- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Bacterial Microcolonies in Gel Beads for High-throughput Screening

Published: Vol 8, Iss 13, Jul 5, 2018 DOI: 10.21769/BioProtoc.2911 Views: 10351
Reviewed by: Elizabeth LibbyPierre-Yves ColinBeatrice Li
Original research article
The authors used this protocol in:

Nov 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
High-throughput screening of a DNA library expressed in a bacterial population for identifying potentially rare members displaying a property of interest is a crucial step for success in many experiments such as directed evolution of proteins and synthetic circuits and deep mutational scanning to identify gain- or loss-of-function mutants.
Here, I describe a protocol for high-throughput screening of bacterial (E. coli) microcolonies in gel beads. Single cells are encapsulated into monodisperse water-in-oil emulsion droplets produced with a microfluidic device. The aqueous solution also contains agarose that gelates upon cooling on ice, so that solid gel beads form inside the droplets. During incubation of the emulsion, the cells grow into monoclonal microcolonies inside the beads. After isolation of the gel beads from the emulsion and their sorting by fluorescence activated cell sorting (FACS), the bacteria are recovered from the gel beads and are then ready for a further round of sorting, mutagenesis or analysis. In order to sort by FACS, this protocol requires a fluorescent readout, such as the expression of a fluorescent reporter protein. Measuring the average fluorescent signals of microcolonies reduces the influence of high phenotypic cell-to-cell variability and increases the sensitivity compared to the sorting of single cells. We applied this method to sort a pBAD promoter library at ON and OFF states (Duarte et al., 2017).
Background
Fluorescence activated cell sorting (FACS) has an unmatched screening throughput of > 107 events/h (Davies, 2012). However, sorting of single cells according to their fluorescence by FACS to screen libraries of synthetic circuits (Schaerli and Isalan, 2013) is often hampered by high phenotypic cell-to-cell variability. Alternatively, it is possible to sort small cell colonies (microcolonies) contained in hydrogel beads (Weaver et al., 1991; Sahar et al., 1994; Zengler et al., 2002; Meyer et al., 2015). Beads with a diameter of approximately up to 50 μm can be sorted by FACS (Weaver et al., 1991; Sahar et al., 1994; Zengler et al., 2002; Fischlechner et al., 2014; Duarte et al., 2017). The microcolonies in the beads are monoclonal, if just a single cell per bead is initially encapsulated, and that cell then grows to a microcolony inside the bead. Highly monodisperse gel beads can be produced in water-in-oil emulsion droplets generated on a microfluidic device (Theberge et al., 2010).
This protocol describes the generation of 1% agarose gel beads (diameter ~50 µm) harboring bacterial microcolonies using microfluidics and their sorting by FACS to isolate the variants with the desired properties (Duarte et al., 2017). It is possible to sort for variants that take on different states (e.g., ON and OFF) under different conditions (e.g., different inducer concentrations) by performing multiple sequential rounds of this protocol. With this method, we sorted cells expressing a fluorescent reporter protein, while it could also be amended to screen for other readouts. If combined with a strategy to maintain fluorescent reaction products in the bead (Fischlechner et al., 2014), it can be used to screen for enzyme or pathway activities. Another option is to co-encapsulate sensor cells that fluoresce upon the production of a compound of interest (Meyer et al., 2015). It is also possible to assay cell growth by relying on the light scatter of the microcolonies or by staining them with a fluorescent biomass indicator dye (e.g., staining nucleic acids or proteins) (Weaver et al., 1991). When encapsulating multiple cells per bead, cell-cell interactions could also be screened for. Thus, the described protocol is broadly applicable in biology.
