- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

An Improved Method for Measuring Chromatin-binding Dynamics Using Time-dependent Formaldehyde Crosslinking
Published: Vol 8, Iss 4, Feb 20, 2018 DOI: 10.21769/BioProtoc.2905 Views: 9823
Reviewed by: Gal HaimovichAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2017
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Formaldehyde crosslinking is widely used in combination with chromatin immunoprecipitation (ChIP) to measure the locations along DNA and relative levels of transcription factor (TF)-DNA interactions in vivo. However, the measurements that are typically made do not provide unambiguous information about the dynamic properties of these interactions. We have developed a method to estimate binding kinetic parameters from time-dependent formaldehyde crosslinking data, called crosslinking kinetics (CLK) analysis. Cultures of yeast cells are crosslinked with formaldehyde for various periods of time, yielding the relative ChIP signal at particular loci. We fit the data using the mass-action CLK model to extract kinetic parameters of the TF-chromatin interaction, including the on- and off-rates and crosslinking rate. From the on- and off-rate we obtain the occupancy and residence time. The following protocol is the second iteration of this method, CLKv2, updated with improved crosslinking and quenching conditions, more information about crosslinking rates, and systematic procedures for modeling the observed kinetic regimes. CLKv2 analysis has been applied to investigate the binding behavior of the TATA-binding protein (TBP), and a selected subset of other TFs. The protocol was developed using yeast cells, but may be applicable to cells from other organisms as well.
Keywords: Chromatin immunoprecipitation (ChIP)Background
Transcription initiation is a complicated process that involves the cooperation and coordinated interaction of dozens of proteins on a chromatinized promoter (Kim et al., 2005; Encode Consortium, 2012; Rhee et al., 2012; Dowen et al., 2014). Many studies have investigated the assembly and regulation of the core transcriptional machinery in vitro (Zawel and Reinberg, 1992; Conaway and Conaway, 1993; Roeder, 1996; Hager et al., 2009; He et al., 2013; Cramer, 2014; Luse, 2014; Horn et al., 2016), but it has been more challenging to examine the stochastic nature of these processes in vivo. There are two general approaches used to measure chromatin-binding dynamics in vivo: microscopy and ChIP-based techniques (Coulon et al., 2013; Mueller et al., 2013). Microscopic techniques, such as fluorescence recovery after photobleaching (FRAP) or single molecule tracking (SMT), have high temporal resolution and have provided fundamental insight into chromatin binding dynamics, including results obtained by tracking individual molecules (Larson et al., 2009; Mueller et al., 2013; Morisaki et al., 2014). However, these approaches can be limited by photophysical effects such as photobleaching, and in addition, in the great majority of cases it is not possible to determine the identity of particular single copy loci where the measured interactions occur (Mueller et al., 2013). Alternatively, ChIP-based approaches provide precise chromatin location information. In Competition ChIP (CC), expression of a differentially tagged isoform of the TF of interest is induced and the relative levels of the constitutive and induced forms of the TF are monitored over time, yielding binding kinetic information through measurements of TF turnover at the sites of interest (van Werven et al., 2009; Lickwar et al., 2013). With advancements in modeling of CC data, residence times as short as 1.3 min have been measured (Zaidi et al., 2017). Relative dynamic measurements have also been made by conditional depletion of TFs from the nucleus using the Anchor Away technique (Haruki et al., 2008; Grimaldi et al., 2014), although specific mathematical models of the process have not yet been reported. The CLK method is complementary to these other ChIP-based approaches, exploiting the time dependence of formaldehyde crosslinking to derive binding kinetic parameters as well as fractional occupancy (Poorey et al., 2013). The first iteration of the CLK assay used ‘standard’ crosslinking and quenching conditions (1% formaldehyde and 250 mM glycine, respectively). Additional work has recently yielded experimental conditions that increase the crosslinking rate and improve quenching of the crosslinking reaction (Zaidi et al., 2017). These new conditions have resulted in a more robust method and the ability to model and analyze crosslinking kinetic data with more reliability and confidence.
Materials and Reagents
- Pipette tips
- Reusable cotton-plug top serological (glass) pipettes (can also use single-use plastic pipettes)
10 ml pipettes (Fisher Scientific, catalog number: 13-675M )
25 ml pipettes (Fisher Scientific, catalog number: 13-675N ) - Pipette sterilizing boxes (Fisher Scientific, catalog number: 03-465 )
- Nalgene PPCO centrifuge bottles with sealing closure (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 3141-0500 )
- 50 ml conical tubes 30 x 115 mm (Corning, Falcon®, catalog number: 352070 )
- 2.0 ml microcentrifuge conical screw cap tubes (FastPrep tubes, Fisher Scientific, catalog number: 02-681-344 )
- Microcentrifuge tube screw caps with O-Rings (for FastPrep tubes, Fisher Scientific, catalog number: 02-681-366 )
- Acid washed 425-600 μm glass beads (Sigma-Aldrich, catalog number: G8772 )
- 18 gauge needle (PrecisionGlide, BD, catalog number: 305195 )
- Disposable culture tubes, glass 13 x 100 mm (Fisher Scientific, catalog number: 14-961-27 )
- 1.5 ml microcentrifuge tubes graduated (Fisher Scientific, catalog number: 05-408-129 )
- Whatman filter paper (GE Healthcare, catalog number: 1003-917 )
- Autoradiography film (Genesee Scientific, catalog number: 30-101 )
- 1.5-1.7 ml polypropylene graduated tube with locking lid (Fisher Scientific, catalog number: 02-681-285 )
- Hard-Shell High-Profile 96-well semi-skirted PCR plates (Bio-Rad Laboratories, catalog number: HSS9641 )
- Tubular roll stock (to seal membrane in plastic before developing; Ampac, catalog number: TRS-95125-3 )
- Immobilon-P membrane (PVDF; Merck, catalog number: IPVH00010 )
- Microseal ‘B’ seal seals (Bio-Rad Laboratories, catalog number: MSB1001 )
- Stericup sterile vacuum filter units, 500 ml (Merck, catalog number: SCGPU05RE )
- Yeast strains, wild type (WT) and overexpression (OE) for each TF of interest (see Poorey et al., 2013 and Zaidi et al., 2017 for strain lists)
- Plasmids for TF overexpression and vector control for WT strain (see Poorey et al., 2013 and Zaidi et al., 2017 for plasmid lists)
- Locus specific primers (Invitrogen)
- In-Fusion HD cloning kit (Takara Bio, Clontech, catalog number: 639649 )
- Ice bucket
- 37% formaldehyde (Fisher Scientific, catalog number: F79 )
- Protein Assay Dye Reagent Concentrate (Bio-Rad Laboratories, catalog number: 500-0006 )
- nProtein A Sepharose 4 Fast Flow beads (GE Healthcare, catalog number: 17-5280-01 )
Note: Ab-conjugated beads can be used instead, such as IgG Sepharose 6 Fast Flow (GE Healthcare, catalog number: 17-0969-01 ) and Sepharose 6 Fast Flow beads (GE Healthcare, catalog number: 17-0159-99 ). - QIAQuick PCR purification kit (QIAGEN, catalog number: 28106 )
- iQ SYBR Green Supermix, 500 x 50 μl rxns (Bio-Rad Laboratories, catalog number: 1708882 )
- Instant non-fat dry milk (Carnation)
- Antibody (i.e., α-TBP, monoclonal, Abcam, catalog number: ab61411 )
- Secondary ECL-conjugated antibody (we use Amersham ECL mouse or rabbit IgG HRP-linked whole Ab, GE Healthcare, catalog numbers: NXA931 or NA934V )
- Amersham ECL Prime Western blotting detecting reagent (GE Healthcare, catalog number: RPN2232 )
- Yeast extract (BD, BactoTM, catalog number: 212750 )
- Bacto peptone (BD, BactoTM, catalog number: 211677 )
- Sugar source, i.e.,:
D-(+)-Glucose (Sigma-Aldrich, catalog number: G7021 )
D-(+)-Galactose (Sigma-Aldrich, catalog number: G5388 )
D-(+)-Raffinose (MP Biomedicals, catalog number: 02102797 ) - Difco yeast nitrogen base without amino acids and ammonium sulfate (BD, DifcoTM, catalog number: 233520 )
Note: Yeast nitrogen base without amino acids (with or without sugar source) can be used as an alternative and doesn’t require the addition of ammonium sulfate (i.e., Sigma-Aldrich, catalog number: Y0626 ). - Amino acids:
- Adenine hemisulfate dihydrate (MP Biomedicals, catalog number: 02100195 )
- L-Histidine hydrochloride monohydrate (Acros Organics, catalog number: 411731000 )
- L-Lysine (Fisher Scientific, catalog number: BP386 )
- L-Tyrosine (Acros Organics, catalog number: 140641000 )
- L-Tryptophan (Fisher Scientific, catalog number: BP395 )
