- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

ChIP-seq Experiment and Data Analysis in the Cyanobacterium Synechocystis sp. PCC 6803



Published: Vol 8, Iss 12, Jun 20, 2018 DOI: 10.21769/BioProtoc.2895 Views: 13507
Reviewed by: Modesto Redrejo-RodriguezAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Oct 2017

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Nitrogen is an essential nutrient for all living organisms. In cyanobacteria, a group of oxygenic photosynthetic bacteria, nitrogen homeostasis is maintained by an intricate regulatory network around the transcription factor NtcA. Although mechanisms controlling NtcA activity appear to be well understood, the sets of genes under its control (i.e., its regulon) remain poorly defined. In this protocol, we describe the procedure for chromatin immunoprecipitation using NtcA antibodies, followed by DNA sequencing analysis (ChIP-seq) during early acclimation to nitrogen starvation in the cyanobacterium Synechocystis sp. PCC 6803 (hereafter Synechocystis). This protocol can be extended to analyze any DNA-binding protein in cyanobacteria for which suitable antibodies exist.
Keywords: ChIP-seqBackground
To maintain homeostasis, bacteria frequently need to adjust gene expression in response to environmental changes. Many of these adjustments are controlled by transcriptional factors (TF) that sense metabolic signals and activate or repress target genes. However, reflecting the traditionally laborious tasks necessary to characterize the activity and scope of TFs in vivo, our knowledge of their binding sites in bacteria is still limited. Only recently, the combination of chromatin immunoprecipitation with high-throughput sequencing analysis has opened the door to rapid determination of genome-level regulons. In particular, ChIP-seq uses the capacity of next-generation sequencing (NGS) to identify numerous DNA sequences in parallel. An attractive feature of ChIP-seq, compared to microarrays, is that there is no restriction to certain regions, such as promoter sequences, and the whole genome can be investigated for TF binding sites.
In cyanobacteria, the global regulator for nitrogen assimilation and metabolism is NtcA, a TF belonging to the CRP (cAMP receptor protein) family (Herrero et al., 2001). In Synechocystis, NtcA controls the cellular response to nitrogen availability by binding as a dimer to the promotor or intragenic regions of its target genes containing the consensus sequence GTAN8TAC (Herrero et al., 2001; Giner-Lamia et al., 2017). In the absence of ammonium, NtcA activates the expression of genes for nitrogen assimilation pathways but also acts as a transcriptional repressor of other genes, such as gifA and gifB, which encode for the glutamine synthetase inactivating factors IF7 and IF17 (García-Domínguez et al., 2000).
The protocol detailed herein has been optimized for immunoprecipitation of DNA from Synechocystis cells using antibodies against NtcA, followed by NGS to identify the specific binding sites of NtcA during early acclimation to nitrogen depletion. Following this protocol, we identified 192 genomic regions bound by NtcA (51 in ammonium-replete conditions and 141 after 4 h of nitrogen starvation) (Giner-Lamia et al., 2017). This protocol can be extended to study other TFs in cyanobacteria. Although the bioinformatic component is applicable to any sequenced prokaryote, the wet-lab component needs to be optimized to ensure efficient DNA extraction.
Materials and Reagents
- 2 ml screw-cap conical tubes (Thermo Fisher Scientific, catalog number: 3462 )
- Glass beads, acids-washed 425-600 µm (Sigma-Aldrich, catalog number: G8772-10G )
- 0.5 ml PCR tubes (Eppendorf, catalog number: 0030124537 )
- 1.5 ml tubes (Eppendorf, catalog number: 022363204 )
- 15 and 50 ml FalconTM tubes (Corning, catalog numbers: 352070 )
- DynaMagTM-2 Magnet (Thermo Fisher Scientific, catalog number: 12321D )
- Synechocystis sp. PCC 6803 cells grown on a plate of BG110C-agar (Stanier et al., 1971)
- NH4Cl (Sigma-Aldrich, catalog number: 254134 )
- TES (Sigma-Aldrich, catalog number: T1375 )
- 37% Formaldehyde (Sigma-Aldrich, catalog number: F8775 )
- Glycine (Sigma-Aldrich, catalog number: 50046 )
- NaCl (Sigma-Aldrich, catalog number: S7653-250G )
- EDTA (Sigma-Aldrich, catalog number: E9884 )
- Agarose (NZYTech, catalog number: MB02702 )
- Triton X-100 (Sigma-Aldrich, catalog number: T8787 )
- Sodium deoxycholate (Sigma-Aldrich, catalog number: 30970 )
- Protease inhibitor cocktail tablets SIGMAFAST (Sigma-Aldrich, catalog number: S8820-2TAB )
- NP-40 (Sigma-Aldrich, catalog number: 74385 )
- LiCl (Sigma-Aldrich, catalog number: L9650 )
- Anti-NtcA antibody (Giner-Lamia et al., 2017)
- SDS (Sigma-Aldrich, catalog number: L3771 )
- BSA (Sigma-Aldrich, catalog number: B4287 )
- DNase-free RNase A solution (Thermo Fisher Scientific, catalog number: EN0531 )
- Proteinase K (Thermo Fisher Scientific, catalog number: 25530049 )
- Phenol:Chloroform:Isoamyl alcohol (25:24:1) (Sigma-Aldrich, catalog number: P2069 )
- CaCl2 (Sigma-Aldrich, catalog number: 449709 )
- Glycerol (Sigma-Aldrich, catalog number: G5516 )
- MnCl2·4H2O (Sigma-Aldrich, catalog number: 221279 )
- ZnSO4·7H2O (Sigma-Aldrich, catalog number: Z1001 )
- Na2MoO4·2H2O (Sigma-Aldrich, catalog number: 331058 )