Materials and Reagents
- PTFE tubing with inner diameter of 0.8 mm and outer diameter of 1.6 mm (Cole-Parmer Instrument, catalog number: EW-06407-41 )
- Stainless steel catheter couplers, 20 ga x 15 mm, non-sterile (Instech laboratories, catalog number: SC20/15 )
- CellTrics filters, 50 µm yellow (Sysmex, catalog number: 04-0042-2317 )
- 1.5 ml micro tubes (for example SARSTEDT, catalog number: 72.706.400 )
- Falcon 5 ml round-bottom tubes, disposable, polystyrene (Corning, Falcon®, catalog number: 352054 )
- 1.4 ml Non coded Screw Cap tubes U-bottom Bulk (Micronic, catalog number: MP32062 )
- Adhesive tape
- Aluminium foil
- Kimwipes (KCWW, Kimberly-Clark, catalog number: 34120 )
- Optional: microscope slides for droplets analysis (for example Kova Glasstic Slide 10 With Counting Grids, Kova International, catalog number: 87144E )
- Gloves
- Small resealable plastic bag
- 2 SGE Gas Tight Syringes, Fixed Luer Lock, volume 100 µl (Trajan Scientific, SGE Analytical Science, catalog number: 005229 )
- SGE Gas Tight Syringe, Fixed Luer Lock, volume 5 ml (Trajan Scientific, SGE Analytical Science, catalog number: 008762 )
- Hamilton needles 20 gauge, Kel-F Hub NDL, 2 in, point style 3 (Hamilton, catalog number: 90520 )
- E. coli (or other bacterial) cells harboring the library to be screened
- Ice
- Glycerol (Sigma-Aldrich, catalog number: G5516 )
- Mineral oil (Sigma-Aldrich, catalog number: M5904 )
- 3M Novec 7500 Engineered Fluid (known as HFE-7500 oil) (3M, catalog number: Novec 7500 )
- 5% (w/w) 008-FluoroSurfactant in HFE7500 (Ran Biotechnologies, catalog number: 008-FluoroSurfactant-5wtH-20G ) (Protect from light)
- Ultra-low Gelling Temperature agarose, type IX-A (Sigma-Aldrich, catalog number: A2576 )
- 1H,1H,2H,2H-Perfluoro-1-octanol (PFO), 97% (Sigma-Aldrich, catalog number: 370533 )
- Petri dishes (14 cm) (Thermo Fisher Scientific, NuncTM, catalog number: 249964 )
- Glass beads (2 mm) (Sigma-Aldrich, catalog number: Z273627 )
- Syringe Filters 0.22 µm pore size, 25 mm diameter (Corning, catalog number: 431219 )
- SYTO 9 Green Fluorescent Nucleic Acid Stain (Thermo Fisher scientific, catalog number: S34854 )
- Ammonium sulfate ((NH4)2SO4) (Sigma-Aldrich, catalog number: 09978 )
- Potassium phosphate dibasic (K2HPO4) (Sigma-Aldrich, catalog number: 60356 )
- Potassium phosphate monobasic (KH2PO4) (Sigma-Aldrich, catalog number: P5379 )
- Iron (II) sulfate heptahydrate (FeSO4·7H2O) (Sigma-Aldrich, catalog number: 215422 )
- Potassium hydroxide (KOH) (Sigma-Aldrich, catalog number: 221473 )
- Thiamine hydrochloride (Sigma-Aldrich, catalog number: T1270 )
- Casamino acids (BD, BactoTM, catalog number: 223050 )
- Magnesium sulfate (MgSO4) (Sigma-Aldrich, catalog number: M2643 )
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S3014 )
- Potassium chloride (KCl) (Sigma-Aldrich, catalog number: P9541 )
- Sodium phosphate dibasic (Na2HPO4) (Sigma-Aldrich, catalog number: S3264 )
- Hydrochloric acid (HCl) (Sigma-Aldrich, catalog number: 258148 )
- Sodium hydroxide (NaOH) (Sigma-Aldrich, catalog number: S8045 )
- Tryptone (BD, BactoTM, catalog number: 211705 )
- Yeast extract (BD, BactoTM, catalog number: 212750 )
- Kanamycin sulfate (Sigma-Aldrich, catalog number: K4000 )
- Medium for the bacteria (in our case M63 medium, see Recipes) containing the appropriate antibiotic and inducer concentrations
- 1x phosphate-buffered saline (PBS) (see Recipes)
- LB medium (see Recipes)
- LB-Agar plates containing the appropriate antibiotic (see Recipes)
Equipment
- Microfluidic devices to produce water-in-oil droplets (diameter 20-50 µm)
Note: For example, PDMS devices purchased from Wunderlichips GmbH (As we do. The design of the device is chosen by the customer. If you would like to purchase devices with our design, please refer to this publication when contacting Wunderlichips.). Alternatively, the devices can be prepared as previously described in detail, including the surface modification required to render them hydrophobic (Devenish et al., 2013). The design file of the device used in this study (40 µm flow-focusing channel) is available from the author’s website: (Figure 1).
Figure 1. Design of the microfluidic device used for droplet generation. The device contains an inlet for the oil phase, an inlet for the aqueous phase (bacteria, agarose, medium) and an exit outlet. The droplets are formed at the flow-focusing geometry (picture inset). For this protocol, the channel width at the flow focusing part is 40 µm and the height of the channels is also 40 µm. Using this device, droplets with a diameter of 40-50 µm can be produced. Scale bar = 40 µm. - Syringe pumps (for example Aladdin infusion pump, World Precision Instruments, catalog number: AL300-220 )
- Inverted light microscope (for example Leica Microsystems, model: Leica DM IL LED ) (A conventional microscope is also possible, but the tubing of the microfluidic device might interfere with the optics.)