- Uracil (Affymetrix, catalog number: 23020 )
- L-Leucine (Acros Organics, catalog number: 125121000 )
- L-Methionine (Fisher Scientific, catalog number: BP388 )
- L-Arginine hydrochloride (Fisher Scientific, catalog number: BP372 )
- L-Serine (Fisher Scientific, catalog number: BP393 )
- Valine (Fisher Scientific, catalog number: BP397 )
- L-Threonine (MP Biomedicals, catalog number: 02103053 )
- L-Isoleucine (Fisher Scientific, catalog number: BP384 )
- L-Phenylalanine (Fisher Scientific, catalog number: BP391 )
- L-Cysteine hydrochloride monohydrate (Fisher Scientific, catalog number: BP376 )
- L-Aspartic Acid (Acros Organics, catalog number: 105041000 )
- L-Proline (Fisher Scientific, catalog number: BP392 )
- Adenine hemisulfate dihydrate (MP Biomedicals, catalog number: 02100195 )
- Bacto agar (BD, catalog number: 214010 )
- Glycine, 2 kg (Bio-Rad Laboratories, catalog number: 1610724 )
- Hydrochloric acid (Fisher Scientific, catalog number: A144SI-212 )
- Tris base (Sigma-Aldrich, catalog number: T1503 )
- Ammonium sulfate (Sigma-Aldrich, catalog number: A4418 )
- Magnesium chloride hexahydrate (Sigma-Aldrich, catalog number: M9272 )
- EDTA (Fisher Scientific, catalog number: BP120 )
- Glycerol (Fisher Scientific, catalog number: BP229 )
- β-Mercaptoethanol (Sigma-Aldrich, catalog number: M3148 )
- Protease inhibitors
cOmplete protease inhibitor tablet EDTA-free (Roche Diagnostics, catalog number: 04693132001 )
OR:
Phenylmethylsulfonyl fluoride (Sigma-Aldrich, catalog number: P7626 )
Benzamidine hydrochloride hydrate (Acros Organics, catalog number: 105241000 )
Pepstatin A (Sigma-Aldrich, catalog number: P4265 )
Leupeptin hemisulfate salt (Sigma-Aldrich, catalog number: L8511 )
Chymostatin (Sigma-Aldrich, catalog number: C7268 ) - SDS (Sigma-Aldrich, catalog number: L3771 )
- DTT (Roche Diagnostics, catalog number: 03117006001 )
- Bromophenol blue (Bio-Rad Laboratories, catalog number: 1610404 )
- Coomassie blue
- Methanol (Fisher Scientific, catalog number: A452 )
- Acetic acid
- Sodium chloride (Fisher Scientific, catalog number: S640 )
- Tween 20 (Sigma-Aldrich, catalog number: P5927 )
Note: This product has been discontinued. - HEPES (Fisher Scientific, catalog number: BP310 )
- Triton X-100 (AMRESCO, catalog number: 0694 )
- Sodium deoxycholate (Sigma-Aldrich, catalog number: D6750 )
- Lithium chloride (Sigma-Aldrich, catalog number: L4408 )
- Nonidet P40 (Spectrum, catalog number: N1156 )
Note: This product has been discontinued. - Diethyl pyrocarbonate (DEPC), 97% pure (Acros Organics, catalog number: 170250250 )
- Acrylamide (Bio-Rad Laboratories, catalog number: 1610101 )
- Bis-acrylamide (Fisher Scientific, catalog number: BP171 )
- Ammonium persulfate (APS, Bio-Rad Laboratories, catalog number: 1610700 )
- TEMED (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 17919 )
- YPD media (see Recipes)
- 30% raffinose or galactose (see Recipes)
- SC media (Yeast synthetic media, see Recipes)
- Amino acid mix (see Recipes)
- 3 M glycine quench solution (see Recipes)
- Benoit’s buffer (see Recipes)
- Laemmli buffer (4x sample buffer, see Recipes)
- Coomassie stain (see Recipes)
- TBS (see Recipes)
- TBST (see Recipes)
- 140 mM ChIP lysis buffer (see Recipes)
- 500 mM ChIP lysis buffer (see Recipes)
- LiCl wash buffer (see Recipes)
- 1x TE (see Recipes)
- ChIP elution buffer (see Recipes)
- DEPC H2O (see Recipes)
- 30%/0.8% Bis-acrylamide solution (see Recipes)
- SDS-PAGE running buffer (see Recipes)
- Transfer buffer (see Recipes)
Equipment
- Glass culture tubes and caps, 18 x 150 mm (i.e., disposable borosilicate glass tubes with plain end, Fisher Scientific, catalog number: 14-961-32 and Diamond culture tube caps, 18 mm, Globe Scientific, catalog number: 118154 )
- Flasks (1 L, 250 ml; i.e., Pyrex narrow-neck heavy-duty glass Erlenmeyer flasks, Corning, PYREX®, catalog numbers: 4980-1L (1L) and 4980-250 (250 ml))
- Pipettes (P2, P2, P200, P1000)
- Pipet-Aid (Drummond Scientific, catalog number: 4-000-100 )
- Incubator, 30 °C (i.e., Isotemp CO2 incubator, Fisher Scientific, Fisherbrand, catalog number: 11-676-603 )
- Magnetic stir bars (Fisher Scientific, catalog number: 14-513-52 )
- Stirring hot plate (Fisher Scientific, catalog numbers: 11-510-49SH or SP88850200 )
- Timer
- Sorvall RC 5B centrifuge (GMI, model: Sorvall RC-5B )
- SLA-3000 rotor (Thermo Fisher Scientific, Thermo ScientificTM, model: SLA-3000 , catalog number: 07149)
- Eppendorf 5810 R benchtop centrifuge with plate buckets and 15 ml/50 ml adapters (Eppendorf, model: 5810R , catalog number: 5811000827)
- MP FastPrep-24 Bead beater (MP Biomedicals, model: FastPrep®-24 Classic, catalog number: 116004500 )
- Bunsen burner
- Branson Sonifier 250 with 1/8” microtip probe (Fisher Scientific, catalog numbers: 22-309782 and 22-309796 )
- Eppendorf 5415C or D benchtop centrifuge (Eppendorf, similar is catalog number: 022620304 )
- Shaker, 30 °C (i.e., Eppendorf, New Brunswick Scientific, model: Excella® E25 , catalog number: M1353-0002)
- 4 °C refrigerator
- -20 °C freezer
- -80 °C freezer
- Autoclave
- AutoMixer magnetic stir plate (Fisher Scientific, catalog number: 14-505-21 )
- Ultrospec 2100pro UV/Visible spectrophotometer (Biochrom, model: ULTROSPEC 2100® , catalog number: 80-2112-21)
- PowerPac Basic power supply (Bio-Rad Laboratories, catalog number: 1645050 )
- Mini-PROTEAN tetra cell with casting stand and frames, combs, short plates, and spacer plates (Bio-Rad Laboratories, catalog number: 1658006FC )
- Mini trans-blot electrophoretic transfer cell with holder cassettes, foam pads, and blue cooling unit (Bio-Rad Laboratories, catalog number: 1703930 )
- Shaker (Reliable Scientific, model: 55S )
- Autoradiography cassette (Fisher Biotech, catalog number: FBXC 810 )
- Labquake shaker rotisserie, 32 x 10 to 19 mm with clip bar (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 415110Q )
- Water baths (55 °C and 65 °C)
- MyiQ real-time instrument (Bio-Rad Laboratories, catalog number: 170-9770 )
Software
- ImageJ (NIH)
- Mathematica
- R or R-Studio
Procedure
This version of crosslinking kinetic (CLKv2) analysis is a modified ChIP procedure that yields kinetic measurements for transcription factor binding to specific loci by fitting ChIP data obtained from cells treated with formaldehyde for different periods of time. Before the samples can be collected for kinetic analysis, control experiments should be completed to optimize experimental conditions; the schematic in Figure 1 outlines the general workflow for this process. In this protocol, the required yeast strains are described first (Procedure A), followed by the detailed procedure for CLK data collection (Procedure B). CLK data collection conditions should be validated and may need to be optimized as described in Figure 1. The control experiments used for optimizing the procedure rely on the basic CLK methodology and so are described afterward (Procedure D and E) to minimize redundancy; Procedure C details quantification of the overexpression factor needed for modeling of the data.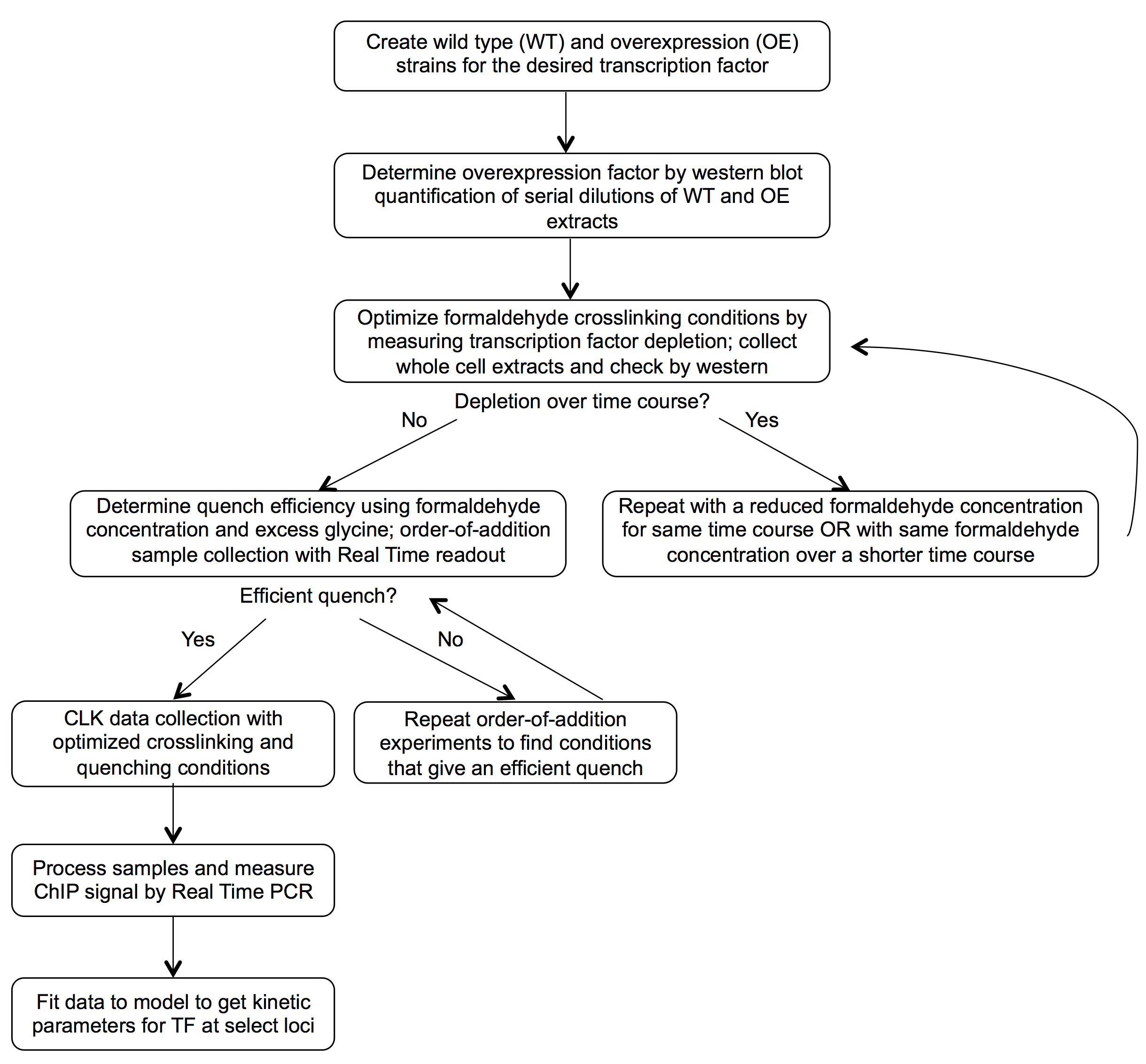
Figure 1. CLKv2 workflow. Control experiments are performed to optimize the assay conditions before collection of CLK data and fitting to the CLK model.
- Yeast strain construction
- For the CLKv2 assay, two strains are used for the analysis of a TF of interest: a wild type (WT) strain and an overexpression (OE) strain. The overexpression strain is isogenic to the WT strain other than driving levels of the TF that are modestly higher (~3-5-fold) over the WT levels. Kinetic analysis of the WT and OE strains in parallel (described below) highly constrains fits of the data by revealing the mass action contribution to the increase in ChIP signal over time. The OE strain can be engineered by introducing into cells an additional copy of the TF gene on a plasmid or by integrating into the genome under control of the native promoter or an appropriate heterologous promoter. If the TF functions as a stable biochemical entity with more than one type of subunit, the OE strain ought to be engineered to drive balanced expression of each subunit, as for example, was done for the analysis of TFIIE (Zaidi et al., 2017). For a detailed description of OE strain and plasmid construction, see Poorey et al., 2013, Zaidi et al., 2017.
- To generate strains for kinetic analysis using an OE plasmid, transform WT S. cerevisiae cells in the strain background of interest with either the OE plasmid construct or an empty vector carrying the same selection marker. Select transformants on appropriate agar plates and restreak cells for single colonies. We typically use strains kept on plates for about one month at 4 °C; archive the WT and OE strains by storage at -80 °C using standard yeast glycerol stock methods (Amberg et al., 2005).