- CuSO4 (PubChem, catalog number: 24462 )
- Co(NO3)2·6H2O (Sigma-Aldrich, catalog number: 239267 )
- MgSO4·7H2O (Sigma-Aldrich, catalog number: 63138 )
- CaCl2·2H2O (Sigma-Aldrich, catalog number: 223506 )
- Citric acid (Sigma-Aldrich, catalog number: 251275 )
- Na2-EDTA (Sigma-Aldrich, catalog number: 27285 )
- Na2CO3 (Sigma-Aldrich, catalog number: S1641 )
- Fe-NH4 citrate (Sigma-Aldrich, catalog number: F5879 )
- Boric acid, H3BO3 (Sigma-Aldrich, catalog number: B6768 )
- 100% freezer-cold ethanol
- MiniElute PCR purification kit (QIAGEN, catalog number: 28004 )
- dsDNA assay kit (Thermo Fisher Scientific, catalog number: Q32851 )
- SsoFastTM EvaGreen® Supermix (Bio-Rad Laboratories, catalog number: 172-5200 )
- PearceTM Protein G Magnetic Beads (Thermo Fisher Scientific, catalog number: 88847 )
- Bradford Protein Assay (Bio-Rad Laboratories, catalog number: 5000001 )
- 5x Tris-buffered saline (TBS) buffer (see Recipes)
- Lysis buffer (see Recipes)
- Block solution (see Recipes)
- Wash buffer 1 (see Recipes)
- Wash buffer 2 (see Recipes)
- 5x IP solution (see Recipes)
- Tris-EDTA (TE) + NaCl Solution (see Recipes)
- Proteinase K solution (see Recipes)
- Trace metal mix A5 (see Recipes)
- Autoclaved BG110C medium liquid (see Recipes) (Stanier et al., 1971)
- Autoclaved BG110C+NH4 medium liquid (see Recipes) (Stanier et al., 1971)
Equipment
- Micropipettes (1,000, 100, 20 and 10 µl)
- 2 L flask and 2 x 1 L flask
- Orbital Shaker (VWR, model: 3600 )
- FastPrep-24 instrument (MP Biomedicals, catalog number: 116004500 )
- Eppendorf Thermomixer R Mixer, 1.5 ml Block (Eppendorf, model: ThermoMixer® R , catalog number: 5355)
- Eppendorf MiniSpin plus® (Eppendorf, model: MiniSpin plus® )
- Eppendorf centrifuge Falcon (Eppendorf, model: 5810R )
- MyCyclerTM Thermal Cycler System (Bio-Rad Laboratories, catalog number: 1709703 )
- Sonicator ultrasonic Processor XL (QSonica, model: XL-2020 )
- Quibit® 2.0 Fluorometer (Thermo Fisher Scientific, model: Quibit® 2.0 )
- CFX Connect Real-Time PCR Detection System (Bio-Rad Laboratories, catalog number: 1855201 )
- HiSeqTM 2000 Sequencing System (Illumina)
- Personal computer with a minimum of 2 GB of RAM and 2 GHz dual-core processor, a minimum of 25-50 GB of hard-drive space
Software
- FastQC (v0.11.5)
(https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) - Bowtie2 (v2.3.4.1)
(http://bowtie-bio.sourceforge.net/bowtie2/index.shtml [Langmead and Salzberg, 2012]) - Samtools (v1.7)
(http://samtools.sourceforge.net [Li et al., 2009]) - DeepTools (v2.0)
(http://deeptools.readthedocs.io/en/latest/content/changelog.html [Ramírez et al., 2016]) - Model-based Analysis of ChIP-Seq (MACS) (v1.4.1)
http://liulab.dfci.harvard.edu/MACS/ [Zhang et al., 2008]) - BayesPeak (v1.22.0)
(http://bioconductor.org/packages/release/bioc/html/BayesPeak.html [Spyrou et al., 2009]) - Integrative Genomics Viewer (IGV) (v2.3)
(http://software.broadinstitute.org/software/igv/ [Robinson et al., 2011]) - ChIPseeker (v1.6.7)
(https://bioconductor.org/packages/release/bioc/html/ChIPseeker.html [Yu et al., 2015])
Procedure
- Preparation of whole-cell extracts for ChIP analysis (Figure 1)

Figure 1. Flowchart showing steps in the ChIP experiment. Transcriptional factor (TF), Antibody (AB).- Start a 50-ml preculture of Synechocystis cells at 0.5 µg Chl/ml from a fresh plate (less than 2 weeks old) in liquid BG110C-NH4 medium at 30 °C under constant illumination (45 µmol photons/m2 sec) on a rotatory shaker.
- Using the preculture (2-3 µg Chl/ml) inoculate a 2 L flask with 500 ml of BG110C-NH4 and continue its incubation under the same conditions until cells reach a chlorophyll concentration of 3-4 µg/ml.
- Split the culture between two autoclaved centrifuge bottles, each containing 250 ml of the original culture for ammonium (NH4+) and nitrogen depletion (-N) treatments. Spin down the cells at 5,000 x g at room temperature for 5 min and discard the supernatants. Wash the pellets twice with 250 ml of BG110C-NH4+ for NH4+, and BG110C for -N treatments. Resuspend the pellets in 250 ml of the corresponding media and transfer the cultures to two 1 L flasks. The cultures were grown as above for 4 h.
- Add 6.75 ml of 37% formaldehyde to both cultures (NH4+ and -N) to reach a final concentration of 1% formaldehyde (for cross-linking). Incubate for 15 min at room temperature with occasional gentle shaking.
- Stop the cross-linking reaction by adding 12.5 ml of 2.5 M glycine to obtain a final concentration of 125 mM and incubate at room temperature for 5 min with occasional gentle shaking.
- Pass the cultures to two autoclaved centrifuge bottles and spin down the cells at 4 °C for 5 min at 5,000 x g. Discard the supernatant (containing formaldehyde) into a suitable waste container. Wash the pellet twice with 10 ml of cold TBS buffer.
- Spin down samples at 4 °C for 5 min at 5,000 x g and discard the supernatant. For each centrifuge bottle, resuspend the cell pellets in 2 ml of cold TBS buffer and distribute the suspension into two screw-cap tubes of 2 ml. Finally, spin down samples again and remove the remaining supernatant with a micropipette. You should have two screw-cap tubes for each treatment (NH4+ and -N)
- Optional: cell pellets can be snap-frozen in liquid nitrogen at this point and stored at -80 °C.