- Fluorescence activated cell sorter (for example BD, model: FACSAriaTM III )
- High-speed camera (for example Teledyne DALSA, model: Genie Nano M640 Mono, catalog number: G3-GM10-M0640 )
Note: Standard cameras are not fast enough to observe/record continuous droplet formation. However, if the exposure time can be adjusted to ~50 µsec, droplet formation can be monitored with single pictures. - Tubing cutter (Cole-Parmer, catalog number: EW-06438-10 )
- 2 Hot/cold compresses (from the pharmacy or grocery store)
- Lab jack (Bochem Instrumente, catalog number: 11020 )
Note: Alternatively some box of the correct height to place the pump with the aqueous syringe at the height of the microscope stage. - -80 °C freezer
- 4 °C fridge
- Set of pipettes covering 0.5-1,000 µl (for example from Gilson)
- Pliers
- Tweezers
- Scissors
- 37 °C incubator
- Autoclave
- Benchtop centrifuge for 1.5 ml tubes
- Spectrophotometer, nanodrop or plate reader to measure the absorbance of bacterial cultures
- Thermoblock for 1.5 ml tubes
Software
- Software to control the high-speed camera (for example Labview or Common vision blox, Stemmer imaging)
- Image processing program, such as ImageJ or Photoshop
- Software to control the FACS (for example BD FACSDIVA)
- Flow cytometry analysis software such as FlowJo (LLC)
Procedure
- Prepare droplet formation
- On the evening before the experiment, prepare pre-cultures of the cells to be sorted by inoculating them into ~5 ml LB containing the appropriate antibiotic (or another suitable medium) and incubate them at 37 °C for 12-18 h in a shaking incubator. Alternatively, the protocol can also be started from glycerol stocks (stored at -80 °C), for example, 15% (v/v) glycerol stocks prepared by adding 420 μl of the overnight culture to 180 μl of 50% (v/v) glycerol.
- Connect the high-speed camera to an inverted microscope (via C-mount).
- Adjust the settings of the high-speed camera. Provide enough light so that the exposure time can be adjusted to the minimum (~50 µsec) and the frame rate to the maximum. Reducing the picture dimension also allows increasing the frame rate.
Note: To observe the droplet formation well, the frequency of the frames should be higher than the frequency of droplet formation. We are typically working at ≥ 600 frames per second. - Place the syringe pumps close to the microscope. In order to keep the tubing short and avoid gelation of the agarose in the tubing, the pump for the syringe with the aqueous solution should be as close as possible to the microscope stage. We used a lab jack to place it at the right height (Figure 2).

Figure 2. Equipment setup for producing microfluidic droplets. A. Overview of the whole setup. B. Close-up view on the microscope stage. - Adjust the diameters in the pumps: 10.3 mm for the 5 ml syringe (oil) and 1.46 mm for the 100 µl syringe (aqueous phase).
- Bend three steel couplers with pliers to introduce a 90° bend in their center.
- Fix the device on the microscope stage. Inspect the device with a 10x objective and focus on the flow focusing part.
- Cut three pieces of tubing (best with a tubing cutter): used for the oil inlet, the aqueous solution inlet, and the outlet respectively. The tubes for the inlets must be long enough to connect the syringes with the device. The one for the oil can be quite long (typically 40 cm in our case). The one for the aqueous solution should be as short as possible to avoid gelation of the agarose in the tube. In our case, it is ≤ 5 cm. The exact length will depend on your microscope setup. The outlet tube is about 9 cm.
Note: As the used plasmids carry an antibiotic resistance gene, we perform this protocol without sterile technique. It is, however, to autoclave the tubing and the steel couplers, and to disinfect the syringes and the device with 70% ethanol. - Connect each tubing to a bent steel coupler by hand (wearing gloves).
- Connect the exit tube to the device by hand (wearing gloves).
- Use scissors to make a small hole in the lid of a 1.5 ml microcentrifuge tubing. Cut the lid off and put it onto another open microcentrifuge tube. Put the exit tube through the small hole into the microcentrifuge tube, which will serve to collect the droplets. The hole in the lid prevents the outlet tubing from falling out. Fix the tube with adhesive tape on the microscope stage and cool it with a small resealable plastic bag full of ice.
- Prepare 5-10 ml solution of fluorinated oil (HFE-7500) with 0.5% 008-FluoroSurfactant by pipetting 0.5-1 ml of 5% 008-FluoroSurfactant in HFE-7500 to 4.5-9 ml HFE-7500. It can be kept for several months at room temperature. Protect the tube from light with aluminum foil.
- Fill the 5 ml syringe with the oil/surfactant. Remove air bubbles.