- For the CLKv2 assay, two strains are used for the analysis of a TF of interest: a wild type (WT) strain and an overexpression (OE) strain. The overexpression strain is isogenic to the WT strain other than driving levels of the TF that are modestly higher (~3-5-fold) over the WT levels. Kinetic analysis of the WT and OE strains in parallel (described below) highly constrains fits of the data by revealing the mass action contribution to the increase in ChIP signal over time. The OE strain can be engineered by introducing into cells an additional copy of the TF gene on a plasmid or by integrating into the genome under control of the native promoter or an appropriate heterologous promoter. If the TF functions as a stable biochemical entity with more than one type of subunit, the OE strain ought to be engineered to drive balanced expression of each subunit, as for example, was done for the analysis of TFIIE (Zaidi et al., 2017). For a detailed description of OE strain and plasmid construction, see Poorey et al., 2013, Zaidi et al., 2017.
- CLK data collection
As mentioned above, the experimental conditions required for analysis of a particular TF should be validated and may need to be optimized as described in Procedure D and E. This section describes the basic CLKv2 procedure as we optimized and recently described (Zaidi et al., 2017).
Cell growth and cell sample collection- Inoculate duplicate 5 ml YPD primary cultures with a colony of the desired WT or OE yeast strains; incubate cultures overnight at 30 °C with shaking.
Note: If starting the primary cultures in selective media, cells may need to grow for ~24 h instead of ~16 h to obtain enough material to start the larger cultures in the next step. - The next day, dilute each primary culture in 450 ml of the desired selective media (SC media with appropriate amino acid drop-out) and incubate with shaking at 30 °C until the OD600 is ~0.8 (~1.3 x 107 cells). Pellet cells by spinning for 7 min at 4,230 x g and 4 °C in Nalgene centrifuge bottles using an SLA-3000 rotor and Sorvall RC 5B centrifuge.
Note: Growing conditions for copper-inducible strains are outlined in Poorey et al., 2013. If the TF of interest is under the control of a GAL-inducible promoter, the cells should be grown in selective media with 2% raffinose until OD600 is ~0.8 is reached; then pellet and resuspend the cells in YEP + 2% galactose and incubate for an hour at 30 °C with shaking. - Resuspend each cell pellet in 450 ml YPD (or desired media); incubate at 30 °C with shaking for about one hour until the OD600 is ~1.0 (~1.9 x 107 cells).
- While the cells are growing, prepare the glycine quench solution. For each time point, 440 ml of 3 M glycine pH 5 is needed. Thus, 4 L of glycine will be required to quench the eight time point samples for one time course; see Recipes for instructions. Pour 440 ml of the 3 M glycine solution into each of eight 500 ml Nalgene centrifuge bottles.
- Pellet cells as in Step 2, then resuspend cells in 90 ml YPD (or desired media). This step yields a cell suspension that is concentrated five-fold over the initial culture.
Note: The cells are concentrated in this way so that in subsequent steps quenching of formaldehyde crosslinking can be made more efficient by dilution of the cell suspension as well as by addition of glycine. - Add a 10 ml glass or plastic pipette to a Pipette-Aid; this will be used for removal of an aliquot of the cell suspension at the first time point. Place flasks with 90 ml culture on a stir plate with a stir bar on medium speed and rapidly add 14 ml 37% formaldehyde to a final concentration of 5%; immediately start the timer. One way to rapidly add formaldehyde is to invert a pre-measured 14 ml aliquot contained in a 15 ml disposable conical tube into the stirring cell suspension. If formaldehyde is added with a Pipette-Aid, then a second Pipette-Aid should be fitted with a 10 ml pipette beforehand for removal of the first time point aliquot.
- At 5 sec, 20 sec, 60 sec (1 min), 120 sec (2 min), 300 sec (5 min), 600 sec (10 min), 900 sec (15 min), and 1,200 sec (20 min) remove a 10 ml aliquot of crosslinked culture with a Pipet-Aid and 10 ml glass pipette and quench by quickly adding the cell aliquot to a bottle containing the prepared glycine solution; cap each bottle immediately after adding the cell suspension and invert a few times to mix.
- Pellet cells by spinning at 4 °C for 7 min at 4,230 x g using an SLA-3000 rotor and Sorvall RC 5B centrifuge.
- Resuspend each cell pellet in 50 ml 4 °C TBS + 300 mM glycine pH 5 and transfer to a 50 ml conical tube; spin for 5 min at 3,220 x g and 4 °C in an Eppendorf 5810 R benchtop centrifuge. Discard supernatant.
- Wash each cell pellet with 50 ml 4 °C TBS; pellet cells as in the previous step.
- Resuspend each cell pellet in 1 ml TBS at 4 °C and transfer each sample to a FastPrep tube.
- Pellet cells by spinning for 2 min at 4 °C and 16,000 x g in an Eppendorf 5810 R benchtop centrifuge. Discard supernatant.
Note: Samples can be stored at -80 °C at this step.
Isolation of fragmented chromatin samples- Resuspend each cell pellet in 600 μl 140 mM ChIP lysis buffer with protease inhibitors added. Add acid washed glass beads to each tube until just above the level of the liquid and screw cap on tightly.
- Bead-beat samples at 4.0 m/sec for 7 cycles, with 45 sec on and one minute off between cycles, using an MP FastPrep-24 Bead beater kept in the cold room.
- Poke a hole in the bottom of each FastPrep tube with a heated (using Bunsen burner flame) 18-gauge needle and then place each tube in a 13 x 100 mm glass tube. Spin for 3 min at 3,220 x g in an Eppendorf 5810 R benchtop centrifuge at 4 °C to transfer the liquid to the bottom of the glass tube.
- Briefly vortex the glass tubes containing the flow through material and transfer each cell suspension to an Eppendorf tube; place tubes on ice.
- Sonicate samples for 7 cycles with 5 pulses/cycle, 30% output, and 90% duty cycles using a Branson Sonifier 250 with microtip probe. Place tubes on dry ice between cycles.
Note: By sonicating one sample after another for each of the 7 cycles, the samples will stay on dry ice long enough to keep them cool but not freeze. It may be necessary to alternate samples between wet and dry ice to keep the pellets from freezing if many samples are sonicated at once. The expected fragment size of the sonicated DNA is ~100-600 bp. The first time the experiment is performed, the size of the sonicated DNA should be checked by running samples on a 1% agarose gel with ethidium bromide followed by imaging with an appropriate system. - Spin tubes for 5 min at 16,000 x g at 4 °C in an Eppendorf 5415C/D benchtop centrifuge.
- Remove the supernatant for each sample to a new 1.5 ml microcentrifuge tube; spin for 20 min at 16,000 x g at 4 °C in an Eppendorf 5415C/D benchtop centrifuge.
- Transfer each supernatant to a new 1.5 ml microcentrifuge tube and quantify protein with a Bradford assay using a 1 μg/μl bovine serum albumin standard and Bradford protein dye as recommended by the manufacturer of the Bradford reagent.
Note: Samples can be stored at -80 °C at this step.
Chromatin immunoprecipitation (ChIP)- Set up ChIP reactions in microcentrifuge tubes with locking lids. For each time point sample, three tubes are needed: immunoprecipitation (IP), mock, and total (input). The IP is done in one step if antibody-conjugated beads are used (a) or two steps if the chromatin extract is incubated first with the antibody followed by incubation with Protein A beads (b).
- For antibody-conjugated beads: First prepare the beads used for the IP and mock samples. The IP beads have the desired antibody conjugated to them; the mock beads are either unconjugated beads of the same type used for the IP, or alternatively, if a tag is used for the TF of interest, chromatin from an otherwise identical untagged strain can be used with the same antibody conjugated beads used for the IP. Aliquot the total volume of beads required to process all the samples (45 μl/sample) by adding to a 1.5 ml microcentrifuge tube. Wash the beads three times with 1 ml 140 mM ChIP lysis buffer, with a quick spin in between washes to recover the beads (~6,000 x g for a few seconds). Resuspend the bead pellets in an equal volume 140 mM ChIP lysis buffer with protease inhibitors to make a 50% slurry. Aliquot 40 μl of the bead slurry into one tube for each IP and mock reaction. Add 1 mg of sample chromatin to each tube on ice and add 140 mM ChIP lysis buffer with protease inhibitors to bring the total volume to 500 μl. For the input samples, aliquot 0.1 mg of the sample chromatin to each tube. Store the input samples at -80 °C; place the IP and mock reaction tubes on a rotating shaker such as the Labquake rotisserie shaker overnight at 4 °C.
- For antibody incubation followed by bead incubation: For the IP samples, aliquot 1 mg of sample chromatin protein into each tube and add antibody for the TF of interest followed by 140 mM ChIP lysis buffer with protease inhibitors to bring the total volume to 500 μl. For the mock reactions, aliquot 1 mg of sample chromatin then 140 mM ChIP lysis buffer with protease inhibitors to bring the volume to 500 μl (no antibody added). For the input samples, aliquot 0.1 mg of the sample chromatin into each tube. Store the input samples overnight at -80 °C; place the IP and mock tubes on a Labquake rotisserie shaker overnight at 4 °C.
Note: The amount of antibody added to the IP varies depending on its concentration and may need to be optimized before processing the CLK samples. Typically, ~5 μg antibody is used for ChIP with 1 mg sample chromatin protein.
- For antibody-conjugated beads: First prepare the beads used for the IP and mock samples. The IP beads have the desired antibody conjugated to them; the mock beads are either unconjugated beads of the same type used for the IP, or alternatively, if a tag is used for the TF of interest, chromatin from an otherwise identical untagged strain can be used with the same antibody conjugated beads used for the IP. Aliquot the total volume of beads required to process all the samples (45 μl/sample) by adding to a 1.5 ml microcentrifuge tube. Wash the beads three times with 1 ml 140 mM ChIP lysis buffer, with a quick spin in between washes to recover the beads (~6,000 x g for a few seconds). Resuspend the bead pellets in an equal volume 140 mM ChIP lysis buffer with protease inhibitors to make a 50% slurry. Aliquot 40 μl of the bead slurry into one tube for each IP and mock reaction. Add 1 mg of sample chromatin to each tube on ice and add 140 mM ChIP lysis buffer with protease inhibitors to bring the total volume to 500 μl. For the input samples, aliquot 0.1 mg of the sample chromatin to each tube. Store the input samples at -80 °C; place the IP and mock reaction tubes on a rotating shaker such as the Labquake rotisserie shaker overnight at 4 °C.
- Skip to the next step if using antibody-conjugated beads. Prepare a 50% slurry of nSepharose Protein A beads in the same way the beads are prepared in Step 1a (Chromatin immunoprecipitation (ChIP)). Aliquot 40 μl of the bead slurry to a new tube for each IP or mock sample. Quick spin the chromatin samples in a benchtop microfuge and transfer each supernatant to a new tube with the nSepharose Protein A bead slurry. Incubate the chromatin-bead samples for 2 h at 4 °C with mixing as in Step 1.
- Quick spin tubes. Wash each bead pellet twice with 1 ml each of the following buffers; quick spin in a microfuge between washes:
- 140 mM ChIP lysis buffer
- 500 mM ChIP lysis buffer
- LiCl wash buffer
- 1x TE pH 8
- 140 mM ChIP lysis buffer
- Remove excess liquid after the last wash and add 75 μl ChIP elution buffer to each tube. Mix beads and elution buffer by tapping the tube several times. Incubate samples at 65 °C for 10 min.