- Start a 50-ml preculture of Synechocystis cells at 0.5 µg Chl/ml from a fresh plate (less than 2 weeks old) in liquid BG110C-NH4 medium at 30 °C under constant illumination (45 µmol photons/m2 sec) on a rotatory shaker.
- Cell Lysis
Note: If tubes with cross-linked cells were stored at -80 °C, it is important to thaw the cell pellets on ice before continuing.- Put the tubes containing cell pellets on ice and resuspend the cells in 500 µl of Lysis buffer (pre-cooled at 4 °C).
- Add 0.5 g of acid-washed glass beads and break cells using 10 bead-beating cycles of 1 min in a FastPrep-24, with 1 min on ice between cycles.
- Spin the tubes at 4,000 x g for 2 min and, carefully collect 90% of the supernatant (lysate) using a micropipette. Mix all collected lysate (approx. 2.7 ml) and divide it into 2 tubes of 2 ml (approx. 1.35 ml per tube).
Note: To avoid contamination of the samples with unbroken cells and glass beads, leave behind 10% of the supernatant. - Sonicate the lysate, 15 cycles (10 sec at 10% amplitude, with 40 sec on ice between cycles) to fragment chromosomal DNA into sequences of sizes between 200 and 400 bp.
Note: This step is critical to retrieve good quality DNA fragments. Duration of sonication and signal amplitude must be adjusted for each apparatus to avoid low or excessive DNA shearing. To optimize this step, we recommend replicating our settings to shear freshly extracted genomic DNA (to avoid wasting valuable sample). Then, load part of the sheared DNA (5-20 µl) on an agarose gel and check whether the DNA fragments are concentrated around 400 bp. Adjust the number of cycles and intensity to compensate for excessive or insufficient shearing. If fragments are mainly > 400 bp, then incrementally increase either amplitude or cycle number, while if they are around < 150 bp, then reduce the number of cycles used. - Centrifuge the sonicated samples at 10,000 x g at 4 °C for 15 min to eliminate cell debris and transfer the supernatant to a clean 1.5 ml microtube.
- To check the length distribution of DNA fragments after shearing, load 20 µl of your sonicated samples in a 1% agarose gel. Your sheared DNA must be concentrated around 200-400 bp.
- Collect 10-20 µl to measure protein concentration of the whole-cell extract, using the Bradford protein assay.
- Whole-cell extracts can be either stored at -20 °C or immediately used for immunoprecipitation.
- Put the tubes containing cell pellets on ice and resuspend the cells in 500 µl of Lysis buffer (pre-cooled at 4 °C).
- Prepare magnetic beads for chromatin immunoprecipitation
- Prepare 20 µl of Protein G magnetic beads per reaction in 1.5 ml tubes (minimum of 2 tubes: one with beads for the immunoprecipitated (IP) sample, and another with beads for the first wash step).
- Add 480 µl of Blocking buffer to each tube to reach a final volume of 500 µl.
- Wash the beads with 500 µl of blocking solution (always use fresh solution) by centrifugation at 1,500 x g for 1 min and discard the supernatant. Repeat the wash twice. Resuspend the beads in 20 µl of Lysis buffer.
- Prepare 20 µl of Protein G magnetic beads per reaction in 1.5 ml tubes (minimum of 2 tubes: one with beads for the immunoprecipitated (IP) sample, and another with beads for the first wash step).
- Chromatin immunoprecipitation and reversion of cross-linking
- Prepare 500 µl of whole-cell extract with a concentration of 4 mg/ml of total protein in Lysis buffer. Transfer 50 µl of the supernatant to a 1.5 ml tube and store at -20 °C. This is the 10% total Input DNA (Figure 1) sample for each ChIP sample.
Note: Input DNA sample control contains cross-linked and sonicated DNA that will not be immunoprecipitated. Input DNA is a very important control in ChIP-seq experiments because it will be used to normalize the signal from ChIP enrichment. It also helps to control for biases in the experimental method by comparing read count enrichment between ChIP and input samples. - Pre-treat cell extracts with 20 µl of magnetic beads washed to reduce unspecific binding of DNA or proteins to magnetic beads. Incubate for 1 h at 4 °C with rotation.
- Collect the beads with the DynaMagTM magnetic stand and pass the supernatant (500 µl) to a clean 1.5 ml tube.
- Add 2-5 µg of antibody to IP samples (depending on the antibody; for commercial antibodies refer to the manufacture’s ChIP-seq protocols).
- Incubate IP samples at 4 °C with rotation overnight (at least 16 h).
- Add 20 µl of pre-washed magnetic beads to each IP sample and incubate for 2 h at 4 °C with rotation.
- After incubation, discard the supernatant carefully using the DynaMagTM magnetic stand and wash the magnetic beads twice with 1.5 ml of lysis buffer with 5 min rotation at room temperature.
- Repeat washing step using Wash buffer 1, Wash buffer 2, and TE buffer.
- Resuspend the magnetic beads in 100 µl of TE buffer containing 20 µg of DNAse-free RNase A, incubate at 37 °C for 30 min. Wash the beads with 1.5 ml of TE buffer.
- To elute the immunoprecipitated material, resuspend the magnetic beads in 100 µl of Elution buffer and incubate at 65 °C for 30 min with occasional vortex rotation (gently, under medium speed).
- Repeat the elution step and combine the two eluates.
- Thaw the input sample on ice. Add 20 µl of 5x elution buffer plus 30 µl of MilliQ water to reach the same buffer concentration as the eluted sample.
- To reverse the cross-linking, incubate the ChIP samples (Antibody-IP and Input) at 65 °C for 5 h.
- Prepare 500 µl of whole-cell extract with a concentration of 4 mg/ml of total protein in Lysis buffer. Transfer 50 µl of the supernatant to a 1.5 ml tube and store at -20 °C. This is the 10% total Input DNA (Figure 1) sample for each ChIP sample.
- DNA purification
- Add 100 µl of MilliQ water to the input sample (to reach 200 µl of volume, as for the IP samples). Add 2 µl of proteinase K to a final concentration of 0.4 µg/µl to all ChIP samples and incubate at 37 °C for 1.5 h.