- Connect the tubing for the oil to the needle of the syringe and fill it completely with oil. Confirm that the oil comes out of the steel coupler (into a Kimwipes tissue) and no air bubbles are left in the tubing.
- Fix the 5 ml syringe with the oil on the pump.
- Connect the tubing with the oil to the device by inserting the steel coupler into the oil inlet of the device.
- Prepare 1-2 ml 2% (w/v) agarose in your medium of choice (M63 in our case), including the appropriate antibiotic. The easiest way to do this is to weigh ~20 mg agarose into a microcentrifuge tube and add the correct amount of medium (1 ml in the case of 20 mg). Melt the agarose by incubating it for at least 15 min in a thermoblock at 60 °C.
Note: I do not use temperatures higher than 60 °C for melting the agarose to prevent potential cell death by elevated temperature in Step A22. - Get bacterial cells from a fresh pre-culture or a glycerol stock. Spin them down (~3,000 x g, 5 min) and resuspend in the medium you are going to use in the experiment.
- Measure the absorbance at 600 nm of the cells with a spectrophotometer, nanodrop or plate reader.
- Calculate the number of cells to be added to get the desired loading of your beads with cells. This depends on the droplet size and follows a Poisson distribution (see Notes for a step by step example of the calculations).
- Prepare ~500 µl of cells at the calculated density in the medium.
- Add 100 µl of the melted agarose (2% (w/v)) to 100 µl of the cells resulting in an aqueous solution containing the cells and 1% (w/v) agarose.
- From now on, you should take care of keeping the agarose solution warm. Work fast to avoid gelation of the agarose and keep it warm with the pre-warmed (e.g., in a 60 °C incubator) hot/cold compress.
- Fill a 100 µl syringe with the aqueous agarose solution. Try to avoid air bubbles. It might be necessary to expel and taking the liquid up several times until there are no air bubbles left.
- Connect the tubing for the aqueous solution to the syringe. Fill the tubing completely with the aqueous solution. Check that the solution comes out of the steel coupler (into a Kimwipes tissue) and no air bubbles are left in the tubing.
- Fix the syringe on the pump. Put a pre-warmed (e.g., in a 60 °C incubator) hot/cold compress on to the syringe and the tubing to avoid gelation of the agarose.
- Connect the tubing with the aqueous solution to the device (via the steel coupler).
- On the evening before the experiment, prepare pre-cultures of the cells to be sorted by inoculating them into ~5 ml LB containing the appropriate antibiotic (or another suitable medium) and incubate them at 37 °C for 12-18 h in a shaking incubator. Alternatively, the protocol can also be started from glycerol stocks (stored at -80 °C), for example, 15% (v/v) glycerol stocks prepared by adding 420 μl of the overnight culture to 180 μl of 50% (v/v) glycerol.
- Droplet formation and microcolony growth
- Start the pump for the oil. Use approximately 2,000 μl/h until you see oil entering the device (< 5 min).
- Start the pump for the aqueous solution. Use approximately 150 μl/h until you see droplets forming. Then slow down the flow rate to 80 μl/h. Wait until droplet formation is stable (< 5 min).
Notes:- These are flow rates that we are typically using. Device design and the composition of the aqueous phase influence the optimal flow rates. The chosen flow rates affect droplet size and frequency of droplet formation.
- Typical reasons for unstable droplet formations are: “jetting” – the aqueous phase goes beyond the flow focusing part of the device and forms irregular droplets (reduce the aqueous flow rate), the agarose has started solidifying into a gel (keep the syringe warmer), the device is blocked by some dust or the PDMS device delaminated from the glass slides around the channels or inlets (change device).
- These are flow rates that we are typically using. Device design and the composition of the aqueous phase influence the optimal flow rates. The chosen flow rates affect droplet size and frequency of droplet formation.
- Take some pictures of the droplets in the device, so that you can determine their size (see Notes 1-2).
- Let the device run until you have produced as many droplets as you want. From time to time replace the hot/cold compress with a warm one. Ensure that the syringes do not run out of liquid.
Note: With these flow rates we typically have a frequency of around 340 Hz, i.e., we produce 1.2 x 106 droplets/h. Seventy-four percent of them will not contain any cell. Some beads will also be lost during the process. - If you want multiple aliquots of the same sample (e.g., to test different incubation times), it is easier and more accurate to change the collection tube after a specific amount of time (e.g., 15 min for each aliquot) than later having to pipette aliquots of the emulsion. I suggest keeping one aliquot as a negative control without incubation.
- If you want to make a second sample, let the device run while preparing the second sample in a second 100 μl syringe (+ a fresh aqueous inlet and outlet tubing).
- Remove the exit tubing and the collection tube. The emulsion remaining in the tubing can be recovered by pushing air through it with a syringe.
