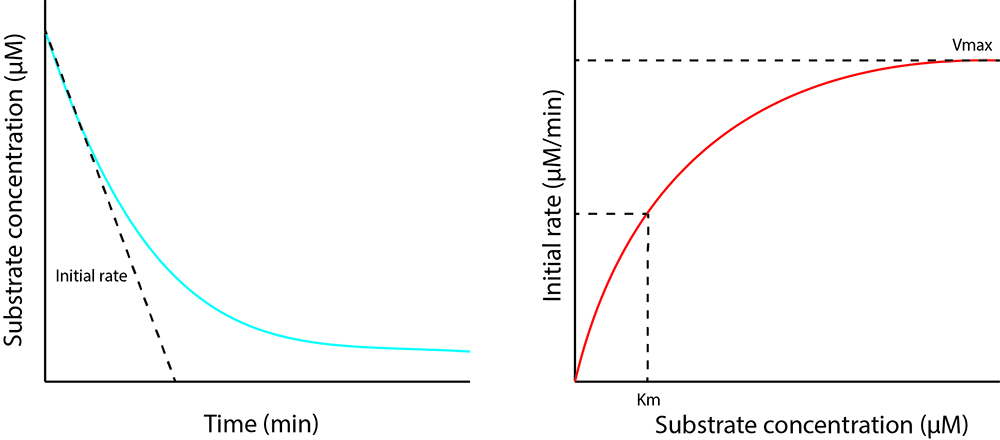
Figure 1. Enzyme kinetic graphs. The left graph shows the output of the assay, which gives the change in substrate concentration over time. The initial rate is determined from this by finding the gradient of the first part of the reaction which is linear. In the right graph, the initial rates of all the substrate concentrations tested are plotted to give the Michaelis-Menten curve which is hyperbolic. This allows determination of the kinetic parameters Vmax and Km.- Crotonylate H3.1 as described in Part I Procedure A.
- To prepare enough crotonylated H3 for 90 assays, use more of each component to give a total volume of 230 µl as described in Table 4.
Table 4. Histone H3 crotonylation reaction set-up with a larger volume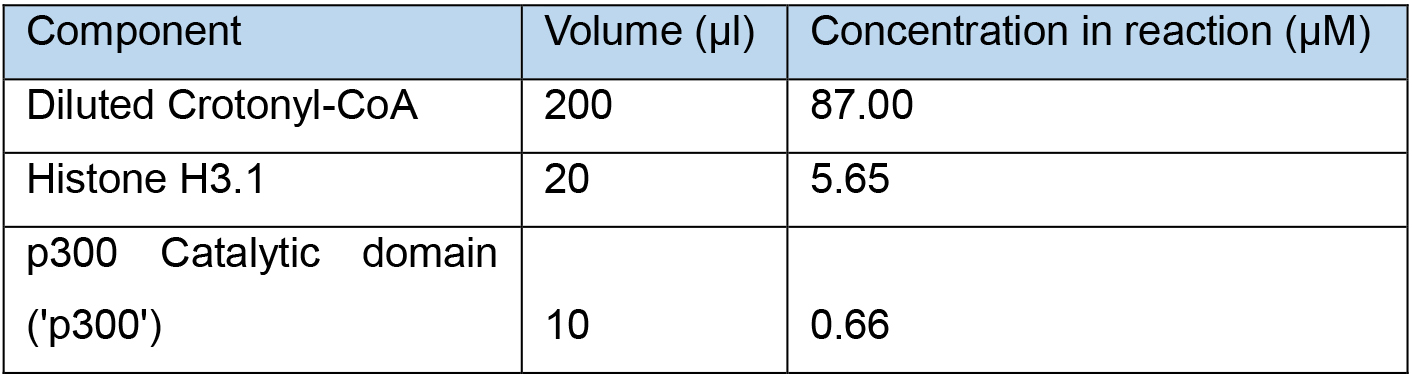
- Incubate in a thermocycler at 30 °C for 2 h.
- Transfer the tube to another thermocycler at 65 °C and incubate for 5 min to stop the reaction.
- Place tube on ice.
- To prepare enough crotonylated H3 for 90 assays, use more of each component to give a total volume of 230 µl as described in Table 4.
- On ice, dilute the reaction mix in HDAC assay buffer to give 1:4, 1:6, 1:8, 1:10 and 1:12 dilutions of the crotonylated histone H3.
Note: Table 5 describes how to do this serially whilst using the minimal quantity of starting reaction mix. However, this can be done in a different way if required. At least 260 µl is needed for each dilution of the crotonylated histone H3 for the following steps.
Table 5. Preparation of different dilutions of the crotonyl-H3 reaction mix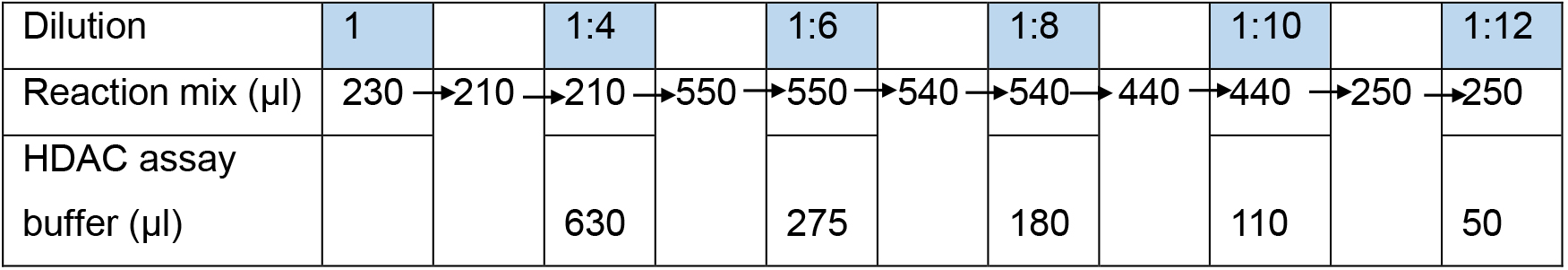
- Prepare three 5-strip tubes numbered 0 min and labeled 1:4, 1:6, 1:8, 1:10 and 1:12 (first row of Figures 2A, 2B, or 2C in blue).