- Quick spin tubes in a microfuge and transfer the supernatant to a new locking lid tube.
- Add another 75 μl aliquot of ChIP elution buffer to each bead pellet. Mix by flicking the tube as above. Incubate samples again at 65 °C for 10 min.
- Quick spin the tubes in a microfuge and combine the supernatants in one tube. Incubate samples overnight at 65 °C.
- Thaw the input samples for a minute or two on the bench. Add 150 μl ChIP elution buffer, mix by briefly vortexing, and incubate the tubes overnight at 65 °C along with the IP and mock samples.
- The next day, clean up the DNA samples using the QiaQuick PCR purification kit by following the instructions provided by the manufacturer. Briefly, first, add 750 μl Buffer PB to each tube; mix and apply 450 μl of the sample to a column, then spin for 2 min at 9,300 x g using an Eppendorf 5415D benchtop centrifuge at room temperature.
- Apply the remainder of sample to each column and spin as in the previous step.
- Apply 750 μl Buffer PE to each column; spin as in the previous step.
- Discard the flow-through and then spin each column again to remove all residual ethanol.
- Place each column in a new microcentrifuge tube then apply 50 μl of 55 °C DEPC H2O to each column. Let the tubes sit on the bench for 1-2 min, then spin as in the previous step. Discard columns and either freeze samples at -80 °C or immediately perform real-time PCR analysis.
Real-time PCR quantitation
A standard curve is generated from serial dilutions of the input sample, which is then used to determine the ChIP levels for the IP and mock samples. The relative ChIP signal, described below, can be calculated for each time point and plotted before model fitting the data. It is critical to quantify the ChIP signal using a standard curve; using the threshold cycle number directly (for example) to quantify ChIP levels will not provide an accurate estimate of the quantity of the ChIP material unless it accounts for the log relationship with input.- First, make a dilution series of input chromatin. Thaw the input samples on the benchtop for a few minutes, then quick spin in a microfuge and place on ice. We refer to the undiluted input as ‘125x’; five-fold dilutions of this material are made to generate 25x, 5x, and 1x input samples for the real-time PCR standard curve. To make the 25x standard, take 2 μl of the undiluted input and add to 8 μl DEPC H2O. Mix, quick spin, and make a dilution of the 25x sample in the same way to yield a 5x sample. With the 5x sample, make a 1x sample in the same way. Keep dilutions of input chromatin on ice.
- Prepare the real-time PCR reaction mix by adding 10 μl iQ SYBR Green Supermix (2x) for each reaction and forward and reverse primers to a final concentration of 0.2 μM; adjust the volume to 19 μl with DEPC H2O. As described below, we run technical triplicates for each sample, and the standards are run in duplicate. Make enough mix for a control reaction containing no sample as well.
- Real-time PCR reactions are run in wells of plates designed for the instrument. Pipette 19 μl of the reaction mix into each well. There will be 17 wells required for each time point: 3 for each IP, mock, and 5x input and 2 for each of the four standards. See Figure 2 below for an example plate set-up.
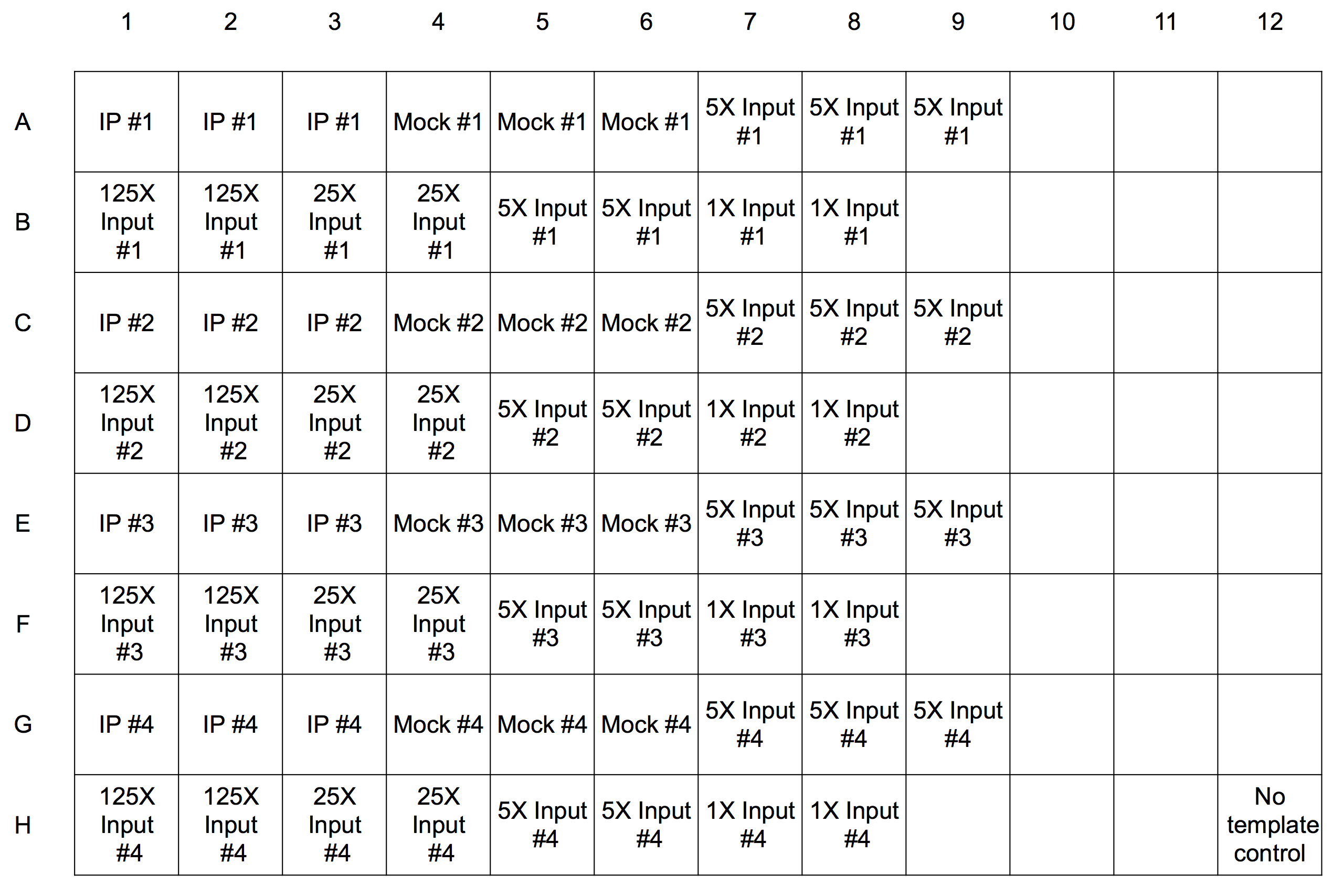
Figure 2. Template for real-time PCR plate for four time point samples. Each experimental sample is run in triplicate (rows A, C, E, and G) and each of the four standards is run in duplicate (rows B, D, F, H). The same master mix is used for each reaction and one well (H12) has no sample added as a negative control. - Thaw the IP and mock samples, then quick spin in a microfuge and place on ice. Add 1 μl of sample to each well as indicated except for the control well. We include the 5x input sample as an unknown as well as a standard; its estimated value as an unknown establishes how well the standard curve captures the ChIP signal quantitatively.
- Seal the plate with film and spin for 3 min at 1,810 x g/4 °C in an Eppendorf 5810 R benchtop centrifuge.
- Run the plate in a MyiQ or related instrument using a protocol optimized for the primer set, and with a melting curve to verify that a single product species is generated. The standard quantities are set to values of 125, 25, 5, and 1.
- To determine the relative ChIP signal at each time point, subtract the average mock signal from the average IP signal and divide by the average estimated 5x input signal. Plot the average of two data sets for each time point along with the standard deviation. The average values will be used for the next step of model fitting. A representative plot is shown in Figure 3.
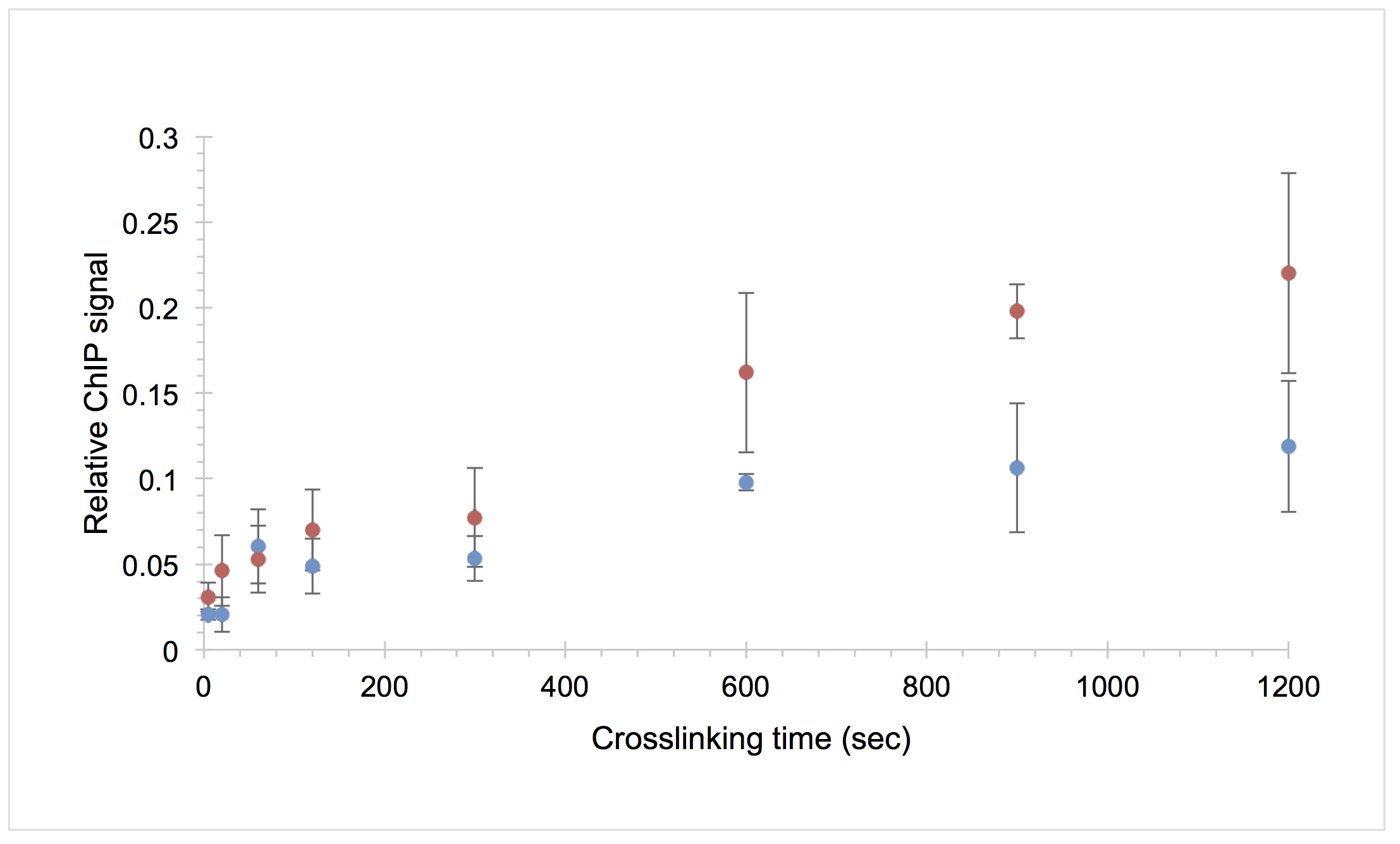
Figure 3. Example CLKv2 data for the TATA-binding protein (TBP) interaction with the URA1 locus. The average relative ChIP signal of two biological replicates is shown for the WT (blue circles) and OE (red circles) strains versus formaldehyde crosslinking time; the error bars represent the standard deviation. This is the raw data used to generate the model fit in Figure 8B in Zaidi et al., 2017.