- Turn off the pump for the aqueous solution and reduce the oil flow rate to about 200 μl/h. Replace the syringe and tubing for the aqueous phase. Increase the oil rate again to 2,000 μl/h and start the aqueous solution again at 150 μl/h until you have droplets and lower it to 80 μl/h. Keeping the oil running reduces the risk of blocking the device.
Note: Occasionally, blocking can happen since some dust enters the device and blocks the flow-focusing part of the device. You can try to remove it by pushing the oil syringe by hand to flush it out of the device. - Keep the emulsion samples on ice until all samples are ready.
- Remove as much fluorinated oil (bottom layer) as you can without losing emulsion. Collect the fluorinated oil in a separated tube for recycling.
Note: The fluorinated oil with the surfactant can be recycled and re-used multiple times. Simply filter it through a 0.22 µm syringe filter. - To prevent evaporation, I recommend overlaying the emulsion samples with a thin layer of mineral oil.
- Incubate the samples at 37 °C (or at the temperature of choice) in a thermoblock or incubator for the chosen amount of time without shaking. Keep the negative control sample on ice in a 4 °C fridge.
Notes:- The incubation time depends on the cells and medium. For the M63 medium, we incubate overnight, for LB medium 4-8 h is enough. The first time it might be worth making a time course by incubating aliquots for different amounts of time and analyzing them on the flow cytometer (see Figure 2 in Duarte et al., 2017).
- Growth in the emulsion is limited by the amount of medium provided in the droplet. A way to increase cell growth is to break the emulsion before incubation. The beads can then be incubated in ample growth medium and oxygen leading to higher cell density than when incubated in the emulsion. The disadvantage of this strategy is that all beads share the same medium. It is, for example, not possible to screen for cells that excrete a quorum sensing molecule that then activates a network in the cells from the same bead. The chemical would also enter neighboring beads, where the molecule is not produced. In this form of incubation, it is also more likely that some cells escape the beads and then grow outside the bead in the growth medium. These free bacteria can then decrease sorting purity and a filtering step where the beads are separated from the free bacteria might become necessary.
- The incubation time depends on the cells and medium. For the M63 medium, we incubate overnight, for LB medium 4-8 h is enough. The first time it might be worth making a time course by incubating aliquots for different amounts of time and analyzing them on the flow cytometer (see Figure 2 in Duarte et al., 2017).
- After incubation, cool the emulsion on ice for at least 15 min. This is to solidify the gel beads.
- Start the pump for the oil. Use approximately 2,000 μl/h until you see oil entering the device (< 5 min).
- Recover gel beads
- If you would like to look at your droplets using (fluorescence) microscopy, you can pipette them onto a microscope slide.
- Pipette 500 µl phosphate buffered saline (PBS) to the emulsion layer.
- Add 20-50 μl 1H,1H,2H,2H-perfluorooctanol (PFO) to the emulsion.
- Shortly vortex (5 sec) the tube and briefly centrifuge (10 sec, 2,500 x g).
- The emulsion (white) should be gone now. If there is still some emulsion left, repeat the last two steps (i.e., add more PFO, vortex and centrifuge).
- You have now three phases in the tube. From bottom to top: fluorinated oil and PFO, PBS containing the beads and mineral oil (if added) (Figure 3). Transfer the PBS phase to a fresh 1.5 ml microcentrifuge tube by pipetting. Try to get as much of it as possible without taking any oil. If you have also transferred some oil, transfer the PBS again into a fresh 1.5 ml microcentrifuge tube. The oil tends to stick to the tube walls.
Note: Dispose of the mineral oil and the fluorinated oil containing the PFO separately as chemical waste. - If you need to stain the cells (e.g., with Syto 9), you can do that now. If they are already fluorescent, you can proceed to the next step.

Figure 3. Three phases after breaking the emulsion
- If you would like to look at your droplets using (fluorescence) microscopy, you can pipette them onto a microscope slide.
- Sort gel beads
- To prevent clogging of the FACS, filter the samples through a 50 μm CellTrics mesh into the 1.4 ml screw cap tubes.
- Using tweezers, place the 1.4 ml tubes with the samples into the 5 ml round-bottom tubes which will fit to the flow cytometer. Like this, you have a minimal sample volume that cannot be sorted at the bottom of the tube.
- Analyse/sort the beads on the flow cytometer. Many universities will have a facility to help you with this.
- Use a 100 μm nozzle and the laser and filters suitable for detecting the fluorescence of your cells (488 nm laser, 530/30 nm filter for GFP).