- Prepare 15 more 5-strip tubes numbered 0.5, 1, 2, 4 or 10 (minutes) and labeled 1:4, 1:6, 1:8, 1:10 and 1:12 (the following five rows in Figure 2 in red).
- On ice, add 14 μl of the 1:4, 1:6, 1:8, 1:10 or 1:12 diluted reaction mix to each one. The tubes are kept on ice until HDAC1 is added.
- To one strip of reactions labeled 0 min (in Figure 2 in blue), add 1 μl HDAC assay buffer.
- Vortex briefly by holding both sides of the strip.
Note: The tubes may be spun down or liquid carefully tapped to the bottom if there are bubbles or liquid on the sides or lid of the tube. - Incubate immediately in a thermocycler at 30 °C for 1 min.
Note: If HDAC1 is added to a reaction it would not be zero time, but would run for a few seconds until the reaction could be stopped. However, a negative control is needed to provide a baseline level. To make sure that no change in substrate concentration is seen without enzyme, we incubated a reaction without HDAC1 for 2 h and saw no reduction in crotonylated H3. Therefore, 1 min was chosen as this is easy to perform. - Transfer to a thermocycler at 95 °C for 1 min.
- Repeat Steps B3f to B3i with the two other strips of tubes labelled 0 min.
- Dilute the HDAC1 stock solution 1:4 in HDAC assay buffer, i.e., 20 µl HDAC1 and 60 µl HDAC assay buffer.
- To each tube in one strip of reactions labeled 0.5 min (in Figure 2 in red), add 1 μl of diluted HDAC1 (for 0.03 µM final concentration).
- Vortex briefly by holding both sides of the strip and briefly spin or tap liquid down if required.
- Incubate immediately in a thermocycler set to 30 °C for 30 sec.
Note: It will take about 20-30 sec to add the HDAC and vortex. If time points shorter than 30 sec are needed, reduce the amount of enzyme rather than doing shorter reaction times as these will be inaccurate. - Transfer the strip to a thermocycler at 95 °C for 1 min to stop the reaction.
- Repeat Steps B3l-B3o with the other 0.5 min strips and then with the 1, 2, 4 and 10 min strips in stages, varying the incubation at 30 °C accordingly.
Note: It is necessary to start the reactions in stages so that the time points can be exact. - Organize the tubes so that you have replicate sets of reactions labeled A, B and C. The tubes can be frozen at this point or the experiment continued as described in Part II Procedure B.
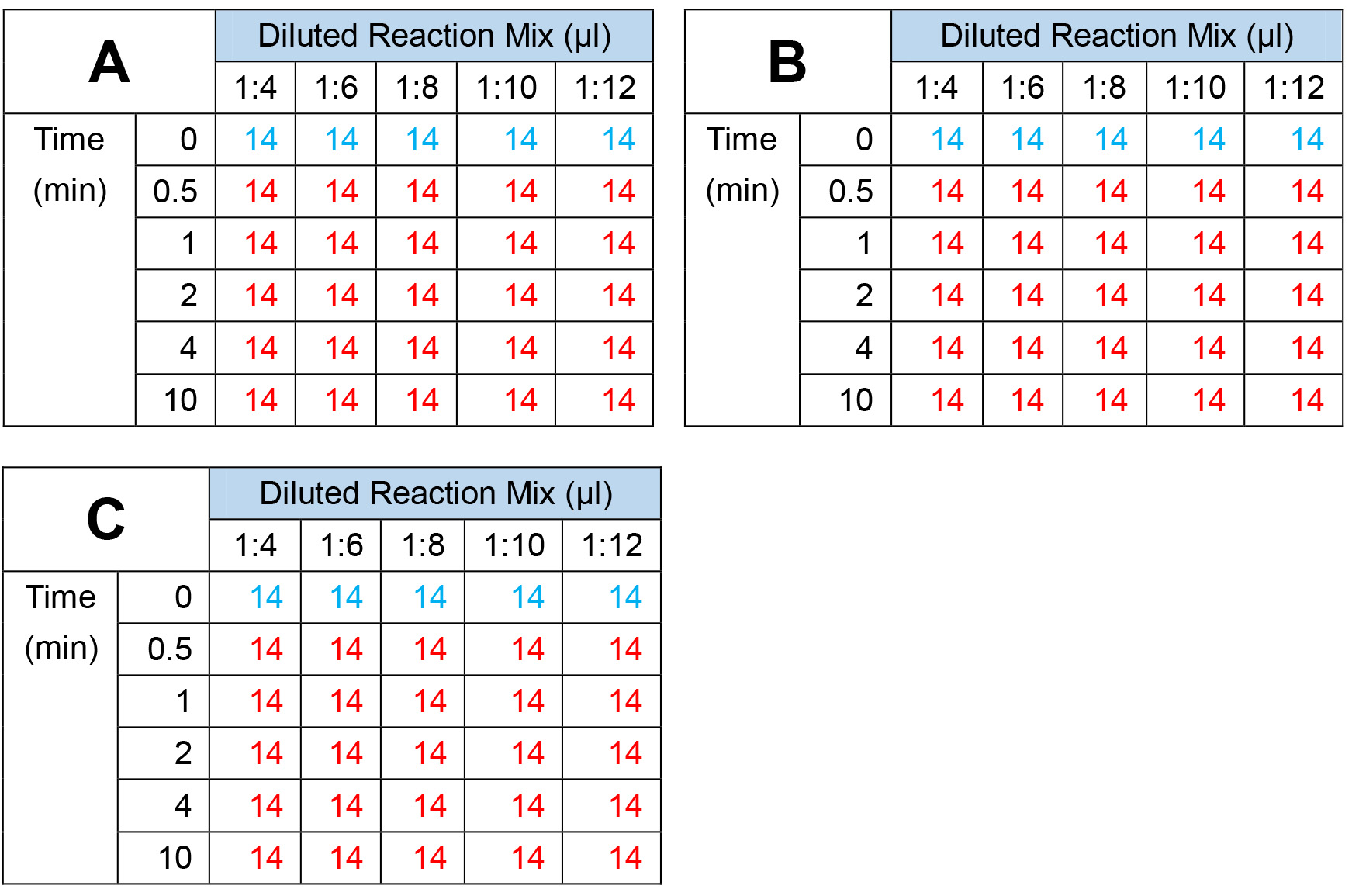
Figure 2. Setup of reactions. The setup of all 90 reactions is shown, organized into replicate number, time and dilution of reaction mix.