- Inoculate duplicate 5 ml YPD primary cultures with a colony of the desired WT or OE yeast strains; incubate cultures overnight at 30 °C with shaking.
- Transcription factor overexpression value quantification
The overexpression factor is the level of the TF in the OE strain relative to the WT strain and is required for fitting the CLKv2 model to the formaldehyde incubation time-dependent ChIP data. This section describes how to estimate the overexpression factor by performing Western blots using extracts from the WT and OE strains.
Cell growth and cell sample collection
This protocol is similar to the detailed instructions in Procedure B, Cell growth and cell sample collection, with changes to the cell culture volume and wash buffers used as indicated below. In addition, the formaldehyde crosslinking and quenching steps are not necessary and therefore not performed for this section.- Inoculate each of two 5 ml YPD primary cultures with a colony of the desired WT and OE yeast strains; incubate cultures overnight at 30 °C with shaking.
- The next day, dilute each primary culture in 50 ml of the desired selective media and incubate with shaking at 30 °C until the OD600 is ~0.8. Pellet cells in 2 x 50 ml conical tubes by spinning for 5 min at 3,220 x g and 4 °C in an Eppendorf 5810 R benchtop centrifuge.
- Resuspend each cell pellet in 50 ml YPD (or desired media); incubate at 30 °C with shaking for about one hour until the OD600 is ~1.0.
- Pellet cells as in Step 2.
- Combine cell pellets for each sample and resuspend in 50 ml 4 °C TBS and transfer to a 50 ml conical tube; spin for 5 min at 3,220 x g and 4 °C. Discard supernatant.
- Wash cell pellets with 10 ml 4 °C Benoit’s buffer with protease inhibitors and β-mercaptoethanol; pellet cells as in the previous step.
- Resuspend cell pellets in 1 ml Benoit’s buffer (with protease inhibitors and β-mercaptoethanol) and transfer each sample to a FastPrep tube.
- Pellet cells by spinning for 2 min at 4 °C in an Eppendorf 5810 R benchtop centrifuge. Discard supernatant.
Note: Samples can be stored at -80 °C at this step.
Preparation of whole cell extract
This protocol is similar to the detailed instructions in Procedure B, Isolation of fragmented chromatin extract samples, with changes to the cell pellet resuspension volume, the buffer used, and the omission of sonication.- Resuspend cell pellets in 300 μl Benoit’s buffer with protease inhibitors and β-mercaptoethanol. Add acid washed glass beads to each tube until just above the liquid. Tap tubes on the lab bench a few times to remove air bubbles.
- Bead-beat samples for 7 cycles, with 45 sec on and one minute off between cycles, using an MP FastPrep-24 Bead beater kept in the cold room.
- Poke a hole in the bottom of each FastPrep tube with a heated (using Bunsen burner flame) 18-gauge needle and then place each tube in a 13 x 100 mm glass tube. Spin for 3 min at 3,220 x g at 4 °C to transfer the liquid to the bottom of the glass tube.
- Briefly vortex the glass tubes containing the flow through material and transfer each cell suspension to an Eppendorf tube.
- Place samples on ice for 30 min.
- Spin tubes for 5 min at 16,000 x g at 4 °C.
- Remove the supernatant for each sample to a new 1.5 ml microcentrifuge tube; spin for 30 min at 16,000 x g at 4 °C.
- Transfer each supernatant to a new 1.5 ml microcentrifuge tube and quantify protein with a Bradford assay as described in Procedure B.
Note: Samples can be stored at -80 °C at this step.
Quantification of TF levels by Western blotting- Pour an SDS-PAGE denaturing gel of appropriate concentration (8-14%) with 30%/0.8% Bis-acrylamide solution.
- Run the SDS-PAGE gel using, for example, a Bio-Rad mini-PROTEAN tetra cell filled to the corresponding marking with SDS-PAGE running buffer. Load a 2x serial dilution of sample for each strain on the same gel starting with 20 μg; the other amounts loaded will be 10 μg, 5 μg, and 2.5 μg. Each sample is mixed with Laemmli loading buffer and heated for 5 min at 95 °C to denature proteins before loading. Run the gel at 120 V for an appropriate amount of time to resolve polypeptide bands in the size range of interest.
Note: More blots may need to be performed with different dilutions if the factor overexpression value is high. - Transfer proteins from the gel to an Immobilon-P (PVDF) membrane overnight in transfer buffer using a Bio-Rad mini trans-blot electrophoretic system (or a related system) at 30 V and 4 °C.
Note: Visually inspect the membrane the next day before blocking to make sure the ladder has transferred onto the membrane. - The next day, block the membrane with an appropriate solution. We typically use 5% nonfat milk/TBST for 1 h on a shaker at room temperature.
- Incubate the membrane with primary antibody, typically an hour or more, and then wash the blot 4 times in TBST for 5 min per wash.
- Incubate the membrane with secondary antibody (incubation time is typically 1 h but may need to be optimized depending on the antibody), and then wash 4 times in TBST for 5 min per wash.
Note: An ECL-conjugated HRP-linked secondary antibody can be used for Western development on film. However, fluorescently labeled antibodies, such as those conjugated with Cy2/3/5, are preferred as the fluorescence intensity can be quantified directly and fluorescence imaging has a wider dynamic range than film. - Mix 500 μl of Solution A and 500 μl of Solution B from the ECL-Prime kit and spread evenly over blot; seal blot in plastic and expose to film.
Note: A fluorescent secondary antibody can also be used instead of the chemiluminescent secondary, such as provided with the ECL-Plex kit. In this case, the blot can be scanned and quantified using a Typhoon scanner or other fluorescence imaging system and a digital copy of the image is saved. - Scan the film using a standard color scanner; save images as TIFF files. As Western blots are notoriously difficult to quantify, we typically perform and quantify multiple Western blot images with multiple film exposure times. It is also a good idea to run gels with a dilution series of the extract to assess how the detection system responds to known changes in the relative level of the TF on the blot.
- Using ImageJ software, open the TIFF image and quantify the protein band(s) of interest in each lane and normalize to a loading control by drawing a box around each band with the rectangular selection tool and measuring the intensity inside each box. Determine the overexpression factor by dividing the quantified OE band by the corresponding WT band; do this for each dilution and average the values to get one OE factor. We estimate the OE value by averaging the effects observed in at least two biological replicate sets of samples.
- Inoculate each of two 5 ml YPD primary cultures with a colony of the desired WT and OE yeast strains; incubate cultures overnight at 30 °C with shaking.
- Measure the effect of formaldehyde crosslinking on the levels of the TF in the soluble protein pool
An assumption in the CLK model is that the levels of the unbound TF do not change with increasing formaldehyde incubation time (Poorey et al., 2013). Before CLK analysis can be performed, it is therefore critical to measure the TF protein levels over the experimental time course to determine whether the TF is depleted from the nuclear pool of soluble proteins. Either whole cell extract or chromatin samples can be used; we’ve found for a number of TFs that either type of extract yields very similar results (Zaidi et al., 2017). The protocols for Procedure D and E are based on the CLKv2 data collection methods in Procedure B.
Cell growth and cell sample collection
This protocol is similar to the detailed instructions in Procedure B, Cell growth and cell sample collection, with changes to the cell culture volume and wash buffers used; a smaller volume is required for this optimization experiment and a different buffer is used for whole cell extract preparation.- Inoculate each of two 5 ml YPD primary cultures with a colony of the desired WT yeast strain; incubate cultures overnight at 30 °C with shaking.
- The next day, dilute each primary culture in 300 ml of the desired selective media and incubate with shaking at 30 °C until the OD600 is ~0.8. Pellet cells by spinning for 7 min at 4,230 x g and 4 °C.
- Resuspend each cell pellet in 300 ml YPD (or desired media); incubate at 30 °C with shaking for about one hour until the OD600 is ~1.0.
- Prepare the glycine quench solution. 4 L of glycine will be required to quench the four time point samples in duplicate; see Recipes for instructions. Pour 440 ml of the 3 M glycine solution into each of 8 x 500 ml Nalgene centrifuge bottles.
- Pellet cells as in Step 2, then resuspend each pellet in 60 ml YPD (or desired media).
- Place each flask with 60 ml culture on a stir plate with a stir bar, then remove a 10 ml aliquot from each culture using a Pipet-Aid and glass pipette and quench in a bottle of 440 ml 3 M glycine for the zero minute control samples. Invert the bottles a few times to mix. Turn the stir bar speed to medium. To the remaining 50 ml culture, add 7.8 ml 37% formaldehyde, yielding a final concentration of 5%.
- At 5, 10, and 15 min (or other time points as needed), quickly remove a 10 ml aliquot of crosslinked culture and quench by adding to a 440 ml aliquot of glycine; cap and invert the bottle a few times to mix.
- Pellet cells by spinning at 4 °C for 7 min at 4,230 x g.
- Resuspend each cell pellet in 50 ml 4 °C TBS and transfer to a 50 ml conical tube; spin for 5 min at 3,220 x g and 4 °C. Discard supernatant.
- Wash cell pellets with 10 ml 4 °C Benoit’s buffer with protease inhibitors and β-mercaptoethanol; pellet cells as in the previous step.
- Resuspend cell pellets in 1 ml Benoit’s buffer (with protease inhibitors and β-mercaptoethanol) and transfer each sample to a FastPrep tube.
- Pellet cells by spinning for 2 min at 4 °C in an Eppendorf 5810 R benchtop centrifuge. Discard supernatant.
Note: Samples can be stored at -80 °C at this step.
Preparation of whole cell extract
This section of the protocol is identical to the detailed instructions in Procedure C, Preparation of whole cell extract.
Quantification of TF levels by Western blotting
This protocol is similar to the detailed instructions under Procedure C, Quantification of TF levels by Western blotting, except for the amount of sample loaded and quantification output.- Run an SDS-PAGE denaturing gel of appropriate concentration (8-14%). Load 15 μg protein for each sample after mixing with Laemmli loading buffer and heating for 5 min at 95 °C to denature proteins. Run the gel at 120 V for an appropriate amount of time.
- Transfer proteins from the gel to an Immobilon-P (PVDF) membrane overnight at 30 V and 4 °C.
- The next day, block the membrane with an appropriate solution optimized for the antibody of interest.
- Incubate the membrane with primary antibody and then wash the blot 4 times in TBST for 5 min per wash.
- Incubate the membrane with secondary antibody (incubation time is typically 1 h but may need to be optimized depending on the antibody), and then wash 4 times in TBST for 5 min per wash.