- Extract DNA with 200 µl phenol:chloroform:isoamyl alcohol (25:24:1) by vortexing for 1 min and centrifuging at 10,000 x g for 10 min at 4 °C. Transfer the upper phase from each extraction to a clean 1.5 ml tube.
- Repeat extractions twice with 200 µl chloroform:isoamyl alcohol (24:1).
- Add 53 µl of 7.5 M NH4AcO and 2 volumes (500 µl) of freezer-cold ethanol. Incubate for at least 2 h at -20 °C (best results are achieved, when stored overnight).
- Centrifuge at 10,000 x g for 30 min at 4 °C.
- Remove the supernatant and wash twice with 1,000 µl of freezer-cold 70% ethanol.
- Air-dry the samples and resuspend the pellet in a convenient volume of nuclease-free MilliQ water (25-50 µl).
- Measure the quantity and quality of the ChIP DNA samples using the Quibit® 2.0 and the Quibit dsDNA assay kit following the instructions provided by the manufacturer.
- To check whether enrichment of the potential or well-known TF binding regions was achieved during the immunoprecipitation, DNA of specific genomic regions can be amplified by quantitative Real-Time PCR (qRT-PCR). To carry out this assessment, add 2.5 pg of IP and Input DNA samples to a 0.5 ml tube per genomic region (locus to study) and perform qRT-PCR using a CFX connect RT-PCR machine and ssoFast EvaGreen Supermix kit.
Note: This step is optional. If information about well-known targets of the TF analyzed is available, then we encourage researchers to analyze the IP DNA by qRT-PCR prior to library construction. In our study, two well-known NtcA binding promoters (glnA and glnB) were analyzed (Giner-Lamia et al., 2017). - Use a minimum of 10 ng of IP and Input DNA samples for library preparation, using the Illumina TruSeq ChIP-seq DNA sample preparation kit v.2, as recommended in the kit manual.
Note: If the yield of IP DNA recovered was low, then the resulting IP DNA samples from different experiments can be pooled using a DNA purification column (miniElute kit, QIAGEN) to obtain > 10 ng IP DNA samples.
- Add 100 µl of MilliQ water to the input sample (to reach 200 µl of volume, as for the IP samples). Add 2 µl of proteinase K to a final concentration of 0.4 µg/µl to all ChIP samples and incubate at 37 °C for 1.5 h.
Data analysis
In this section, we provide an example of ChIP-seq analysis prepared as a tutorial, using a subset of the NtcA original data. The files contain only 1% of total reads obtained from NtcA ChIP-seq experiments (Giner-Lamia et al., 2017) to ensure faster computational time. All material necessary for this tutorial can be found on the GitHub website (https://github.com/ginerorama/NtcA_bio-protocols_tutorial). Although only the command lines for nitrogen depletion ChIP-seq files are described in this tutorial, the intermediate files for both nitrogen depletion (-N) and nitrogen replete (NH4+) conditions are available on the GitHub tutorial page. A flowchart outlining bioinformatic ChIP-seq analysis, as described in this tutorial, is given in Figure 2.

Figure 2. Flowchart of the ChIP-seq bioinformatic analysis pipeline
Note: In this tutorial, we assume familiarity with basic shell commands required to work with a terminal interface.
- Quality control analysis of the sequencing reads using FastQC. Before analyzing your sequences, you should always carry out quality control of the raw sequence data to identify potential artifacts. The FastQC (Figure 3) software contains different analysis modules including: (i) Per base sequencing quality (the higher the score the better the base call; in any case the lower quartile for any base should be higher than 10); (ii) Per base sequence content (this should show a non-random distribution of the nucleotide at each base; differences between A and T, or G and C should not be greater than 10% for any position); and (iii) Duplicate sequences (non-unique sequences should not constitute more than 20% of the total sequences). More information on FastQC modules is available at https://www.bioinformatics.babraham.ac.uk/projects/fastqc/Help/.
Note: Although FastQC can be run using command line execution, it also has a graphical user interface that facilitates analysis for researchers not familiar with command line programs.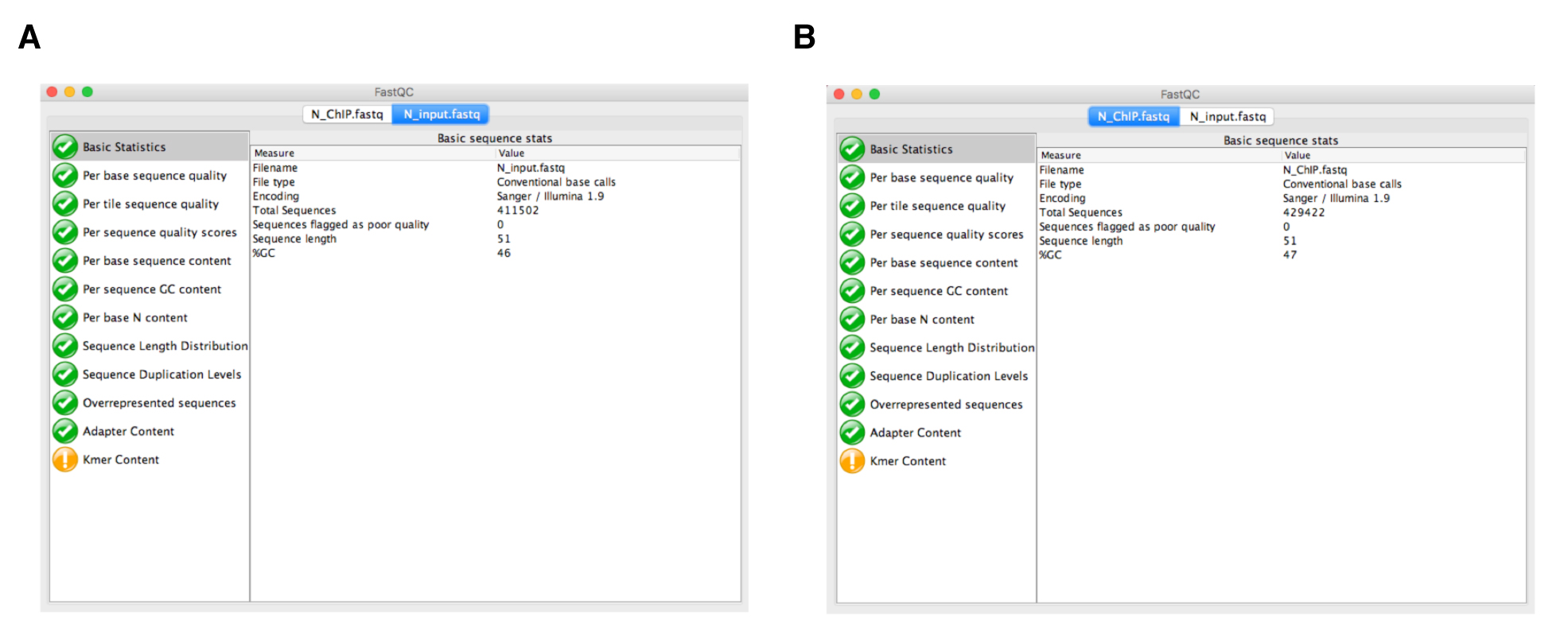