- Crotonylate H3.1 as described in Part I Procedure A.
- Decrotonylation assay with different concentrations of HDAC1
Part II: Revealing the decrotonylation result using antibody-based approaches
- Western blot
Western blot is suited to assays with a low number of individual reactions and provides clear bands of defined molecular weight, showing how the reaction progressed.- Open a pre-cast SDS-PAGE gel and rinse the wells with Milli-Q water to rinse away the preserving solutions and bubbles in the wells.
- Assemble the gel in the electrophoresis tank with 200 ml of 1x MES buffer in the inner chamber and fill the outer chamber half full with 1x MES buffer.
Note: Make sure that the inner tank is not leaking. Use a buffer dam if there is only one gel in the tank. MES buffer should be fresh each time in the inner chamber but can be used up to three times in the outer chamber. Placing the tank on ice may improve the quality of the run. - Load 5 µl of pre-stained protein marker in the first well of the pre-cast SDS-PAGE gel.
- Load 8 µl of each sample in the subsequent wells.
- Run at 100-150 V, 250 mA for 1-1.5 h until the blue dye front reaches the bottom of the gel and/or the different sized bands of the marker have separated.
- Rinse gel in 1x TGS buffer briefly and cut off the wells from the top of the gel.
- Soak blotting paper in transfer buffer and place onto the semi-dry transfer machine.
- Soak a piece of nitrocellulose membrane that is the same size as the blotting paper in transfer buffer and layer on top of the blotting paper taking care not to introduce bubbles in between the layers.
- Layer the gel on top of the stack, positioning the marker so that it is parallel to the vertical side of the membrane and so that the blue line, showing how far the samples have reached, is above the membrane.
Note: Again take care not to introduce bubbles in between the layers. - Trim the gel so that it is the same size as the membrane.
Note: When many gels are run, it is useful to cut a corner or corners of the membrane to distinguish them. Pen is not advised as it will run. - Place another piece of blotting paper soaked in transfer buffer on top.
- Roll a round tube, such as a 10 ml pipette, firmly from one side to the other on top of the stack to remove any remaining bubbles.
- Transfer proteins onto the membrane for 45 min at 15 V, 270 mA.
Note: One hour is recommended when two or three stacks are in the transfer machine. - Place the membrane in a 50 ml Falcon tube and rinse membrane in TBS-T for a few seconds.
- Block by adding 15 ml TBS-T BSA and incubate with gentle agitation at room temperature for 1 h.
- In a 5 ml Eppendorf tube, prepare a 5 ml solution of anti-H3K18 crotonyl antibody at 1:2,000 to 1:5,000 dilution (depending on lot number of antibody) in TBS-T BSA.
- Pour the used TBS-T BSA solution into another tube, which should be stored at 4 °C for later use and add the solution of primary antibody to the membrane containing tube.
- Incubate overnight at 4 °C with gentle agitation (rocking or rolling).
- Wash three times briefly (5-10 sec) with 10 ml TBS-T.
- Wash twice for 15 min with 15 ml TBS-T and gentle agitation.
- Wash once for 15 min with 15 ml TBS-T BSA.
Note: The TBS-T BSA from the previous day can be re-used. - In a 5 ml Eppendorf tube, prepare a 5 ml solution of anti-rabbit antibody coupled with horseradish peroxidase (HRP) at 1:10,000 dilution in TBS-T BSA.
- Discard the TBS-T BSA and incubate in anti-rabbit antibody for 1 h at room temperature.
- Wash three times briefly (5-10 sec) with 10 ml TBS-T and then wash three times for 15 min with 15 ml TBS-T.
- Remove the membrane from the falcon tube and place on a clean, flat surface e.g., on a large weigh boat.
- Pipette 1 ml of ECL chemiluminescent substrate (1:1 ratio of A and B), making sure that all of the membrane is covered and leave for 1 min.
- Dab off excess ECL by touching the edge of the membrane against a paper towel and place on a sheet of transparent acetate (or some other transparent sheet) in a developing cassette.
- Layer another acetate sheet on top, taking care not to introduce bubbles.
