- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

FRAP: A Powerful Method to Evaluate Membrane Fluidity in Caenorhabditis elegans

Published: Vol 8, Iss 13, Jul 5, 2018 DOI: 10.21769/BioProtoc.2913 Views: 10350
Reviewed by: Tugsan TezilKelly H. OhAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Sep 2017
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


Abstract
FRAP (Fluorescence Recovery After Photobleaching) is probably the most direct method to investigate the dynamics of molecules in living cells. Here, we describe FRAP to quantify membrane fluidity in C. elegans. Using FRAP, we have shown that cold, glucose and exogenous saturated fatty acids can decrease the fluidity of cellular membranes in certain mutants.
Keywords: FRAPBackground
Biological membranes, defining features of all cells, are primarily composed of phospholipids (Van Meer et al., 2008). The fatty acid species present in the phospholipid bilayer greatly influence its properties. For example, a high saturated fatty acid content increases membrane rigidity while a high unsaturated fatty acid content promotes fluidity (Pilon, 2016). The fatty acid composition of cellular membranes often shows clear correlations with the composition of dietary fats, which can be incorporated directly into phospholipids (Abbott et al., 2012; Dancy et al., 2015). However, considering that wide variations in diets exist, cells must have regulatory mechanisms that monitor and adjust membrane composition and achieve the desired membrane properties, such as fluidity, curvature, thickness, etc. Using FRAP on worms that express a prenylated GFP in intestinal cells, we have previously shown that mutants lacking PAQR-2, a homolog of the mammalian adiponectin receptors, have reduced membrane fluidity upon cultivation in the presence of glucose or on diets rich in saturated fatty acids (Svensk et al., 2016; Devkota et al., 2017). During FRAP, fluorescent molecules in a specified region are photobleached using a high-power laser and subsequent recovery of the bleached region is recorded and quantified (Reits and Neefjes, 2001). Here we describe a detailed FRAP protocol to study the fluidity of membranes in C. elegans.
Materials and Reagents
- Petri dish (60 x 15 mm) with cams (SARSTEDT, catalog number: 82.1194.500 )
- Microscope frosted slides 25 x 75 x 1 mm (Thermo Fisher Scientific, catalog number: 2951-001T )
- Microscope cover glass 22 x 22 mm (VWR, catalog number: 631-1570 )
- 0.2 μm filter (polyethylenesulfone membrane, VWR, catalog number: 28145-501 )
- C. elegans strains QC114 {etEx2 [(pQC09.6) glo-1p::GFP::ras-2 CAAX + (pRF4) rol-6(su1006)]} and QC129 (paqr-2[tm3410]) (Caenorhabditis Genetics Center/CGC, University of Minnesota, USA)
- E. coli OP50 strain (CGC, University of Minnesota, USA)
- Levamisole (Sigma-Aldrich, catalog number: L9756 )
- Potassium phosphate monobasic (KH2PO4) (Sigma-Aldrich, catalog number: 795488 )
- Potassium phosphate dibasic (K2HPO4) (Sigma-Aldrich, catalog number: P2222 )
- Sodium Chloride (NaCl) (Sigma-Aldrich, catalog number: 31434 )
- Bacto Peptone (BD, catalog number: 211677 )
- Agar Powder (VWR, catalog number: 20767.298 )
- Cholesterol (Sigma-Aldrich, catalog number: C8667 )
- Magnesium Sulfate (Sigma-Aldrich, catalog number: M2643 )
- Calcium Chloride (Sigma-Aldrich, catalog number: C8106 )
- Agarose (Sigma-Aldrich, catalog number: A9539 )
- α-D-glucose (Sigma-Aldrich, catalog number: 158968-500G )
- Ethanol (95%, Solveco, catalog number: 1000 )
- Phosphate buffer (see Recipes)
- M9 buffer (see Recipes)
- Nematode Growth Media (NGM) (see Recipes)
- Agarose pads (see Recipes)
- Levamisole solution (see Recipes)
Equipment
- Standard incubators for worm maintenance (cooled incubator by Panasonic/Sanyo, model: MIR 254 )
- 40x water immersion objective
- Stereomicroscope (Leica Microsystems, model: Leica MZ75 )
- Laser Scanning Confocal Microscope (ZEISS, model: LSM 700 )
Software
- Zen 2012 SP5 software (ZEISS)
- Microsoft Excel for Mac 2011
Procedure
- C. elegans strain and maintenance
The transgenic strain QC114 carries the etEx2 extrachromosomal transgene that supports expression of a membrane-associated prenylated GFP in intestinal cells (Mörck et al., 2009) and is available from the C. elegans Genetics Center (CGC; MN; USA). The etEx2 transgene should be maintained by picking rollers and can be easily crossed into any mutant or genetic background of interest; the example below assumes that etEx2 has been crossed into a mutant background such as paqr-2(tm3410) where membrane fluidity is clearly reduced by dietary SFAs (Svensk et al., 2016; Devkota et al., 2017).