Figure 3. Quality control analysis using FastQC. The modular set of analyses carried out by FastQC in both N_input.fastq (A) and N_ChIP.fastq (B) files are marked in green, indicating that sequencing data are correct. - Alignment of the reads to the genome. The reference genome for Synechocystis sp. PCC 6803 can be downloaded from the National Center for Biotechnology Information (NCBI) Genomes Database, available at https://www.ncbi.nlm.nih.gov/assembly. It has GenBank assembly accession number: GCA_000009725.1; RefSeq: NC_009911.1. In our case, the genome file is NC_009911.1.fasta. This genome file is also available on the GitHub tutorial page. For each sample, we map the FastQ files containing the sequence reads to the reference genome using the bowtie2 program. To do this, we need to create an index of our reference genome using the bowtie2-build function of Bowtie2; bowtie2-build outputs a set of six files with the suffixes (.1.bt2, .2.bt2, .3.bt2, .4.bt2, .rev.1.bt2, and .rev.2.bt2). These files constitute the index. The original genome sequence Fasta file is no longer used by Bowtie2, once this index is built. Now, we can run Bowtie2 using the default parameters. The output file from Bowtie2 is in a Sequence Alignment Map format (.SAM) and contains all the alignment information. The generic command lines for index generation and mapping in bowtie2 are as follows:
Genome index generation:
:$ bowtie2-build path_to_genome_reference genome_index
Arguments:
path_to_genome_reference: the system path to the file containing the reference genome downloaded from NCBI in fasta format.
Genome_index: The basename of the index files. (Name.1.bt2, Name.2.bt2, Name.3.bt2, Name.4.bt2, Name.rev.1.bt2, and Name.rev.2.bt2)
Thus, the index for Synechocystis genome is generated by the command:
:$ bowtie2-build NC_009911.1.fasta Synechocystis
Genome alignment:
:$ Bowtie2 -x basename_genome_index -U sequence_reads.fastQ
-S alignment_file.sam
Arguments:
-x basename_genome_index: The basename of the index for the reference genome. In this case, the basename is the name of any of the index files, not including the final 1.bt2 /. or rev.1.bt2 /.
-U sequence_reads.fastQ: File containing the unpaired reads to be aligned.
-S alignment_file.sam: File to write and save SAM alignments to.
In our example, we would use the following command line to align two ChIP-samples using bowtie2:
:$ Bowtie2 -x Synechocystis -U N_ChIP.fq -S N_ChIP.sam
:$ Bowtie2 -x Synechocystis -U N_Input.fq -S N_Input.sam - SAM to BAM. To analyze our alignment reads, we need to transform the format of the SAM file obtained from Bowtie2 to work more efficiently with the aligned reads. SAM format files are very large files and have to be converted into a Binary Alignment Map (.BAM) format. A BAM file is a binary encoded version of the SAM file that contains the same information, but is typically of smaller size. It is accepted by most programs to analyze the alignment data, once it has been sorted and indexed.
To convert the SAM format into BAM format, we use Samtools.
The generic command lines to transform a SAM file into a sorted BAM file in Samtools are:
:$ samtools view -bS alignment_file.sam > alignment_file.bam
:$ samtools sort alignment_file.bam > alignment_file_sorted
:$ samtools index alignment_file_sorted.bam
Arguments:
- alignment_file.sam: name of the alignment SAM file generated by bowtie2.
- alignment_file.bam: name of the BAM file generated.
- alignment_file_sorted: name of the final sorted BAM file generated.
Thus, the command lines to convert both N_ChIP.sam and N_Input.sam into N_ChIP_sorted.bam and N_Input_sorted.bam, respectively, are:
SAM to BAM conversion:
:$ samtools view -bS N_Input.sam > N_Input.bam
:$ samtools view -bS N_ChIP.sam > N_ChIP.bam
Sorting BAM files:
:$ samtools sort N_Input.bam > N_Input_sorted.bam
:$ samtools sort N_ChIP.bam > N_ChIP_sorted.bam
Finally, the index files are generated using samtools index:
:$ samtools index N_Input_sorted.bam
:$ samtools index N_ChIP_sorted.bam
Note: Samtools index will generate .bai files that must be placed in the same folder as the sorted bam files. Otherwise, programs like Bamcoverage or IGV will not load the sorted bam files. - BAM file normalization. BAM files are still large files and inspection of these files using a genome browser like IGV demands high memory usage on a personal computer. To solve this problem, we used the Bamcoverage utility from the Deeptools2 (v2.0) suite. This tool takes an alignment of reads or fragments as input (BAM file) and generates a coverage track (bigWig or bedGraph) as output. The bigWig files are smaller than BAM files, facilitating the simultaneous loading of multiple ChIP-seq tracks in IGV (Figure 4). In addition, Bamcoverage normalizes all the ChIP-seq files (using different methods, i.e., Reads Per Kilobase per Million mapped reads; RPKM) necessary to compare the enriched peaks from samples with different sequencing depths (i.e., different numbers of reads). The bigWig normalized files generated by Bamcoverage can be loaded into IGV to inspect and analyze the NtcA binding peaks (Figure 4). IGV requires to load the genomes in a special format file. For this tutorial, the Synechocystis genomes files in IGV format (pcc6803.genome.fasta and pcc6803.genome.fasta.fai) are available on the GitHub website. Please refer to the user guide on the IGV webpage to know how create a genome file for IGV.