- Seal edges with tape, close the lid and avoid exposing membrane to light.
- In a dark room, place x-ray film on membrane for 15 sec to 5 min depending on the signal.
- Pass the exposed film into the automatic developer and wait for it to come out at the other side.
- There may be lot-to-lot variation between antibodies, therefore it is good practice to validate the specificity of the anti-H3K18cr antibody (or whatever anti-acyl antibody used), e.g., using peptide dot blots.
- While we have not tested other image capturing systems as alternatives for the ECL/X-ray film setup, it is likely that such systems that can capture chemiluminescence are not only practical, but potentially even better suited than the use of x-ray films.
- Open a pre-cast SDS-PAGE gel and rinse the wells with Milli-Q water to rinse away the preserving solutions and bubbles in the wells.
- Dot blot
Dot blot is more appropriate than Western blot when multiple reactions (more than 15) need to be analyzed. It allows all the reactions to be put on a single membrane, so that their spot intensities can be directly compared. Reactions must be spotted in quadruplicate to allow for variation in pipetting or spreading of the solution on the membrane. This protocol is described as required for Part I Procedure B3.- Cut one a piece of nitrocellulose membrane for each of the A, B and C sets of reactions.
Note: Cut a slightly larger piece that you think you need and trim it later. A typical size for a 10 x 12 spot grid once trimmed is 5.5 x 7.5 cm. - Pipette 2 μl of the reactions using a P2 micropipette in quadruplicate in grid format as shown in Figure 3.
Note: Hold the pipette steady and dispense liquid at an even rate to reduce variability in spots. Use a pencil to mark the grid as you go so that you can put the spots in a line. The consistency and speed of spotting may be improved by using a vacuum-based dot blot apparatus such as Bio-dot® from Bio-Rad. However, we have not tested such a device.
Figure 3. Layout of reaction spots on a nitrocellulose membrane for dot blot assay. Placing the spots in this ordered way makes quantification easier later on. - Allow spots to dry fully. This takes 5-10 min.
- Rinse briefly in transfer buffer and then with TBS-T.
- Continue with the Western blot protocol (Part II Procedure A) from Step A15, blocking the membrane.
Note: It can be useful to use a larger volume of antibody as the pieces of membrane are bigger for this dot blot. Ten to fifteen milliliters is recommended for both primary and secondary antibody. The membrane pieces may be too large for a falcon tube, so a suitably sized tray could be used instead.
- Cut one a piece of nitrocellulose membrane for each of the A, B and C sets of reactions.
Data analysis
- Quantify each spot or band
- Scan films to a computer using an Epson scanner and its software. Select the following parameters: positive film, 8-bit grayscale and 400 dpi. Scan it and save it as a TIFF file, including the date, experiment and exposure time in the file name.
- Choose an appropriate exposure time for each substrate concentration where the spots are not too faint or overexposed.
Note: If the spot is too faint it will be hard to distinguish over background. If the spot is over-exposed (at saturation), it reaches the maximum intensity possible in that area and differences between spots will not be observed. Different exposure times can be chosen for each substrate concentration so that the most suitable exposure is chosen for each. - In a suitable program such as Adobe Illustrator or Photoshop, add a square of black and a square of white. Save it as a TIFF file.