Note: The transgene is in the form of an extrachromosomal array but this is not a concern since the experiment is performed only in the GFP-positive worms (Figure 1B) (Video 1).- Grow transgenic worms at 20 °C on 60 mm plates pre-seeded with approximately 200 μl of an overnight culture of E. coli OP50, which was maintained on LB plates and kept at 4 °C (re-streaked every 6-8 weeks) (Stiernagle, 2006).
- Bleach the gravid hermaphrodites and allow the eggs to hatch overnight in M9 buffer (Stiernagle, 2006; He, 2011). Transfer the synchronized L1 larvae to new plates containing 20 mM glucose. Prepare 20 mM glucose plates using filter sterilized (0.2 μm filter is used) and freshly prepared stock solution of 1 M glucose, which is added to cooled but still molten (~60 °C) NGM after autoclaving; seed these plates with E. coli OP50 as above.
Note: Glucose is readily converted into SFAs by the dietary E. coli, resulting in membrane rigidification in the paqr-2 mutant (Devkota et al., 2017). The control and paqr-2 mutant have the same membrane fluidity on normal culture plates lacking glucose. - After 16 h of incubation at 20 °C, mount the transgenic worms (rollers) on 2% agarose pads in a drop of 100 mM levamisole (dissolved in water; to paralyze the worms), and apply a cover glass.
Notes:- The worms are usually in the L2 stage after the overnight incubation; at this early stage, the etEx2 transgene is expressed strongly and marks clearly the intestinal plasma membranes.
- Mount 20-25 transgenic worms (rollers) per agarose pad in 1-1.5 μl of 100 mM levamisole. This step is critical to avoid worm twitching or movement.
- The worms are usually in the L2 stage after the overnight incubation; at this early stage, the etEx2 transgene is expressed strongly and marks clearly the intestinal plasma membranes.
- Grow transgenic worms at 20 °C on 60 mm plates pre-seeded with approximately 200 μl of an overnight culture of E. coli OP50, which was maintained on LB plates and kept at 4 °C (re-streaked every 6-8 weeks) (Stiernagle, 2006).
- FRAP configuration: Confocal microscope
- Turn on the microscope, laser and computer. Launch the Zeiss Zen 2012 software. In the Locate tab, click on Microscope Control. Choose bright field or fluorescence. Choose green (FSet10 wf) filter for fluorescence. The entire experiment is performed with 40x water immersion objective (Numerical Aperture 1.1).
- Go to the Acquisition tab to begin a new FRAP configuration. Use Smart Setup, choose EGFP dye and 488 nm laser.
- Make sure the objective is a 40x water immersion objective. For continuous confocal scanning, use the frame mode with 12-bit intensity resolution over 256 x 256 pixels (digital zoom 4x) and a pixel dwell time of 1.58 μsec. Use the unidirectional scanning and set pinhole to 1 Airy Unit (Figure 1A).
- For photobleaching, choose time series, bleaching and the regions tab. Choose appropriate shape and size of the bleaching region. This protocol is based on a circular region with a 7-pixel radius. The entire experiment consists of 200 cycles and each cycle runs for 242.04 msec (Time Series tab). Bleaching can be set using 20 iterations using 50% laser power transmission. Set up to take 10 scans before starting the photobleaching. Under these settings, approximately 60-70% of the signal is bleached.
- Save the configuration.
- Turn on the microscope, laser and computer. Launch the Zeiss Zen 2012 software. In the Locate tab, click on Microscope Control. Choose bright field or fluorescence. Choose green (FSet10 wf) filter for fluorescence. The entire experiment is performed with 40x water immersion objective (Numerical Aperture 1.1).
- Microscopy and data acquisition
- In the Locate tab, under the microscope control, use the bright field to find a worm. It is important that the worms are well immobilized: any movement will compromise FRAP measurement.