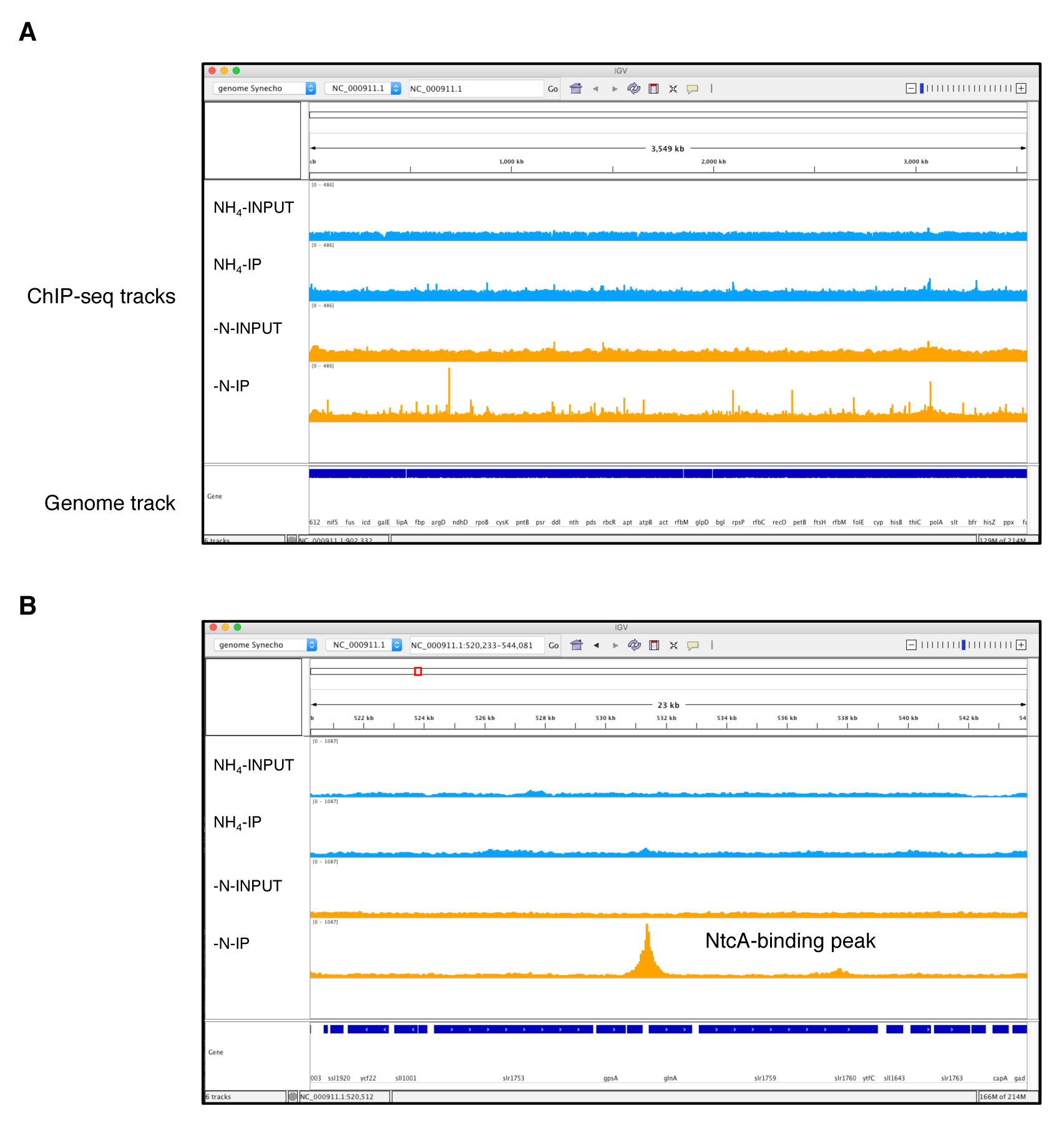
Figure 4. NtcA ChIP-seq data visualization generated using Integrated Genomics Viewer. The two IP samples (NH4+ and -N) and their respective input samples are represented by four separate tracks. The y-axis of each track represents the normalized coverage of sequenced DNA fragments. A. Complete genome coverage of the NtcA ChIP-seq. B. Zoomed chromosomal regions around an NtcA peak located within the glnA promoter region for -N treatment, which is absent for NH4+ treatment, and in both input samples.
An example of Bamcoverage usage:
:$ bamCoverage -b alignment_file.bam -o coverage_file.bw
-normalizeUsing
-b alignment_file.bam: BAM file to process (sorted)
-o coverage_file.bw: ouput file in bigWig format.
-normalizeUsing: It is possible to normalize the number of reads per bin using four different methods: CPM = Counts Per Million mapper reads, BPM = Bin Per Million mapped reads, RPGC = reads per genomic content, and RPKM.
The command line to normalize our ChIP-seq data using the RPKM method is given by:
:$ bamCoverage -b N_Input_sorted.bam -o N_Input.bw
-normalizeUsingRPKM
:$ bamCoverage -b N_ChIP_sorted.bam -o N_ChIP.bw
-normalizeUsingRPKM - Peak calling. Peak calling is carried out using two programs, MACS and the Bioconductor R package BayesPeak. Both programs work without sequence files for Input DNA, using regional counts from IP as a background. When Input DNA sample is available, they compare IP with input sample to identify enrichment. This procedure leads to better sensitivity and specificity than using IP sample alone. For both programs, the previously generated BAM files from IP and Input libraries are used as input files. MACS is a very popular peak finder that can be run using a command line interface in Linux or on a MAC computer. Here, we show a standard analysis with MACS, using a command line. To use the BayesPeak package, please refer to user information available on the Bioconductor webpage https://bioconductor.org/packages/release/bioc/html/BayesPeak.html).