Note: When ImageJ gives the quantitative peaks for an image selection it shows the highest peak at the top of the window regardless of whether this peak is high in other selections. This makes comparison difficult between spots when they cannot be selected together (for example when there are too many spots). To avoid this problem, an area of black is included which gives the maximum possible value, and an area of white is included which gives the minimum possible value. This means that all values are relative to this and different selections can be compared. - Open the image in ImageJ.
- Select an area of spots using the rectangle tool including the area of white and black.
- On the analyze tab, under the gels section, choose ‘select first lane’ (shortcut: Ctrl+1) as shown in Figure 4.
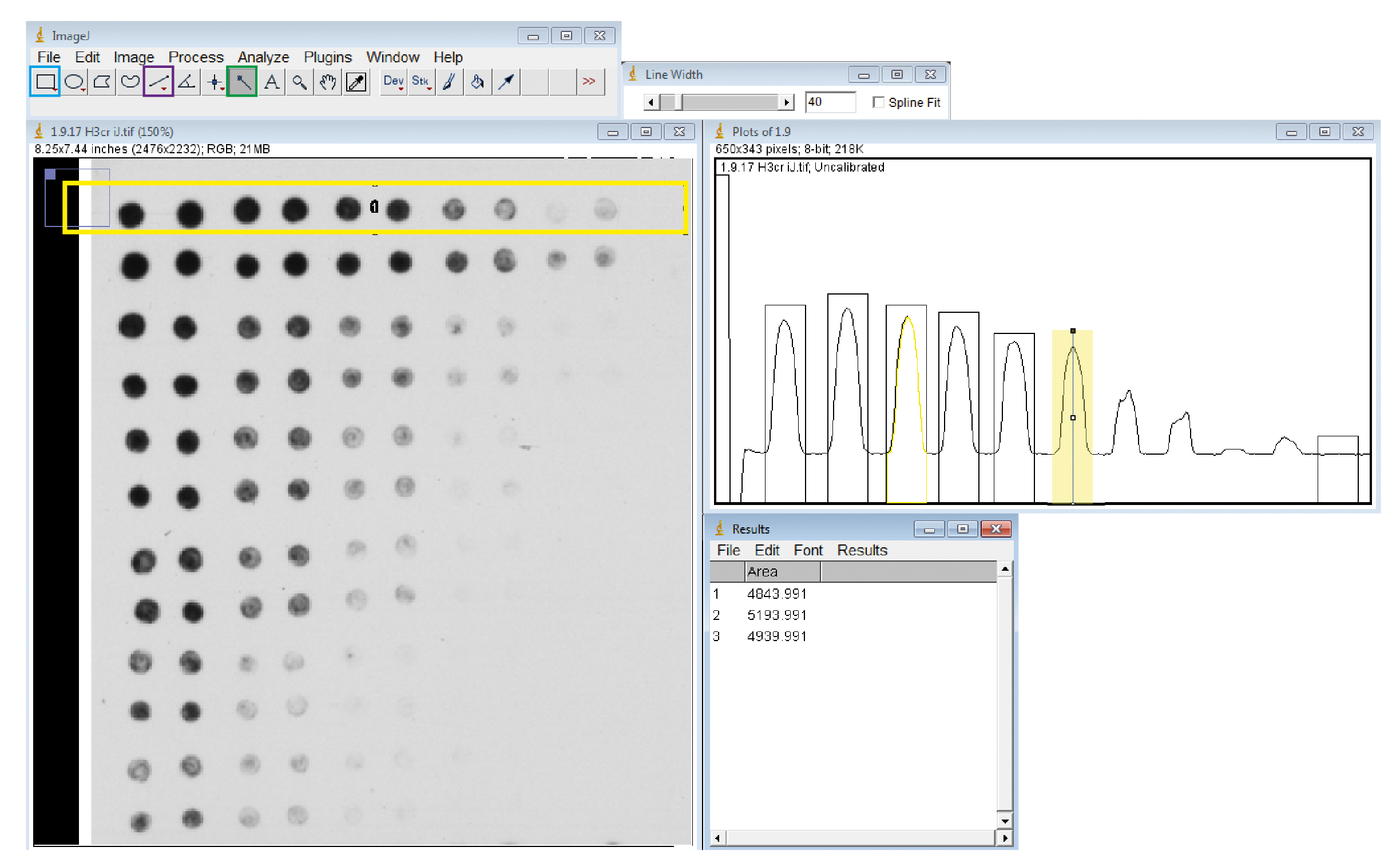
Figure 4. Determination of spot intensity using ImageJ software. Once the image is opened in ImageJ, a horizontal line of bands can be selected using the rectangle tool (blue box). Once this selection is plotted, the individual peaks corresponding to different spots can be selected using the line tool (purple box). The wand tool (green box) is then used to show the values of selected peaks in the results tab. In the image above, the first two peaks have been selected, the third has been highlighted with the wand tool, and the sixth peak is being selected with the line tool (these are combined screenshots to show many operations at once). - In the same section, choose ‘plot lanes’ (shortcut: Ctrl+3).
- Double-click the line tool and type a suitable value in the window to set it to the appropriate width, then select peaks corresponding to a spot or band.
Note: The value only changes when you type the number, not when you press enter. Try out a few widths to find one that fits the peak well and then use this for all of the selected peaks. A value of 50 to 60 (no units given) usually fits the peaks from a spot and 70 to 100 for a band. Control/command-Z can be used to remove a selection if it is in the wrong place. - With the same width setting, select an area without a peak which corresponds to the background.
- Using the wand tool, click on each peak area in turn and the values will come up in a results window.
- Select these, copy and paste the values into Excel.
- Attribute a reaction name to each value according to its position on the membrane.
- Repeat Steps 5 to 12 for the other rows.
Note: Keep track of the order that you analyze spots and remember that there are four replicates for each reaction which are split into two rows.
- Scan films to a computer using an Epson scanner and its software. Select the following parameters: positive film, 8-bit grayscale and 400 dpi. Scan it and save it as a TIFF file, including the date, experiment and exposure time in the file name.
- Calculate the initial reaction rate for each substrate concentration in Excel
This is only applicable to Part I B3 where multiple time points and substrate concentrations were measured.- Subtract values for each spot by the background value.
- Calculate the average of the values for the four spots for each reaction.
- Calculate the value of each time point relative to the zero time point.
Note: For each dilution of substrate, divide each time point by the zero time point so that zero time is 1 and other values are relative to this. - Convert dilutions of substrate to µM of crotonyl-H3. This assumes that the 5.65 µM of H3.1 was fully crotonylated at lysine 18 in the first reaction, which has been tested in Figure 5.
Notes:- Multiply each relative value by the concentration of modified histone in the reaction mix, as shown in Table 6, to convert spot intensity to substrate concentration.
- We assume this because we also spotted 0.45 to 45 µM of a synthetic crotonylated H3K18cr peptide (a short section of amino acids around the K18 position) and found that the preparation of 5.65 µM crotonylated H3 had an intensity in between 4.5 and 9 µM of crotonyl-peptide as shown in Figure 5. The peptide was diluted in HDAC assay buffer.