- The width of the sideward scatter (SSC-W) versus the height of the forward scatter (FSC-H) is the combination of scatter parameters that separates the beads best from other particles (free bacteria, agarose debris, dust) or electronic noise of the machine. Therefore, first gate all beads (with and without microcolonies) on the SSC-W – FSC-H dot-plot (Figure 4A). For separating beads with and without microcolonies, we use the SSC-H – FSC-H dot plot (Figure 4B).
- The thresholds on the FSC-H and SSC-H can be set so that particles smaller than the beads are not recorded.
- Measure your controls (beads not incubated, beads with WT cells…).
- On the fluorescence histogram (Figure 4C) or the FSC-H–fluorescence dot plot set the gate for the beads with the fluorescence of interest, for example the 5% of beads with the highest fluorescence.
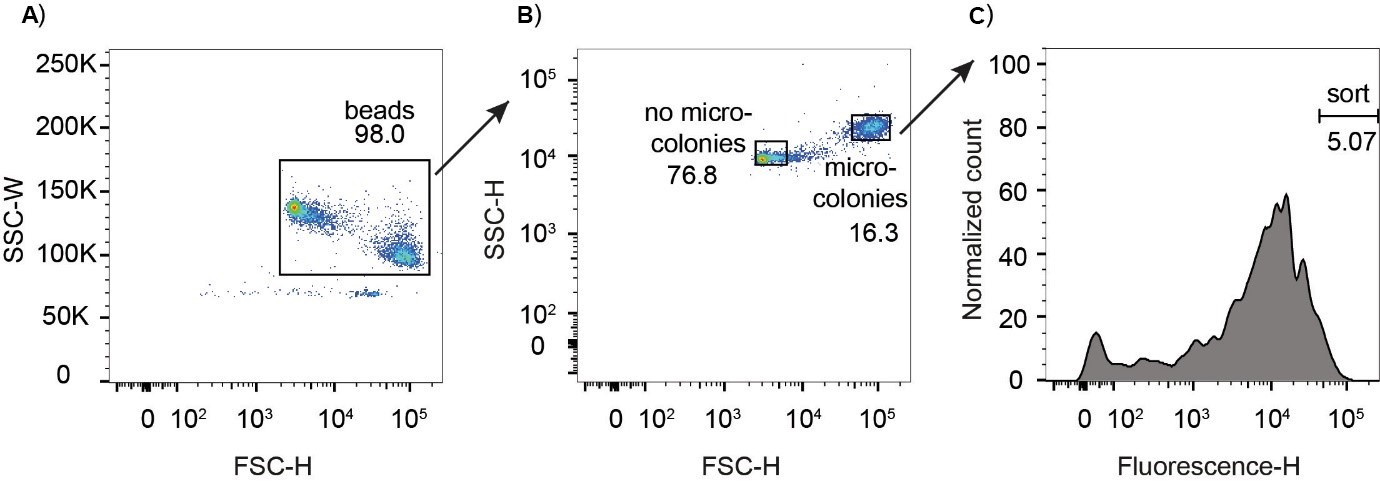
Figure 4. Gating strategy. A. Width of the sideward scatter (SSC-W) versus the height of the forward scatter (FSC-H) is the combination of scatter parameters that separates the beads best from other particles (free bacteria, agarose debris, dust) or electronic noise of the machine. Therefore, we first gate all beads (with and without microcolonies) on the SSC-W – FSC-H dot-plot. B. We then plot the gated beads in the SSC-H – FSC-H dot plot for gating the beads with microcolonies. The negative control should only contain the beads without microcolonies. C. We then plot the fluorescence of the beads containing microcolonies as histogram and set the gate for sorting, for example the 5% brightest beads. - Sort them into a 1.5 ml tube containing 200 µl of LB medium.
Note: The 50 µm agarose beads actually break open when sorted with the 100 µm nozzle. This is convenient for recovering the bacteria from the beads, as no additional agarose digest and/or melting has to be performed. If it is important for your experiments to keep the beads intact, they can be strengthened before the sorting, for example with a layer of polyacrylamide (Spencer et al., 2016). - Plate the beads out on pre-warmed LB-agar plate (supplemented with the appropriate antibiotic) and incubate overnight at 37 °C. We typically plate out each sorted sample on two big LB-agar plates (14 cm Ø) and typically recover > 10x more colonies than the number of beads collected.
- The colonies from the plates can be individually picked or resuspended all together in medium using glass beads. The resuspended libraries can be stored as glycerol stocks (15% v/v) at -80 °C. Samples can be analyzed, e.g., by sequencing, (single cell) flow cytometry, plate reader experiments etc. The glycerol stocks can be used to start another round of this protocol.
- To prevent clogging of the FACS, filter the samples through a 50 μm CellTrics mesh into the 1.4 ml screw cap tubes.