- Mix 500 μl of Solution A and 500 μl of Solution B from the ECL-Prime kit and spread evenly over blot; seal blot in plastic and expose to film.
- Scan the film.
- Using ImageJ software, open the TIFF image and quantify the protein band(s) of interest in each lane and normalize to the zero minute time point and/or a loading control. We estimate the effects of formaldehyde incubation time on soluble TF levels by averaging the effects observed in at least two biological replicate sets of samples.
Note: The relative change in protein level with crosslinking time will guide the implementation of the assay in the next steps. See Zaidi et al., 2017 for a range of results with different factors. If the protein of interest is not depleted with increasing formaldehyde incubation time, then the CLKv2 assay can be conducted using this concentration of formaldehyde and over this range of formaldehyde incubation times. Alternatively, if the protein of interest is depleted from extracts with increasing formaldehyde incubation time, the options are to test a different (reduced) concentration of formaldehyde and/or to confine the crosslinking time course to the period in which the factor is not depleted. If the formaldehyde concentration is reduced, effort should be made to determine the maximum usable concentration in order to maximize the crosslinking rate, such as treating cells with a titration of formaldehyde concentrations at a later time point to find the optimal condition.
- Inoculate each of two 5 ml YPD primary cultures with a colony of the desired WT yeast strain; incubate cultures overnight at 30 °C with shaking.
- Validate the quenching conditions
Collection of order-of-addition samples
It is important to verify that the quenching conditions quantitatively block formaldehyde crosslinking, and in this way ensure that the ChIP signals truly reflect the yield of crosslinked material following incubation with formaldehyde for a given time. We have found that the glycine quench protocol reported here effectively quenches 5% formaldehyde under the conditions described; the experiments in this section should be conducted to validate that this is true in your hands and under your specific conditions.
This protocol is similar to the detailed instructions in Procedure B, Cell growth and cell sample collection, with changes to the cell culture volumes.- Inoculate duplicate 5 ml YPD primary cultures with a colony of the desired WT yeast strain; incubate cultures overnight at 30 °C with shaking.
- The next day, dilute each primary cultures in 200 ml of the desired selective media and incubate with shaking at 30 °C until the OD600 is ~0.8. Pellet cells by spinning for 7 min at 4,230 x g and 4 °C as described above.
- Resuspend each cell pellet in 200 ml YPD (or desired media); incubate at 30 °C with shaking for about one hour until the culture OD600 is ~1.0.
- Prepare the glycine quench solution. 3 L of glycine will be required to quench the three time point samples in duplicate; see Recipes for instructions. Pour 440 ml of the 3 M glycine solution into each of 6 x 500 ml Nalgene centrifuge bottles.
- Pellet cells as in Step 2, then resuspend pellets in 60 ml YPD (or desired media). Transfer 10 ml culture into each of three 500 ml flasks.
- Place each flask on a stir plate with a stir bar on medium speed.
- Collect samples as follows:
- No formaldehyde: add the 440 ml glycine solution to one flask with 10 ml culture and mix for a few seconds; pour solution and cells back into the bottle, cap the bottle and invert a few times to mix thoroughly. This sample can remain on the bench at room temperature while the other samples are processed.
- Glycine before formaldehyde: to another flask containing a cell sample, add all 440 ml 3 M glycine pH 5 from the prepared bottle. Mix for a few seconds on a stir plate, then add 1.56 ml 37% formaldehyde (5% final concentration) and allow crosslinking to proceed for 8 min at room temperature on a stir plate. Pour the entire sample back into the bottle.
- Formaldehyde followed by glycine: to the last flask containing a cell sample, add 1.56 ml 37% formaldehyde (5% final concentration); mix by thoroughly swirling, and incubate on a stir plate for 8 min at room temperature. (The formaldehyde incubation time can be adjusted; 8 min is recommended here as it is in the regime in which the ChIP signal is typically still dependent on formaldehyde incubation time, i.e., not saturated, but incubation for several minutes is easy to handle with high temporal precision.) Add 440 ml 3 M glycine pH 5 to the sample and mix for a few minutes. Pour entire solution back into the bottle.
Note: Two sets of samples are collected, but it is easiest to collect one set of samples first (a, b, c) and then the second set if both sets are collected on the same day.
- No formaldehyde: add the 440 ml glycine solution to one flask with 10 ml culture and mix for a few seconds; pour solution and cells back into the bottle, cap the bottle and invert a few times to mix thoroughly. This sample can remain on the bench at room temperature while the other samples are processed.
- Pellet cells by centrifugation as in Step 2.
- Resuspend each cell pellet in 50 ml 4 °C TBS + 300 mM glycine pH 5 and transfer to a 50 ml conical tube; spin for 5 min at 3,220 x g and 4 °C. Discard supernatant.
- Wash each cell pellet with 50 ml 4 °C TBS; pellet cells as in the previous step.
- Resuspend each cell pellet in 1 ml TBS at 4 °C and transfer each sample to a FastPrep tube.
- Pellet cells by spinning for 2 min at 4 °C and 16,000 x g. Discard supernatant.
Note: Samples can be stored at -80 °C at this step.
Isolation of fragmented chromatin samples
This section of the protocol is identical to the detailed instructions in Procedure B, Isolation of fragmented chromatin samples.
Chromatin immunoprecipitation (ChIP)
This section of the protocol is identical to the detailed instructions in Procedure B, Chromatin immunoprecipitation (ChIP).
Real-time PCR
This protocol is identical to the detailed instructions under Procedure B, Real-time PCR quantification.
To determine the relative ChIP signal at each time point, subtract the average mock signal from the average IP signal and divide by the average estimated 5x input signal. We typically plot the results as a bar graph and perform t-tests to assess statistically significant differences in comparing different samples. With an efficient quench, the ‘glycine only’ and ‘glycine before formaldehyde’ ChIP signals should be quantitatively indistinguishable (Figure 4).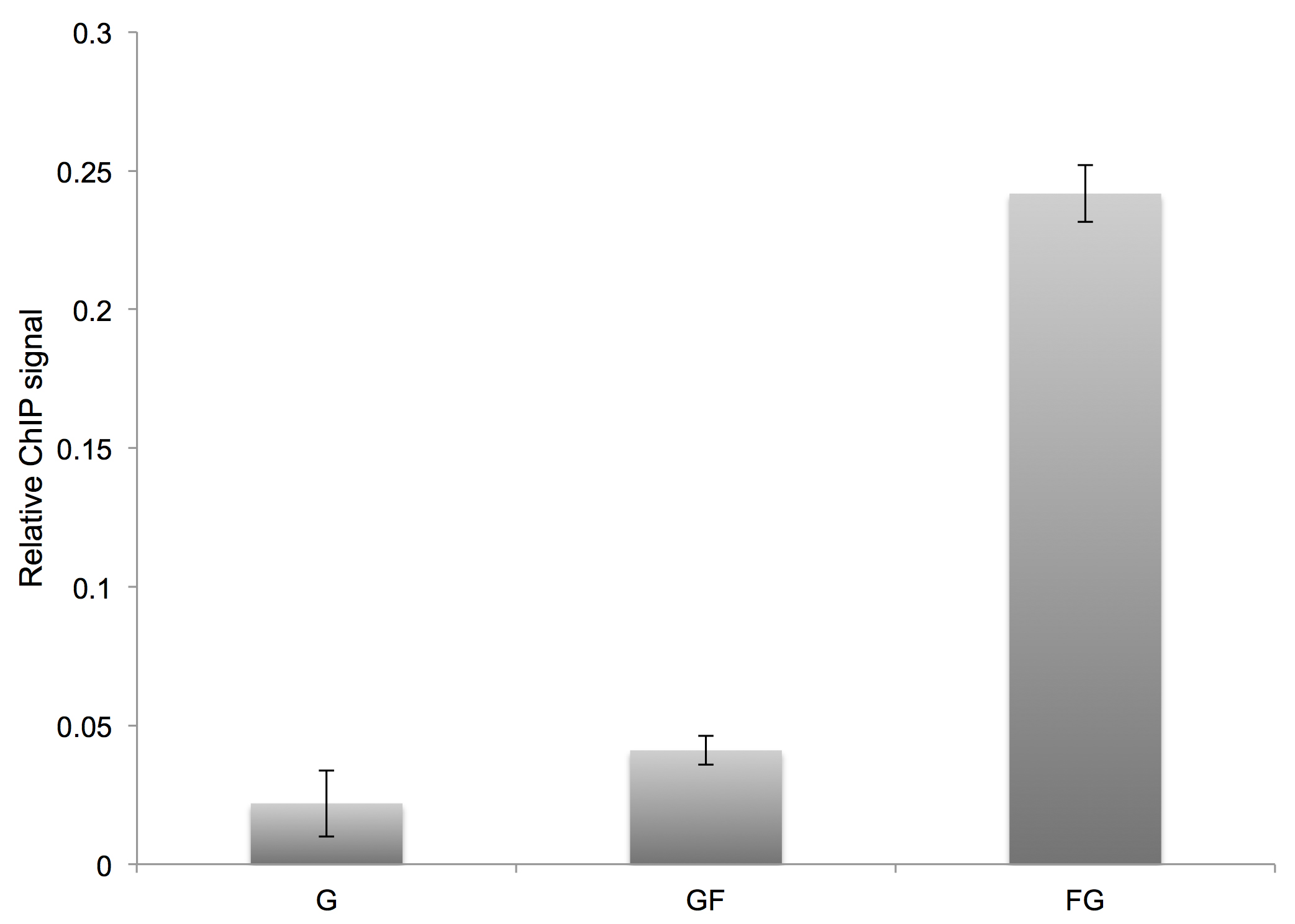
Figure 4. Representative results for an order-of-addition experiment. G = glycine only, GF = glycine quench followed by formaldehyde addition, FG = formaldehyde crosslinking followed by glycine quench. In this example, the relative ChIP signal was calculated following immunoprecipitation of the Gal4 protein and real-time PCR readout for interaction at the GAL3 locus. Error bars represent the standard deviation from two biological replicates. - Inoculate duplicate 5 ml YPD primary cultures with a colony of the desired WT yeast strain; incubate cultures overnight at 30 °C with shaking.
Data analysis
Model fitting of data
- Poorey et al. (2013, Science) developed the mass-action kinetic model that we used to analyze the data generated in that study and extract kinetic parameters. The procedure to fit the data to this model was systematically studied and re-formulated in Zaidi et al. (2017) based on the experimental regimes seen in the experimental data. In the following, we give a brief overview of the quantitative fitting procedure used to estimate kinetic parameters.
- In general, CLKv2 data can be represented by three types of fits: TF-limited, crosslink (XL)-limited, and linear XL-limited (see Figure 5). These categories are determined by the crosslinking rate (kxl*CFH), the overall on-rate (ka*CTF), and the off-rate (kd). In the TF-limited regime, ka*CTF<<kxl*CFH and kd<<kxl*CFH. On the other hand, in the XL-limited regime, ka*CTF>>kxl*CFH and kd>>kxl*CFH. The linear XL-limited regime is a sub-regime of the XL-limited model where the crosslinking rate is slow enough to be comparable to the latest crosslinking time points generated in the experiment (1,200 sec or more). We also observed data for which the on or the off-rate was comparable to the crosslinking rate, which we call the full-model regime.