- In the Acquisition tab, open the saved FRAP configuration (from Step B5). Make sure that the signal is neither saturated nor below the detection level (saturation and lack of fluorescence signal is generally indicated as red and blue spots respectively). This can easily be adjusted using the ‘Master Gain’ and ‘Digital Offset’ function. Note that one has to click on ‘Range Indicator’ tab to visualize the saturation level.
- Select a region of the intestinal cell that is not saturated and use the circular region with a 7-pixel radius to photobleach (Figure 1B).
Note: In our experience, it is not necessary to select a GFP-rich plasma membrane as control region; the effect of bleaching due to scanning seems to affect all regions equally. - Start the continuous scan (Video 1).
 Video 1. Illustration of FRAP. The first twenty cycles of the FRAP experiment. Red circle of 7-pixel radius is the bleaching region.
Video 1. Illustration of FRAP. The first twenty cycles of the FRAP experiment. Red circle of 7-pixel radius is the bleaching region. - After the scanning, click on the ‘Mean ROI’ tab and select a control reference region in the sample that was not bleached. This will be used to compensate for the effect of bleaching on the overall signal.
- Save the image and the table (.txt format) that shows the fluorescent intensity of the entire experiment.
- In the Locate tab, under the microscope control, use the bright field to find a worm. It is important that the worms are well immobilized: any movement will compromise FRAP measurement.
Data analysis
- Open the text file in Excel. All data analysis, graphing and statistics were performed with the Excel program.
- Compensate for the loss of fluorescence due to repetitive scanning: repetitive scanning and imaging reduces the overall fluorescence over time. This is compensated in each FRAP experiment by using a reference region and determining the slope of its decrease in fluorescence over time; typically, the reference data shows a linear decline, and the slope can be calculated by graph fitting. For example, if there is a bleaching rate of 0.25% per second, then each data point in the FRAP measurement area should be divided by 1 - (0.0025 x number of seconds elapsed). This produces the compensated data.
- Set the post-bleach fluorescence as zero: Identify the lowest intensity value (immediately after bleaching) and subtract this value from all intensities, thus setting the post-bleach fluorescence as zero.