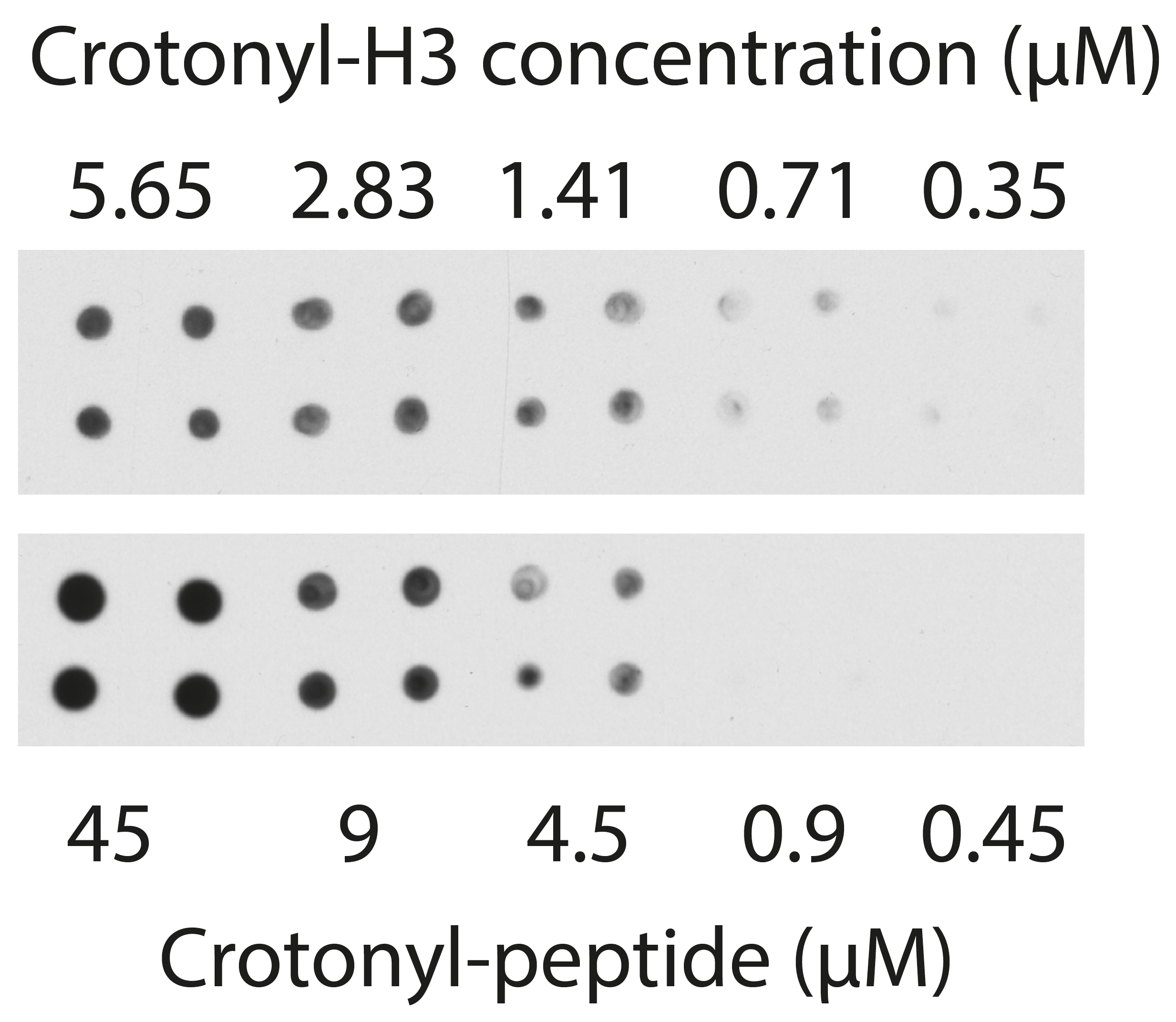
Figure 5. Comparison of ECL intensity of crotonylated histone H3 and the H3K18cr crotonyl-peptide by dot blotting. The H3K18cr peptide sequence was TGGKAPR-Lys(Crotonyl)-QLATKAA-EDA-Btn, EDA is a spacer and Btn indicates C-terminal biotinylation. The intensity of spots for 5.65 µM crotonyl-H3 corresponds to an intensity of crotonyl-peptide between 4.5 and 9 µM.
Table 6. Converting dilutions of crotonyl-H3 reaction mix to concentration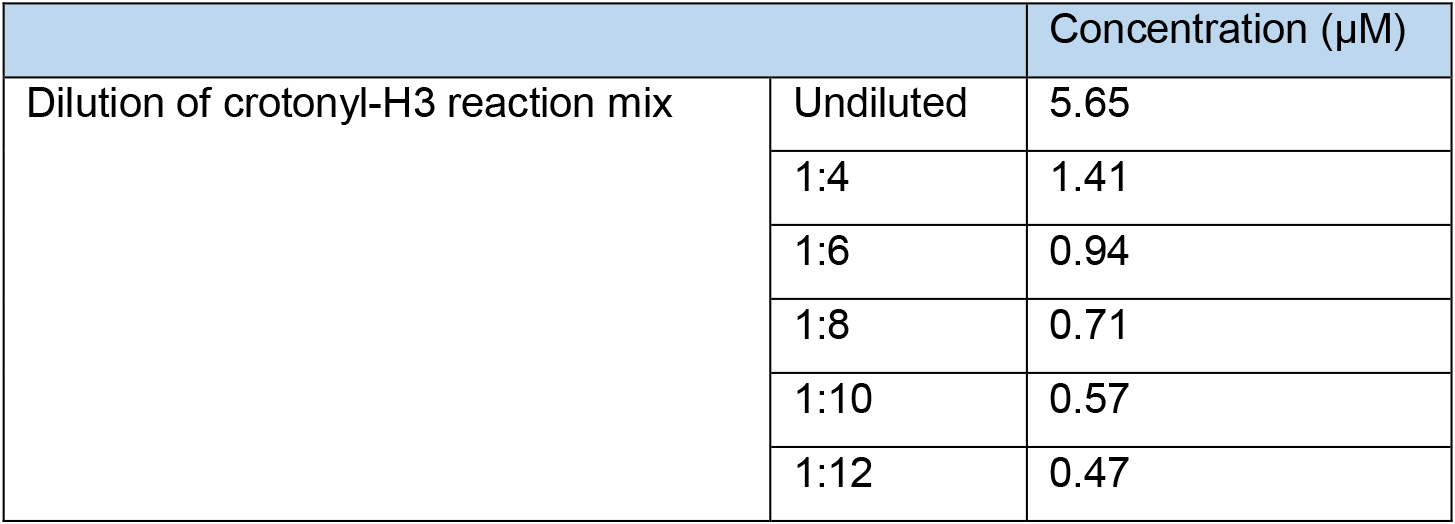
- Multiply each relative value by the concentration of modified histone in the reaction mix, as shown in Table 6, to convert spot intensity to substrate concentration.