An example of peak calling using MACS:
:$ macs14 –t ChIP_alignment_file.bam -c Input_alignment_file.bam
-g genome_size -n outputfile_name --bw –-nomodel --shiftsize
Arguments:
–t ChIP_alignment_file.bam: ChIP-seq treatment BAM file
-c Input_alignment_file.bam: The control or input BAM file
-g genome_size: genome size of your sequenced organism
-n outputfile_name: The name of any of the MACS files generated during the analysis
--bw: band width used to scan the genome for model building. This parameter can be set to the sonication fragment size expected (see Step B4)
--nomodel: This setting is optional. It skips the model building step. This is recommended when applying MACS to ChIP-seq data with broad peaks.
--shiftsize: The shift size in bp.
The command line to analyze our ChIP-seq data with MACS is given by:
:$ macs14 –t N_ChIP_sorted.bam -c N_Input_sorted.bam
-g 3.5e6 -n NtcA_N --bw 200 –-nomodel –-shiftsize 50
MACS will generate four files, including the NtcA_N_peaks.xls file. This file comprises a table in Excel format containing information about the detected peaks, including chromosome name, start position of peak, end position of peak, length of peak region, peak summit position related to the start position of peak region, number of tags in peak region, -10 x log10 (P-value) for the peak region, fold enrichment for this region against a random Poisson distribution with local lambda, and the false discovery rate (FDR) as a percentage. In our case, a total of 95 binding peaks were detected. The other three files generated by MACS are: NtcA_N_peaks.bed (BED format file containing the peak locations), NtcA_N_summits.bed (BED format file containing the summit locations for called peaks), and NtcA_N_negative_peaks.xls (a tabular file containing information about negative peaks). Negative peaks are called by swapping the ChIP-seq and control channel. In our case study, zero negative peaks were called. A detailed explanation of all setting options available for MACS can be found at https://github.com/taoliu/MACS/blob/macs_v1/README.rst). - Peak annotation. To retrieve the nearest genes around the binding peaks obtained with MACS and to annotate the genomic region of each peak, we used the Bioconductor R package ChIPseeker. It supports annotation of ChIP peaks and provides tools to visualize ChIP peak coverage as well as profiles of peaks binding to transcriptional start site (TSS) regions. To use ChIPseeker, it is necessary to install the Bioconductor package GenomicFeatures, which uses TxDb objects to store transcript metadata. These objects include the maps of 5’ and 3’ untranslated regions (UTRs) and protein coding sequences (CDS) for a set of mRNA or DNA sequences associated with the genome. Here, we will create a TxDb object, based on Synechocystis GTF (General Feature Format, which consists of one line per feature, each containing nine columns of data, plus optional track definition lines). This genomic feature file is available on the GitHub tutorial page.
The following commands are executed in R to create a genomic feature file using GenomicFeatures:
#Install GenomicFeatures and ChIPseeker
source("https://bioconductor.org/biocLite.R")
biocLite("GenomicFeatures")
biocLite("ChIPseeker")
#Creating a TxDb using makeTranscriptDbFromGFF function from GenomicFeatures
library(GenomicFeatures)
setwd(path to tutorial files in your computer)
txdb <- makeTxDbFromGFF(‘NC_000911.1.gff’, format=’gff’)
genes <- genes(txdb)
Now, we can annotate the peaks using the annotatePeak function in ChIPseeker. We will use the BED file generated by MACS in the peak calling analysis (see above). The function annotatePeak requires a peak-containing object (peaks in bed format), a TSS range region (in our case: −300 bp and +300 bp from the TSS) and the Synechocystis TxDb object created above. The command lines to annotate the peaks are given by:
#peak annotation using ChIPseeker.
library(ChIPseeker)
peakfile = ‘NtcA_N_peaks.bed’ #bed file generated by MACS and located in the same directory as the R working directory
peakAnno <- annotatePeak(peakfile, tssRegion=c(-300,300), TxDb=txdb)
write.table(peakAnno,file = ‘N_annotated_peaks.txt’, sep=’\t’)
The output file from ChIPseeker contains the position, the strand, and the distance from peak to the TSS of the nearest genes. The genomic region of the peaks is also reported in the annotation column (Promoter, 5’ UTR, 3’ UTR, exon, intron, downstream, intergenic).
Notes
All software mentioned in this article can be readily installed and run on computers with Linux (Ubuntu version 14 or higher) or Mac OS (Mac OSX 10.6 or higher). They require a minimum of 2 GB of RAM and 2 GHz dual-core processor for genomes of the size of Synechocystis (i.e., 3.6 Mb). To store all files generated during the analysis, depending of the number of experimental samples, a minimum of 25-50 GB of hard-drive space is essential. However, to perform the tutorial described in our data analysis section, only 1 GB of hard-drive space is required. Most of the software can also be executed within Windows, but require a lengthier installation process. Alternatively, some of the bioinformatic tools listed above are offered by online platforms, such as Galaxy (https://usegalaxy.org/).