Data analysis
To analyze and plot the flow cytometry data recorded during the sorting, we use the program FlowJo and apply the gating strategy as described in Figure 4.
Analysis of the libraries and/or individual clones, e.g., by sequencing, (single cell) flow cytometry and plate reader experiments will indicate if you enriched for the desired properties and whether another round of sorting is required. See Duarte et al. (2017) for examples to analyze the libraries.
Notes
Calculation of the cell numbers:
- Determine the diameter of droplets
- Take a picture of the microscope ruler with a microscope camera.
- Use the measuring tool of Photoshop (or a similar program) to determine how many pixels correspond to 1 µm.
- Take pictures of the droplets (as a monolayer). Do not use coverslips as this might distort the diameter measurement.
- Measure their diameters (d) in pixels.
- Calculate the diameter in µm.
- Take a picture of the microscope ruler with a microscope camera.
- Calculate the volume (V) of your droplets
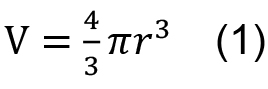
r: radius
1 m3= 1,000 L
Example: d = 50 µm → r = 25 x 10-6 m → V = 6.5 x 10-14 m3 = 6.5 x 10-11 L - Choose how many cells/droplet you want
Cells are distributed into droplets according the Poisson distribution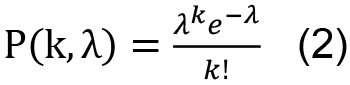
e=2.71828 (Euler’s number)
λ: average number of cells/droplet
k: number of cells in a droplet (0, 1, 2, 3, 4…)
Poisson distribution calculators can be found online, for example here: http://stattrek.com/online-calculator/poisson.aspx.
An example: λ = 0.3 → P(0, 0.3) = 0.74; P(1, 0.3) = 0.22; P(2, 0.3) = 0.033; P(3, 0.3) = 0.003.
This means, if on average we have 0.3 cells/droplet we will have 74% empty droplets, 22% droplets with 1 cell, 3.3% droplets with 2 cells and 0.3% droplets with 3 cells.
The average of 0.3 cells/droplet is a good balance between empty droplets and droplets with more than one cells. - Convert optical density at 600 nm (OD600) to cell density
- To correlate the measured OD600 with your cell density, you need to calibrate your instrument for your cell type.
- Here is an example of a detailed protocol how to do this:
http://delrio.dcccd.edu/jreynolds/microbiology/2421/lab_manual/counts.pdf. - For E. coli, a rough estimate is that OD600 of 1 corresponds to 5 x 108 -1 x 109 cells/ml.
- To correlate the measured OD600 with your cell density, you need to calibrate your instrument for your cell type.
- Calculate your cell dilutions
Example:- We have droplets with a 50 µm diameter.
- We want an average of 0.3 cell/droplet.
- We calibrated that the OD600 of 1 corresponds to 5 x 108 cells/ml.
- We measured the OD600 of our culture to be 0.2.
- Using equation (1) we calculate the volume of our droplets to be 6.5 x 10-11 L, this means, we have 1.5 x 1010 droplets/L and 1.5 x 107 droplets/ml of aqueous solution.
- For an average of 0.3 cell we thus need 0.3 x 1.5 x 107 cells/ml = 4.5 x 106 cells/ml.
- With an OD600 of 1 corresponding to 5 x 108 cells/ml, 4.5 x 106 cells/ml correspond to an OD600 of 0.009. This means the final OD600 of our sample should be 0.009. As we measured an OD600 of 0.2 we will need to dilute the cells overall 22-fold. A 2-fold dilution is achieved by mixing the cells with the agarose, hence we prepare a 11-fold dilution.
- These calculations will give you a first indication for your cell density. If the flow cytometry analysis (Figure 4) indicates that too many or too few beads contain cells, the cell density should be accordingly adjusted in the next experiment.
- Your cells might settle down in the syringe and the tubing, causing a time-dependent decrease in the number of cells entering the device and therefore lead to more empty droplets than expected by Poisson distribution. The extend of this effect is dependent on cell type, solution viscosity, tubing length and duration of your experiment. To prevent it, you can use a density matching agent such as Percoll or alginate. See Devenish et al. (2013) for details.
- We have droplets with a 50 µm diameter.