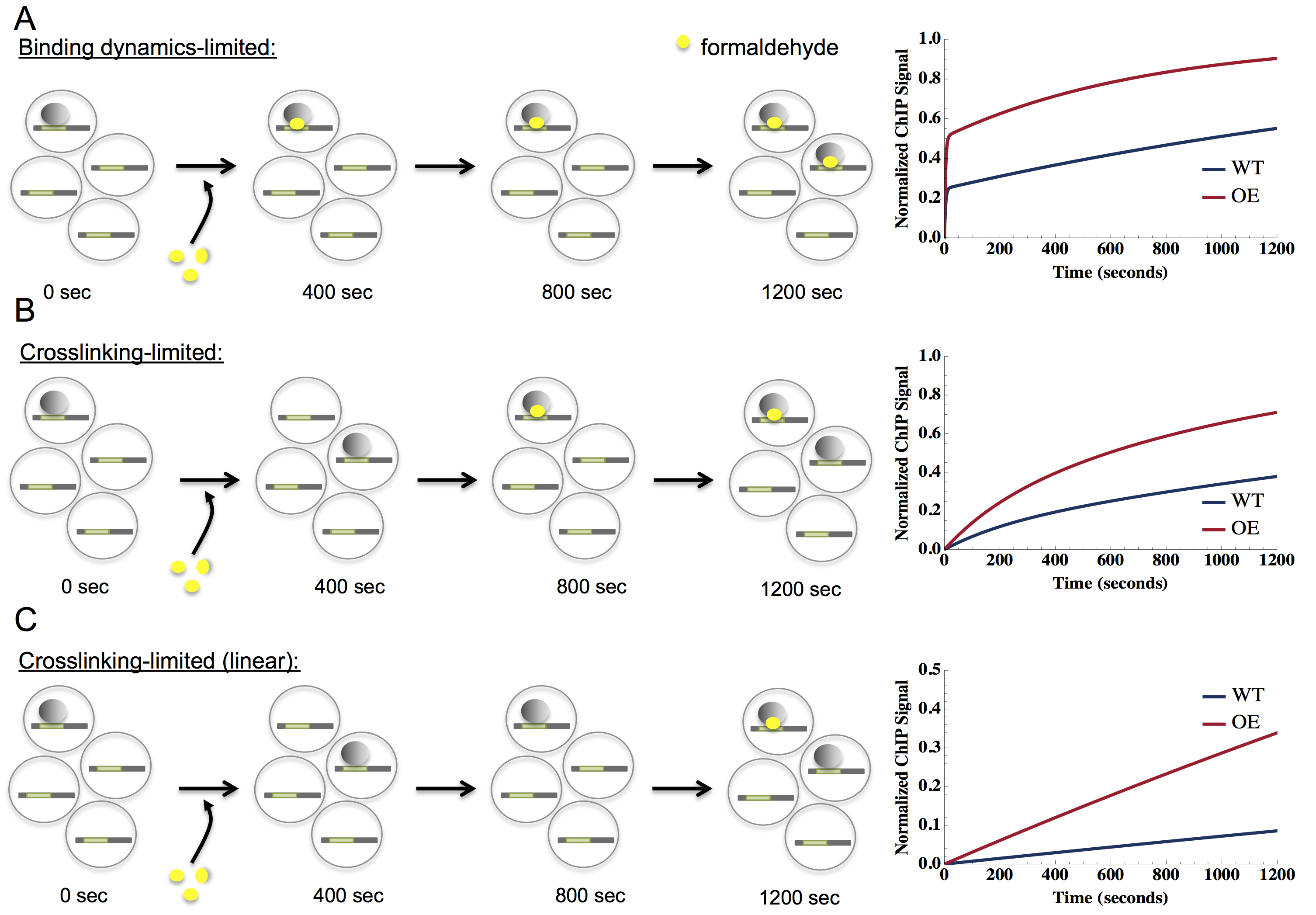
Figure 5. Schematic of potential crosslinking rates and TF-binding dynamics. CLKv2 data usually represent one of three possibilities, which are shown in panels A-C. The left side of the panel represents binding of a factor (gray shaded circles) to its binding site (denoted in green on DNA, symbolized as a thick horizontal line) in a cell (large gray circles). Four cells are shown for each time point. As time progresses from 0 to 1,200 sec, represented by black arrows, factors may become crosslinked to DNA with formaldehyde (yellow circles). The right side of the figure represents the resulting model fit for each data class. A. Data that are binding dynamics limited, where crosslinking occurs quickly (seconds) to capture bound TFs. The data are fit by a model with two distinguishable exponential phases, including an initial steep rise (often manifest as a non-zero y-intercept) followed by a shallow rise, shown in the plot on the right. B. Data that are crosslinking-limited, where crosslinking is slower than TF-binding rates (many minutes, ~600 sec in the panel). A single exponential fits the data, shown on the right with a y-intercept at zero. C. Data that are crosslinking-limited (linear), where the crosslinking rate is much slower than TF-binding rates (crosslinking time scale comparable to or greater than the longest experimental time point, 1,200 sec in the panel), is linear as a function of crosslinking time, shown on the right. - Briefly, data that falls into the TF-limited category shows a quick exponential rise in the first few data points, often appearing as a non-zero y-intercept. The initial rise merges into a second exponential with a shallower slope, eventually flattening out to a saturation level. In this regime, the crosslinking rate (kxl), on-rate (ka), and the off-rate (kd) can be determined; residence time (t1/2), occupancy (θb) and dissociation constant (Kd) can then be calculated. Data that falls into the XL-limited category manifests a single exponential starting from zero that reaches saturation; in this case, crosslinking is slower than TF-binding dynamics. In this regime, ka and kd cannot be independently estimated and only Kd, kxl, and θb are obtained. For both the TF-limited and XL-limited data, we can estimate the saturation of the ChIP signal, which we use to normalize the data such that the late time data saturates around one, in line with the expectation that the crosslinked fraction of sites in a CLK experiment would go to one for long crosslinking times. The linear XL-limited plot is best approximated by a straight line from zero, consistent with the theoretical model for very slow crosslinking. The saturation level cannot be estimated, as the crosslinking rate is much slower than the TF-dynamics with no apparent saturation within the time scale of the experiment. In fact, the crosslinking time scale is greater than that of the last crosslinking time point in the linear XL-limited regime. In this regime, only Kd and θb can be estimated. For XL-limited fits, it is possible in some cases to put an approximate lower bound on ka and kd based on the crosslinking rate, kxl*CFH. Finally, we fit the full model to the data that did not unambiguously fall into any of the above three categories.
- Error in the extracted parameters was estimated by refitting the model to 1,000 iterations of simulated data for each locus. The simulated data for each crosslinking time point was sampled from a Gaussian distribution with the mean given by the numerical fit and the variance given by the mean of the squared residual between the experimental data and the fit.
- An F-test can be used for selecting between different fits when the data does not unambiguously fit into TF-limited or XL-limited regimes. In most cases considered in Zaidi et al., 2017, the simpler comparison of residuals (or adjusted R-squared) was sufficient to select the better fit. One feature that distinguishes the XL-limited regime from the TF-limited regime is the dependence of the former on the concentration of formaldehyde used for crosslinking. Seeing a dependence of the ChIP signal on formaldehyde concentration by repeating the experiment with a different concentration than used in the original experiment corroborates XL-limited dynamics. In addition to numerical error estimates, we assessed the significance of the fits by estimating p-values. For more details, see Zaidi et al., 2017.
- Model fitting routines were programmed in Mathematica for the TF-limited, XL-limited, and full model fits, and in R for the XL-limited linear regime. Numerical parameter errors were also estimated in Mathematica. Custom fitting and error analysis scripts developed specifically for the CLKv2 analysis reported in Zaidi et al., 2017, are available upon request.
Notes
This protocol was developed with Saccharomyces cerevisiae cells using sterile conditions for cell growth. Sterile technique is not required once cell samples have been obtained for crosslinking.
Recipes
- YPD media (1 L)
10 g Bacto yeast extract
20 g Bacto peptone
20 g dextrose (glucose)
Add all components to ddH2O with final volume of 1 L
Autoclave and store at room temperature
Note: For galactose or raffinose media, substitute for dextrose at the same concentration (2%). However, these components should not be autoclaved and 66.7 ml is added from 30% liquid stock after autoclaving (see Recipe 2). - 30% raffinose or galactose (100 ml)
30 g sugar source
Bring volume up to 100 ml with ddH2O in a glass bottle
Microwave bottle for ~30 sec, then place on a stir plate with stir bar on medium speed until sugar is dissolved
Filter sterilize and store at room temperature - SC media (yeast synthetic media) (1 L)
1.2 g yeast nitrogen base without amino acids and ammonium sulfate
5 g ammonium sulfate
1 g amino acid (dropout) mix
Add all components to ddH2O with final volume of 950 ml
Adjust pH to 7 with NaOH, then autoclave. After autoclaving, cool then add sugar to 2% and add back any amino acids that have been left out if using a dropout mix
Store at room temperature
Note: Yeast nitrogen base without amino acids and yeast synthetic drop-out medium supplements are both commercially available and can be used to simplify this recipe. - Amino acid mix
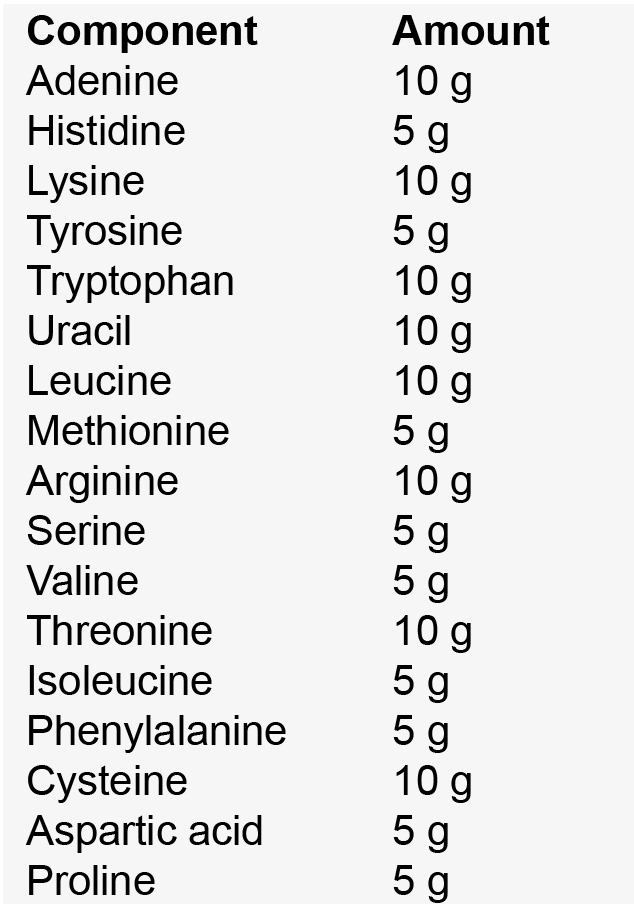
Place all components in a mortar and pestle; grind well then distribute into sterile 50 ml conical tubes
Store at room temperature
Note: To make a dropout mix (for use in selective media), leave out the desired components. - 3 M glycine quench solution
For each 1 L of solution:
225.21 g glycine
~750 ml ddH2O
Place water in a glass beaker on a heated stir plate. Turn stir bar to medium speed and add glycine. 3 M is close to the max solubility of glycine in water, so low heat with mixing is required for about an hour to completely dissolve the glycine. Once glycine is completely dissolved, let it cool and adjust pH to 5 using concentrated HCl. Bring the volume to 1 L with ddH2O. Keep glycine at room temperature once made; cooling it will cause the glycine to precipitate out of solution. If this happens, place glycine back on the heated stir plate with a low heat and stirring until dissolved again - Benoit’s buffer
200 mM Tris-HCl (pH 8.0)
400 mM (NH4)2SO4
10 mM MgCl2
1 mM EDTA
10% glycerol
7 mM β-mercaptoethanol
Protease inhibitors: Roche Complete Protease Inhibitor Cocktail Tablet OR 1.0 mM phenylmethylsulfonyl fluoride*
2.0 mM benzamidine*
2.0 μM pepstatin*
0.6 μM leupeptin*
2.0 μg of chymostatin*
*Note: Per ml of buffer.