- Normalize: determine the average intensities of the five measurements that precede the bleaching. Divide all intensities by that value. Now all intensities should have values between 0 and 1 (except for some of the first five that may be above the average of the first five).
- Plot the data: % of FRAP (normalized) vs. time (sec). In the example shown, FRAP is performed on wild type (N2) and paqr-2 mutant worms cultivated on 20 mM glucose (Figure 1C).
- Determine the plateau after recovery: calculate the average of the last five measurements (assuming that the recovery has plateaued or nearly plateaued by then). This corresponds to the mobile fraction, i.e., the maximum recoverable fluorescence.
- Determine the halftime of recovery (Thalf): identify at what time point the intensity is recovered to half of the maximum recovery (done empirically by checking the data). Do that for each individual experiment within a set, and then get the average value ± standard error of the mean. Use t-test to determine the significance between Thalf values. The example here shows that paqr-2 mutant worms have a reduced mobile fraction and slower recovery than N2 worms when cultivated in 20 mM glucose (Figure 1D).
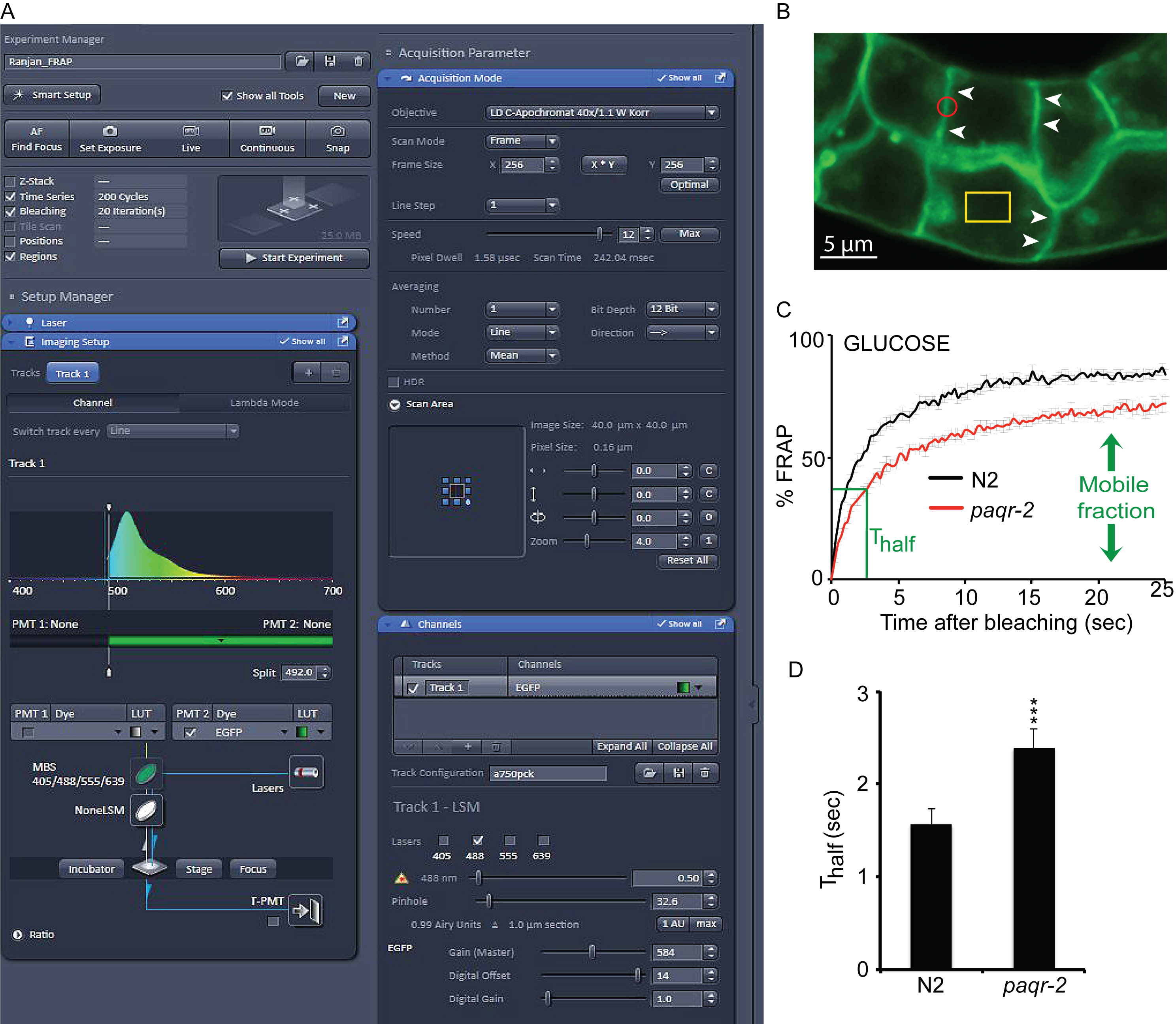
Figure 1. FRAP setup, bleaching and data analysis. A. Screenshot of the ZEN software window showing the confocal setup for bleaching. B. Illustration of the prenylated GFP marker on the plasma membrane of intestinal cells. Arrowheads indicate clear intestinal membranes and the red circle represents the size of a region bleached during FRAP analysis. The yellow rectangle shows the control reference region that is not bleached during the experiment. C. FRAP curve showing the recovery of wild type N2 and paqr-2 mutants grown on 20 mM glucose (error bars show the SEM; n = 6-10 worms). paqr-2 mutants have a slower recovery and a reduced mobile fraction. D. Thalf values showing the time to recover half of the maximum fluorescence. paqr-2 mutants take significantly longer time to recover. Statistics with t-test (two-tailed; equal variance) showing the degree of significance. ***: P < 0.001
Notes
- It is important that the worms do not move during the entire FRAP process. One typically plans on obtaining reliable FRAP measurements for approximately 6-10 worms per experiment, which in practice often requires 15-20 measurement attempts because of worms occasionally twitching or moving slightly.
- We have always done only one FRAP measurement per worm.
- Note that the worms that have been left too long on the agarose pads won’t have the good signal required to perform the experiment. Worms mounted for more than 60 min should not be used.