Recipes
- M63 medium (supplemented with 0.2% (w/v) glycerol, 0.025% (w/v) casamino acids, 0.00005% (w/v) thiamine)
Prepare 5x stock:- Add the following to 800 ml dH2O:
10 g (NH4)2SO4
68 g KH2PO4
2.5 g FeSO4·7H2O - Adjust pH to 7.0 using KOH
- Adjust volume to 1 L with dH2O
- Sterilize by autoclaving
- Add the following:
200 ml 5x stock
0.1 ml 0.5% thiamine hydrochloride
1.25 ml 20% Casamino Acids
10 ml of sterile 20% glycerol
1 ml of sterile 1 M MgSO4
Antibiotic (e.g., 50 µg/ml kanamycin) - Adjust volume to 1 L with dH2O
- Add the following to 800 ml dH2O:
- 1x PBS
- Add the following to 800 ml dH2O:
8 g of NaCl
0.2 g of KCl
1.44 g of Na2HPO4
0.24 g of KH2PO4 - Adjust the pH to 7.4 with HCl
- Add distilled water to a total volume of 1 L
- Sterilize by autoclaving
- Add the following to 800 ml dH2O:
- LB medium
- Add the following to 800 ml dH2O
10 g Tryptone
5 g yeast extract
10 g NaCl - Adjust pH to 7.0 using NaOH
- Adjust volume to 1 L with dH2O
- Sterilize by autoclaving
- Add the following to 800 ml dH2O
- LB-Agar
- Add the following to 800 ml dH2O
10 g Tryptone
5 g yeast extract
10 g NaCl
15 g agar - Adjust pH to 7.0 using NaOH
- Adjust volume to 1 L with dH2O
- Sterilize by autoclaving
- After autoclaving, cool to 50 °C, add antibiotic (if needed, e.g., 50 µg/ml kanamycin), and pour into Petri dishes.
- Let harden, then invert and store at 4 °C
- Add the following to 800 ml dH2O
Acknowledgments
I thank Içvara Barbier for critical reading. I acknowledge funding from the Swiss National Science Foundation (project 31003A_175608). This protocol was adapted from procedures published in (Duarte et al., 2017). The author declares no competing interests.
References
- Davies, D. (2012). Cell separations by flow cytometry. Methods Mol Biol 878: 185-199.
- Devenish, S. R., Kaltenbach, M., Fischlechner, M. and Hollfelder, F. (2013). Droplets as reaction compartments for protein nanotechnology. Methods Mol Biol 996: 269-286.
- Duarte, J. M., Barbier, I. and Schaerli, Y. (2017). Bacterial microcolonies in gel beads for high-throughput screening of libraries in synthetic biology. ACS Synth Biol 6(11): 1988-1995.
- Fischlechner, M., Schaerli, Y., Mohamed, M. F., Patil, S., Abell, C. and Hollfelder, F. (2014). Evolution of enzyme catalysts caged in biomimetic gel-shell beads. Nat Chem 6(9): 791-796.
- Meyer, A., Pellaux, R., Potot, S., Becker, K., Hohmann, H. P., Panke, S. and Held, M. (2015). Optimization of a whole-cell biocatalyst by employing genetically encoded product sensors inside nanolitre reactors. Nat Chem 7(8): 673-678.
- Sahar, E., Nir, R. and Lamed, R. (1994). Flow cytometric analysis of entire microbial colonies. Cytometry 15(3): 213-221.
- Schaerli, Y. and Isalan, M. (2013). Building synthetic gene circuits from combinatorial libraries: screening and selection strategies. Mol Biosyst 9(7): 1559-1567.
- Spencer, S. J., Tamminen, M. V., Preheim, S. P., Guo, M. T., Briggs, A. W., Brito, I. L., D, A. W., Pitkanen, L. K., Vigneault, F., Juhani Virta, M. P. and Alm, E. J. (2016). Massively parallel sequencing of single cells by epicPCR links functional genes with phylogenetic markers. ISME J 10(2): 427-436.
- Theberge, A. B., Courtois, F., Schaerli, Y., Fischlechner, M., Abell, C., Hollfelder, F. and Huck, W. T. (2010). Microdroplets in microfluidics: an evolving platform for discoveries in chemistry and biology. Angew Chem Int Ed Engl 49(34): 5846-5868.
- Weaver, J. C., Bliss, J. G., Powell, K. T., Harrison, G. I. and Williams, G. B. (1991). Rapid clonal growth measurements at the single-cell level: gel microdroplets and flow cytometry. Biotechnology (N Y) 9(9): 873-877.
- Zengler, K., Toledo, G., Rappe, M., Elkins, J., Mathur, E. J., Short, J. M. and Keller, M. (2002). Cultivating the uncultured. Proc Natl Acad Sci U S A 99(24): 15681-15686.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Schaerli, Y. (2018). Bacterial Microcolonies in Gel Beads for High-throughput Screening. Bio-protocol 8(13): e2911. DOI: 10.21769/BioProtoc.2911.
Category
Microbiology > Heterologous expression system > Escherichia coli
Molecular Biology > DNA > Mutagenesis
Systems Biology > Genomics > Screening
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.