Recipes
- 5x TBS buffer (for 1 L)
100 ml of 1 M Tris-HCl (pH 7.5)
150 ml of 5 M NaCl
ddH2O to 1 L
Filter sterile
Store at 4 °C - 1x Lysis buffer (for 100 ml)
10 ml 0.5 M HEPES/KOH, (pH 7.5) (50 mM final)
28 ml 5 M NaCl (140 mM final)
200 µl 0.5 M EDTA (1mM final)
5 ml 20% Triton X-100 (1% final)
2 ml 5% sodium deoxycholate (0.1% final)
EDTA-free protease inhibitor cocktail
MilliQ H2O to 100 ml
Filter sterilize
Store at 4 °C - Block solution (20 ml)
20 ml 1x Phosphate-buffered saline (PBS)
0.1 g Bovine serum albumin (BSA)
Always use fresh solution - 1x Wash buffer 1 (for 100 ml)
10 ml 0.5 M HEPES/KOH, (pH 7.5) (50 mM final)
10 ml 5 M NaCl (500 mM final)
200 µl 0.5 M EDTA (1mM final)
5 ml 20% Triton X-100 (1% final)
2 ml 5% sodium deoxycholate (0.1% final)
EDTA-free protease inhibitor cocktail
MilliQ H2O to 100 ml
Filter sterilize
Store at 4 °C - 1x Wash buffer 2 (for 100 ml)
2.5 ml 1 M Tris-HCl, (pH 8) (10mM final)
2.5 ml 10 M LiCl (250 mM final)
5 ml 10% NP-40 (0.5% final)
10 ml 5% sodium deoxycholate (0.5% final)
MilliQ H2O to 100 ml
Filter sterilize
Store at 4 °C - 5x IP elution solution (2 ml)
500 µl 1 M Tris-HCl (pH 7.5) (250 mM final)
200 µl 0.5 M EDTA (50 mM final)
1 ml 10% SDS (5% final)
MilliQ H2O to 2 ml - TE + NaCl Solution (25 ml)
250 µl 1 M Tris-HCl (pH 7.5) (10 mM final)
50 µl 0.5 M EDTA (1 mM final)
2.5 ml 5 M NaCl (50 mM final) - Proteinase K solution (1 ml)
20 mg Proteinase K (20 µg/µl final)
20 µl 1 M Tris-HCl, pH 7.4 (20 mM final)
1 µl 1 M CaCl2 (1 M final)
625 µl 80% glycerol (50 % final)
Store at -20 °C
Stable only for 6 months - Trace metal mix A5 (1L)
2.86 g H3BO3
0.22 g ZnSO4·7H2O
1.81 g MnCl2·4H2O
0.31 g Na2MoO4·2H2O
0.08 g CuSO4.5H2O
0.05 g Co(NO3)2.6H2O - 100x BG11 (1 L)
7.5 g MgSO4·7H2O
3.6 g CaCl2·2H2O
0.6 g citric acid
0.6 g Fe-NH4 citrate
0.1 g Na2-EDTA
2.0 g Na2CO3
100 ml Trace metal mix A5
ddH2O to 1 L - BG110C (1L)
1 g NaHCO3
0.2 ml 1M K2HPO4
10 ml 100x BG11
ddH2O to 1 L
Autoclave before use - BG110C-NH4 (1 L)
970 ml autoclaved BG110C
10 ml of pre-filtered 1 M NH4Cl (10 mM final)
20 ml of pre-filtered 1 M TES pH 7.5 (20 mM final)
Acknowledgments
This protocol was adapted from Picossi et al. (2014). A sincere thank you to Trudi A. Semeniuk for her diligent proofreading of this protocol. This work was supported by grants from National Portuguese Funding through FCT (Fundaçao para a Ciência e a Tecnologia) projects [PTDC/BIA-MIC/4418/2012, IF/00881/2013, UID/BIM/04773/2013–CBMR, UID/Multi/04326/2013–CCMAR]. MAHP current position is funded by the Australian Government through the Australian Research Council Centre of Excellence for Translational Photosynthesis (CE1401000015). JGL current position is funded by the Spanish Ministry of Economy and Economy and European Regional Development Funds(FEDER) BIO2016-77639-P (MINECO/FEDER), respectively.
Competing interests
None of the authors have any conflicts of interest or competing interests to declare.
References
- García-Domínguez, M., Reyes, J. C. and Florencio, F. J. (2000). NtcA represses transcription of gifA and gifB, genes that encode inhibitors of glutamine synthetase type I from Synechocystis sp. PCC 6803. Mol Microbiol 35(5): 1192-1201.
- Giner-Lamia, J., Robles-Rengel, R., Hernandez-Prieto, M. A., Muro-Pastor, M. I., Florencio, F. J. and Futschik, M. E. (2017). Identification of the direct regulon of NtcA during early acclimation to nitrogen starvation in the cyanobacterium Synechocystis sp. PCC 6803. Nucleic Acids Res 45(20): 11800-11820.
- Herrero, A., Muro-Pastor, A. M. and Flores, E. (2001). Nitrogen control in cyanobacteria. J Bacteriol 183(2): 411-425.
- Langmead, B. and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9(4): 357-359.
- Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R. and Genome Project Data Processing, S. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25(16): 2078-2079.
- Picossi, S., Flores, E. and Herrero, A. (2014). ChIP analysis unravels an exceptionally wide distribution of DNA binding sites for the NtcA transcription factor in a heterocyst-forming cyanobacterium. BMC Genomics 15: 22.
- Ramírez, F., Ryan, D. P., Gruning, B., Bhardwaj, V., Kilpert, F., Richter, A. S., Heyne, S., Dundar, F. and Manke, T. (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44(W1): W160-165.
- Robinson, J. T., Thorvaldsdottir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G. and Mesirov, J. P. (2011). Integrative genomics viewer. Nat Biotechnol 29(1): 24-26.
- Spyrou, C., Stark, R., Lynch, A. G. and Tavare, S. (2009). BayesPeak: Bayesian analysis of ChIP-seq data. BMC Bioinformatics 10: 299.
- Stanier, R. Y., Kunisawa, R., Mandel, M. and Cohen-Bazire, G. (1971). Purification and properties of unicellular blue-green algae (order Chroococcales). Bacteriol Rev 35(2): 171-205.
- Yu, G., Wang, L. G. and He, Q. Y. (2015). ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31(14): 2382-2383.
- Zhang, Y., Liu, T., Meyer, C. A., Eeckhoute, J., Johnson, D. S., Bernstein, B. E., Nusbaum, C., Myers, R. M., Brown, M., Li, W. and Liu, X. S. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9(9): R137.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Giner-Lamia, J., Hernández-Prieto, M. A. and Futschik, M. E. (2018). ChIP-seq Experiment and Data Analysis in the Cyanobacterium Synechocystis sp. PCC 6803. Bio-protocol 8(12): e2895. DOI: 10.21769/BioProtoc.2895.
Category
Microbiology > Microbial genetics > DNA > DNA sequencing
Molecular Biology > DNA > DNA-protein interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.