Recipes
- Phosphate buffer
- Mix 66 ml of 1 M sterile K2HPO4 and 434 ml of 1 M sterile K2HPO4 (pH 6.0)
- Store at room temperature
- Mix 66 ml of 1 M sterile K2HPO4 and 434 ml of 1 M sterile K2HPO4 (pH 6.0)
- M9 buffer
- Dissolve 3.0 g KH2PO4, 6.0 g Na2HPO4 and 5 g NaCl in distilled H2O and adjust volume to 1 L. Sterilize by autoclaving
- Allow to cool then add 1 ml of sterile 1 M MgSO4
- Store at room temperature
- Dissolve 3.0 g KH2PO4, 6.0 g Na2HPO4 and 5 g NaCl in distilled H2O and adjust volume to 1 L. Sterilize by autoclaving
- Nematode Growth Media (NGM)
- Add 3 g NaCl, 2.5 g Bacto peptone and 20 g agar in 975 ml distilled water. Sterilize by autoclaving
- Add 1 ml cholesterol (5 mg/ml in EtOH; this is never autoclaved), 1 ml 1 M MgSO4, 1 ml 1 M CaCl2 and 25 ml 1 M phosphate buffer (pH 6.0); the latter two solutions are autoclaved separately then used as stock solutions kept at room temperature
- Pour around 10 ml media per Petri plates and let them dry overnight
- Store the plates at 4 °C
- Add 3 g NaCl, 2.5 g Bacto peptone and 20 g agar in 975 ml distilled water. Sterilize by autoclaving
- Agarose pads
- Microwave 2% agarose in M9 until molten
- Place a drop on a glass slide that is flanked by two others that have about 0.5 mm of regular lab tape onto them
- Place a fourth slide across the first three, so as to flatten the drop to a thickness of about 0.5 mm
- Slide off the top slide once the agarose has cooled, leaving behind a fresh, flat agarose pad
- Store in a humid box to prevent the pad from drying up
- Microwave 2% agarose in M9 until molten
- Levamisole solution
- Weigh 2.04 g of levamisole
- Add to 100 ml distilled water
- Mix until fully dissolved
- Weigh 2.04 g of levamisole
Acknowledgments
We thank the Center for Cellular Imaging at University of Gothenburg for microscopy support. This work was funded by Vetenskaprådet, Cancerfonden, Carl Trygger Stiftelsen, Diabetesfonden and Kungliga Vetenskaps och Vitterhets-Samhället. The authors declare no conflicts of interest or competing interests.
References
- Abbott, S. K., Else, P. L., Atkins, T. A. and Hulbert, A. J. (2012). Fatty acid composition of membrane bilayers: importance of diet polyunsaturated fat balance. Biochim Biophys Acta 1818(5): 1309-1317.
- Dancy, B. C., Chen, S. W., Drechsler, R., Gafken, P. R. and Olsen, C. P. (2015). 13C- and 15N-labeling strategies combined with mass spectrometry comprehensively quantify phospholipid dynamics in C. elegans. PLoS One 10(11): e0141850.
- Devkota, R., Svensk, E., Ruiz, M., Stahlman, M., Boren, J. and Pilon, M. (2017). The adiponectin receptor AdipoR2 and its Caenorhabditis elegans homolog PAQR-2 prevent membrane rigidification by exogenous saturated fatty acids. PLoS Genet 13(9): e1007004.
- He, F. (2011). Synchronization of worm. Bio-protocol Bio101: e56.
- Mörck, C., Olsen, L., Kurth, C., Persson, A., Storm, N. J., Svensson, E., Jansson, J. O., Hellqvist, M., Enejder, A., Faergeman, N. J. and Pilon, M. (2009). Statins inhibit protein lipidation and induce the unfolded protein response in the non-sterol producing nematode Caenorhabditis elegans. Proc Natl Acad Sci U S A 106(43): 18285-18290.
- Pilon, M. (2016). Revisiting the membrane-centric view of diabetes. Lipids Health Dis 15(1): 167.
- Reits, E. A. and Neefjes, J. J. (2001). From fixed to FRAP: measuring protein mobility and activity in living cells. Nat Cell Biol 3(6): E145-147.
- Stiernagle, T. (2006). Maintenance of C. elegans. WormBook: the online review of C. elegans biology. The C. elegans Research Community 1-11.
- Svensk, E., Devkota, R., Stahlman, M., Ranji, P., Rauthan, M., Magnusson, F., Hammarsten, S., Johansson, M., Boren, J. and Pilon, M. (2016). Caenorhabditis elegans PAQR-2 and IGLR-2 protect against glucose toxicity by modulating membrane lipid composition. PLoS Genet 12(4): e1005982.
- Van Meer, G., Voelker, D. R. and Feigenson, G. W. (2008). Membrane lipids: Where they are and how they behave. Nat Rev Mol Cell Biol 9(2): 112-124.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Devkota, R. and Pilon, M. (2018). FRAP: A Powerful Method to Evaluate Membrane Fluidity in Caenorhabditis elegans. Bio-protocol 8(13): e2913. DOI: 10.21769/BioProtoc.2913.
Category
Cell Biology > Cell imaging > Confocal microscopy
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.