- Plot substrate concentration against time for each crotonylated histone concentration as shown in Figure 6.
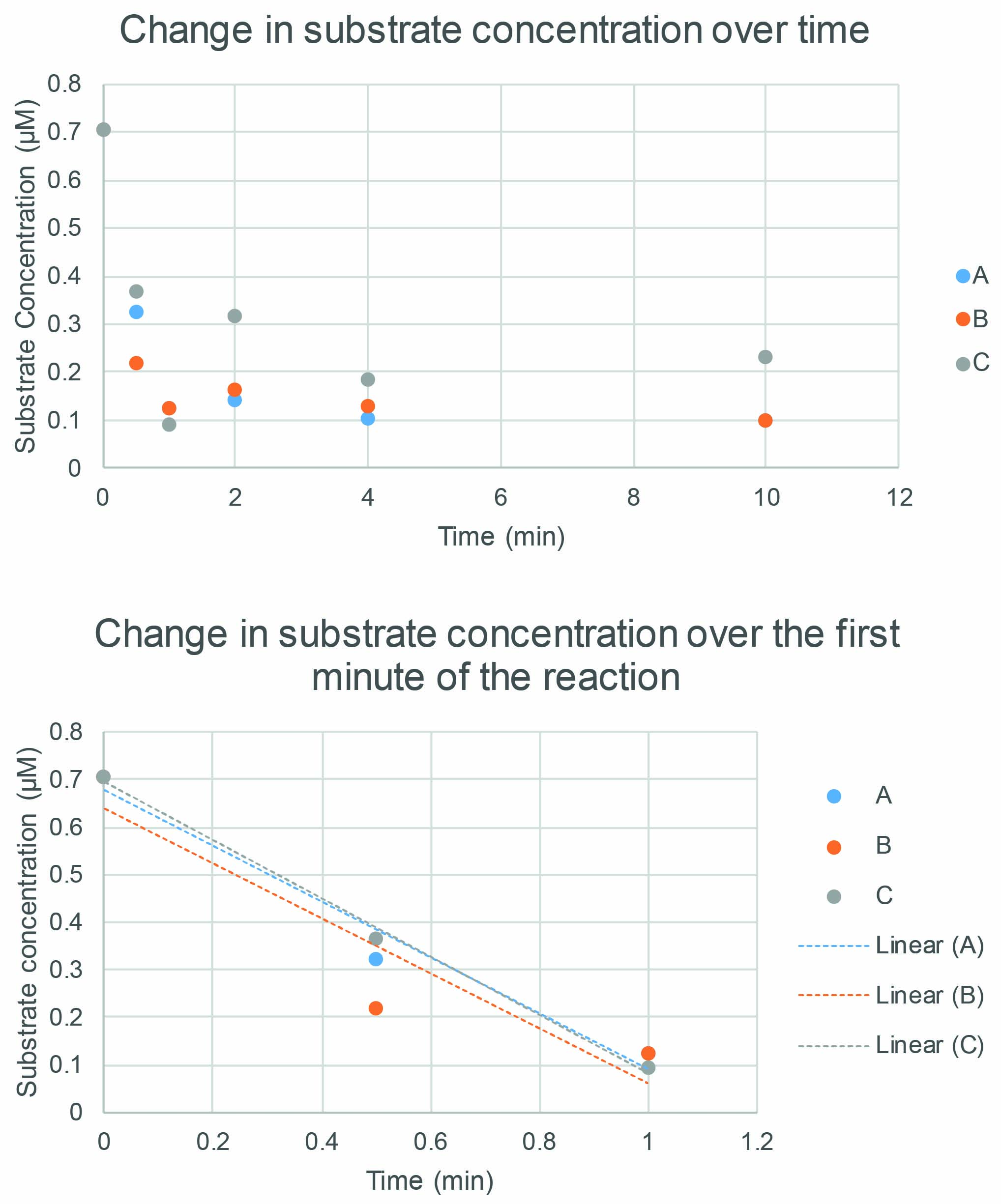
Figure 6. Determination of initial rate by plotting substrate concentration against time. Plotting the substrate concentration for each time point and replicate allows us to see how the reaction has progressed. In the first graph (upper panel), the reaction is fastest during the first minute, so only these time points are plotted in the second graph (lower panel) which now shows a linear relationship and allows the initial rate (the gradient of the line) to be determined. Three replicates: A, B and C. - Determine duration for the initial rate of the reaction before it starts to slow and fit a linear regression line for just these points.
Note: In the set of reactions shown in Figure 6, the initial rate occurs in the first thirty seconds to first minute of the reaction. Choose whichever matches the data points closest. - Take the gradient of the line for each of A, B and C replicates, this gives the rate.
- Repeat for the other substrate concentrations.
- Create a table of substrate concentration against rate and transfer to GraphPad Prism.
- Subtract values for each spot by the background value.
- Calculate kinetic constants using GraphPad Prism version 7
- Plot the initial rate of reaction against substrate concentration.