Add Tris-glycerol components to ddH2O with desired final volume (usually make 500 ml at a time)
Adjust pH to 8 and filter sterilize. Store at 4 °C
Take out an aliquot of buffer for experiments. Add β-mercaptoethanol and protease inhibitors. Once β-mercaptoethanol is added, that aliquot can only be used for the day and must be remade fresh the next time it’s needed. Store the aliquot on ice/at 4 °C while using
Note: If the protease inhibitor tablet is used, 25 ml buffer needs a ½ tablet and 50 ml takes a whole tablet. - Laemmli buffer (4x sample buffer)
0.2 M Tris-HCl pH 6.8
40% glycerol
277 mM SDS
200 mM DTT
3 mM bromophenol blue
Add all components to ddH2O; aliquot into 1.5 ml microcentrifuge tubes and store at -20 °C - Coomassie stain
1.2 g Coomassie blue
300 ml methanol
60 ml acetic acid
240 ml H2O
Add all components; store at room temperature - TBS
50 mM Tris-HCl pH 7.5
300 mM NaCl
Add all components to ddH2O and bring to the desired volume (usually make 20 L at a time in a carboy)
Store at room temperature - TBST
50 mM Tris-HCl pH 7.5
300 mM NaCl
0.05% Tween 20
Add all components to ddH2O and bring to the desired volume (usually make 20 L at a time in a carboy)
Store at room temperature - 140 mM ChIP lysis buffer
50 mM HEPES pH 7.5
140 mM NaCl
1% Triton X-100
0.1% sodium deoxycholate
Protease inhibitors: Roche Complete Protease Inhibitor Cocktail Tablet OR 1.0 mM phenylmethylsulfonyl fluoride*
2.0 mM benzamidine*
2.0 μM pepstatin*
0.6 μM leupeptin*
2.0 μg of chymostatin*
*Note: Per ml of buffer.
Add all components except protease inhibitors to ddH2O with desired final volume (usually 500 ml at a time)
Filter sterilize the solution and store at room temperature
Take out an aliquot of buffer for experiments. Add protease inhibitors. Aliquot can be used the same day that protease inhibitors are added but a new aliquot must be remade fresh the next time it’s needed. Store the aliquot on ice/at 4 °C while using
Note: If the protease inhibitor tablet is used, 25 ml buffer needs a ½ tablet and 50 ml takes a whole tablet. - 500 mM ChIP lysis buffer
50 mM HEPES pH 7.5
500 mM NaCl
1% Triton X-100
0.1% sodium deoxycholate
Add all components to ddH2O with desired final volume (usually make 500 ml at a time)
Filter sterilize. Store at room temperature - LiCl wash buffer
10 mM Tris pH 8.0
250 mM LiCl
0.5% NP-40
0.5% sodium deoxycholate
1 mM EDTA
Add all components to ddH2O with desired final volume (usually make 500 ml at a time)
Filter sterilize. Store at room temperature - 1x TE
10 mM Tris-HCl pH 8.0
1 mM EDTA
Add all components to ddH2O with desired final volume (usually make 500 ml at a time)
Filter sterilize. Store at room temperature - ChIP elution buffer
50 mM Tris-HCl pH 8.0
1% SDS
10 mM EDTA
Add all components to ddH2O with desired final volume (usually make 10 ml at a time)
Store at room temperature. Solution should be remade each time a new experiment is done - DEPC H2O
1 ml diethyl pyrocarbonate
1,000 ml H2O
Mix DEPC with H2O in a screw cap bottle; incubate at room temperature in a hood for ~2 h with occasional stirring
Aliquot into 100 ml bottles, then autoclave
Store at room temperature for up to 12 months - 30%/0.8% Bis-acrylamide solution
For 500 ml:
150 g acrylamide
4 g Bis-acrylamide
Add all components to ddH2O with desired final volume (usually make 500 ml at a time)
Filter sterilize
Store at 4 °C in a foil-wrapped bottle - SDS-PAGE running buffer
192 mM glycine
25 mM Tris
0.1% SDS
Add all components to ddH2O and bring to the desired volume (usually make 20 L at a time in a carboy)
Store at room temperature - Transfer buffer
25 mM Tris
192 mM glycine
15% methanol
Add all components to ddH2O and bring to the desired volume (usually make 20 L at a time in a carboy)
Store at room temperature
Acknowledgments
This work was supported by grants from the National Institutes of Health (R01 GM55763 to D.T.A, R21 GM110380 to S.B and D.T.A, and NCI Cancer Training Grant T32 CA009109-38 to E.A.H.) and a Wagner Fellowship to E.A.H. The protocols in this manuscript have been adapted from those reported in Poorey et al., 2013, Viswanathan et al., 2014 and Zaidi et al., 2017. The authors declare that they have no conflicts of interest or competing interests with the contents of this article.
References
- Amberg, D. C., Burke, D., and Strathern, J. N. (2005). Methods in Yeast Genetics: A Cold Spring Harbor laboratory course manual. Cold Spring Harbor Laboratory Press.
- Encode Consortium, E. P. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489(7414): 57-74.
- Conaway, R. C. and Conaway, J. W. (1993). General initiation factors for RNA polymerase II. Annu Rev Biochem 62: 161-190.
- Coulon, A., Chow, C. C., Singer, R. H. and Larson, D. R. (2013). Eukaryotic transcriptional dynamics: from single molecules to cell populations. Nat Rev Genet 14(8): 572-584.
- Cramer, P. (2014). A tale of chromatin and transcription in 100 structures. Cell 159(5): 985-994.
- Dowen, J. M., Fan, Z. P., Hnisz, D., Ren, G., Abraham, B. J., Zhang, L. N., Weintraub, A. S., Schujiers, J., Lee, T. I., Zhao, K. and Young, R. A. (2014). Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell 159(2): 374-387.
- Grimaldi, Y., Ferrari, P. and Strubin, M. (2014). Independent RNA polymerase II preinitiation complex dynamics and nucleosome turnover at promoter sites in vivo. Genome Res 24(1): 117-124.
- Hager, G. L., McNally, J. G. and Misteli, T. (2009). Transcription dynamics. Mol Cell 35(6): 741-753.
- Haruki, H., Nishikawa, J. and Laemmli, U. K. (2008). The anchor-away technique: Rapid, conditional establishment of yeast mutant phenotypes. Mol Cell 31(6): 925-932.
- He, Y., Fang, J., Taatjes, D. J. and Nogales, E. (2013). Structural visualization of key steps in human transcription initiation. Nature 495(7442): 481-486.
- Horn, A. E., Kugel, J. F. and Goodrich, J. A. (2016). Single molecule microscopy reveals mechanistic insight into RNA polymerase II preinitiation complex assembly and transcriptional activity. Nucleic Acids Res 44(15): 7132-7143.
- Kim, T. H., Barrera, L. O., Zheng, M., Qu, C., Singer, M. A., Richmond, T. A., Wu, Y., Green, R. D. and Ren, B. (2005). A high-resolution map of active promoters in the human genome. Nature 436(7052): 876-880.
- Larson, D. R., Singer, R. H. and Zenklusen, D. (2009). A single molecule view of gene expression. Trends Cell Biol 19(11): 630-637.
- Lickwar, C. R., Mueller, F. and Lieb, J. D. (2013). Genome-wide measurement of protein-DNA binding dynamics using competition ChIP. Nat Protoc 8(7): 1337-1353.
- Luse, D. S. (2014). The RNA polymerase II preinitiation complex. Through what pathway is the complex assembled? Transcription 5(1): e27050.
- Morisaki, T., Muller, W. G., Golob, N., Mazza, D. and McNally, J. G. (2014). Single-molecule analysis of transcription factor binding at transcription sites in live cells. Nat Commun 5: 4456.
- Mueller, F., Stasevich, T. J., Mazza, D. and McNally, J. G. (2013). Quantifying transcription factor kinetics: at work or at play? Crit Rev Biochem Mol Biol 48(5): 492-514.
- Poorey, K., Viswanathan, R., Carver, M. N., Karpova, T. S., Cirimotich, S. M., McNally, J. G., Bekiranov, S. and Auble, D. T. (2013). Measuring chromatin interaction dynamics on the second time scale at single-copy genes. Science 342(6156): 369-372.
- Rhee, H. S. and Pugh, B. F. (2012). Genome-wide structure and organization of eukaryotic pre-initiation complexes. Nature 483(7389): 295-301.
- Roeder, R. G. (1996). The role of general initiation factors in transcription by RNA polymerase II. Trends Biochem Sci 21(9): 327-335.
- van Werven, F. J., van Teeffelen, H. A., Holstege, F. C. and Timmers, H. T. (2009). Distinct promoter dynamics of the basal transcription factor TBP across the yeast genome. Nat Struct Mol Biol 16(10): 1043-1048.
- Viswanathan, R., Hoffman, E. A., Shetty, S. J., Bekiranov, S. and Auble, D. T. (2014). Analysis of chromatin binding dynamics using the crosslinking kinetics (CLK) method. Methods 70(2-3): 97-107.
- Zaidi, H., Hoffman, E. A., Shetty, S. J., Bekiranov, S. and Auble, D. T. (2017). Second-generation method for analysis of chromatin binding with formaldehyde-cross-linking kinetics. J Biol Chem 292(47): 19338-19355.
- Zawel, L. and Reinberg, D. (1992). Advances in RNA polymerase II transcription. Curr Opin Cell Biol 4(3): 488-495.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Hoffman, E. A., Zaidi, H., Shetty, S. J., Bekiranov, S. and Auble, D. T. (2018). An Improved Method for Measuring Chromatin-binding Dynamics Using Time-dependent Formaldehyde Crosslinking. Bio-protocol 8(4): e2905. DOI: 10.21769/BioProtoc.2905.
- Zaidi, H., Hoffman, E. A., Shetty, S. J., Bekiranov, S. and Auble, D. T. (2017). Second-generation method for analysis of chromatin binding with formaldehyde-cross-linking kinetics. J Biol Chem 292(47): 19338-19355.
Category
Molecular Biology > DNA > DNA-protein interaction
Biochemistry > Protein > Interaction > Crosslinking
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.