- In the analysis section, choose non-linear regression.
- Select enzyme kinetics-substrate versus velocity, choose Michaelis-Menten and click ‘OK’.
- This will give you Vmax, the maximum initial rate for that enzyme concentration when all active sites are occupied. It will also give you Km, the substrate concentration required to give half of the maximum rate.
- Select the enzyme kinetics-substrate versus velocity section again but this time choose Kcat. In the constrain tab by the parameter name ‘Et’, constrain ‘constant equal to’ and input the concentration of enzyme used into the value box, in our case this is 0.03, corresponding to the 0.03 µM of HDAC1 used.
Note: An example of such an analysis is shown in Figure 7b of Fellows et al. (2018), depicting an analysis of the comparative kinetics of HDAC1 decrotonylation and deacetylation.
- Plot the initial rate of reaction against substrate concentration.
Recipes
- Histone acylation buffer
50 mM Tris-HCl pH 8
50 mM KCl
0.1 mM EDTA
0.01% Tween 20
10% Glycerol
1 mM DTT
Aliquot and store at -20 °C until use - HDAC Assay buffer
25 mM Tris-HCl pH 7.5
50 mM KCl
1 mM MgCl2
1 µM ZnSO4
Aliquot and store at -20 °C until use - 1x MES buffer
50 ml 20x RunBlue MES buffer
950 ml distilled or Milli-Q water - 1x TGS buffer
100 ml 10x Tris/glycine/SDS electrophoresis buffer (Bio-Rad)
900 ml distilled or Milli-Q water - Transfer Buffer (recipe for 1 L buffer)
5.8 g Tris
2.9 g Glycine
200 ml Methanol
800 ml distilled or Milli-Q water - Tris-buffered saline (TBS)
302.5 g Tris
438.0 g NaCl
1 L distilled or Milli-Q water - TBS with tween 20 (TBS-T)
2.5 ml of 20% Tween 20 in 1 L of TBS - TBS-T with 3% (w/v) bovine serum albumin (TBS-T BSA)
Dissolve 6 g of BSA in 200 ml TBS-T and store at 4 °C
Acknowledgments
This work was supported by an MRC DTP studentship to RF, BBSRC and MRC funding to PVW. We thank Tabitha Rücker and Karolina Doubkova for critical reading of the manuscript. The authors declare no conflicts of interest. This is an elaborated method based on the work published by Fellows et al. (2018).
References
- Bannister, A. J. and Kouzarides, T. (2011). Regulation of chromatin by histone modifications. Cell Res 21(3): 381-395.
- Castillo, J., Lopez-Rodas, G. and Franco, L. (2017). Histone post-translational modifications and nucleosome organisation in transcriptional regulation: some open questions. Adv Exp Med Biol 966: 65-92.
- Fellows, R., Denizot, J., Stellato, C., Cuomo, A., Jain, P., Stoyanova, E., Balazsi, S., Hajnady, Z., Liebert, A., Kazakevych, J., Blackburn, H., Correa, R. O., Fachi, J. L., Sato, F. T., Ribeiro, W. R., Ferreira, C. M., Peree, H., Spagnuolo, M., Mattiuz, R., Matolcsi, C., Guedes, J., Clark, J., Veldhoen, M., Bonaldi, T., Vinolo, M. A. R. and Varga-Weisz, P. (2018). Microbiota derived short chain fatty acids promote histone crotonylation in the colon through histone deacetylases. Nat Commun 9(1): 105.
- Pengelly, A. R., Copur, O., Jackle, H., Herzig, A. and Muller, J. (2013). A histone mutant reproduces the phenotype caused by loss of histone-modifying factor Polycomb. Science 339(6120): 698-699.
- Sabari, B. R., Tang, Z., Huang, H., Yong-Gonzalez, V., Molina, H., Kong, H. E., Dai, L., Shimada, M., Cross, J. R., Zhao, Y., Roeder, R. G. and Allis, C. D. (2015). Intracellular crotonyl-CoA stimulates transcription through p300-catalyzed histone crotonylation. Mol Cell 58(2): 203-215.
- Sabari, B. R., Zhang, D., Allis, C. D. and Zhao, Y. (2017). Metabolic regulation of gene expression through histone acylations. Nat Rev Mol Cell Biol 18(2): 90-101.
- Wegener, D., Hildmann, C., Riester, D. and Schwienhorst, A. (2003a). Improved fluorogenic histone deacetylase assay for high-throughput-screening applications. Anal Biochem 321(2): 202-208.
- Wegener, D., Hildmann, C. and Schwienhorst, A. (2003b). Recent progress in the development of assays suited for histone deacetylase inhibitor screening. Mol Genet Metab 80(1-2): 138-147.
- Wegener, D., Wirsching, F., Riester, D. and Schwienhorst, A. (2003c). A fluorogenic histone deacetylase assay well suited for high-throughput activity screening. Chem Biol 10(1): 61-68.
- Wei, W., Liu, X., Chen, J., Gao, S., Lu, L., Zhang, H., Ding, G., Wang, Z., Chen, Z., Shi, T., Li, J., Yu, J. and Wong, J. (2017). Class I histone deacetylases are major histone decrotonylases: evidence for critical and broad function of histone crotonylation in transcription. Cell Res 27(7): 898-915.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Fellows, R. and Varga-Weisz, P. (2018). In vitro Enzymatic Assays of Histone Decrotonylation on Recombinant Histones. Bio-protocol 8(14): e2924. DOI: 10.21769/BioProtoc.2924.
Category
Biochemistry > Protein > Posttranslational modification
Molecular Biology > Protein > Deacylation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.