- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Quantification of Extracellular Double-stranded RNA Uptake and Subcellular Localization Using Flow Cytometry and Confocal Microscopy
Published: Vol 8, Iss 12, Jun 20, 2018 DOI: 10.21769/BioProtoc.2890 Views: 7651
Reviewed by: Ivan ZanoniLaura CampisiKristofor K. Ellestad
Original research article
The authors used this protocol in:

Sep 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Double-stranded RNA is a potent pathogen-associated molecular pattern (PAMP) produced as a by-product of viral replication and a well-known hallmark of viral infection. Viral dsRNAs can be released from infected cells into the extracellular space and internalized by neighboring cells via endocytosis. Mammals possess multiple pattern recognition receptors (PRRs) capable of detecting viral dsRNAs such as endosomal toll-like receptor 3 (TLR3) and cytosolic RIG-I-like receptors (RLRs) which lead to the production of type I interferons (IFNs). Thus, intracellular localization of viral dsRNA can provide insight into the downstream signaling pathways leading to innate immune activation. Here, we describe a quantitative method for measuring extracellular dsRNA uptake and visualizing subcellular localization of internalized dsRNA via flow cytometry and confocal microscopy respectively.
Keywords: Double-stranded RNABackground
Double-stranded RNAs (dsRNAs) are a common by-product of viral replication and are potent activators of antiviral immunity via the production of type I interferon (IFN) and other pro-inflammatory cytokines (Nellimarla and Mossman, 2014). Viral dsRNAs are sensed within endosomes by TLR3 (Matsumoto et al., 2003) or in the cytosol by the RIG-I-like receptors (RLRs), RIG-I and MDA-5 (Kato et al., 2006). During lytic infections, these dsRNAs can be released into the extracellular space where they bind surface receptors on neighboring cells, such as class A scavenger receptors (SR-A) and Raftlin, and are subsequently internalized via clathrin-mediated endocytosis (Itoh et al., 2008; DeWitte-Orr et al., 2010; Watanabe et al., 2011; Dansako et al., 2013).
In our previous study, we found out that the protein SID1 transmembrane family member 2 (SIDT2) localizes to late endosomes and lysosomes and that loss of SIDT2 leads to subcellular accumulation of the synthetic dsRNA analog, poly(I:C), while not affecting initial endocytosis-mediated internalization (Nguyen et al., 2017). To do so, we developed and utilized flow cytometry and confocal microscopy-based approaches to quantitatively measure poly(I:C) uptake and subcellular localization respectively in vitro. In this protocol, we describe a further refinement of these assays to allow for high-throughput assessment of internalization and subcellular localization of different dsRNAs. These methods allow for further dissection of dsRNA trafficking during viral infection and the downstream effects of these dsRNAs on innate immune signaling.
Materials and Reagents
- 8 well microscope slide (ibidi, catalog number: 80826 )
- 24 well tissue culture plate (Corning, Falcon®, catalog number: 353047 )
- Sterile filtered pipette tips (0.5 µl to 1,000 µl) (Corning, Axygen, catalogue numbers: TF-300-L-R-S , TF-20-L-R-S , TF-200-L-R-S and TF-1000-L-R-S )
- 10 cm culture dishes (Corning, Falcon®, catalog number: 353003 )
- 10 ml centrifuge tubes (SARSTEDT, catalog number: 62.9924.284 )
- 1.5 ml microcentrifuge tubes (Sigma-Aldrich, catalog number: EP0030120086 )
- 1.2 ml Micro Titertube (Thermo Fisher Scientific, Quality Scientific Plastics, catalog number: 845-Q )
- Serological pipettes, individually wrapped, 10 ml (Corning, Falcon®, catalog number: 356551 )
- Mammalian cell line of interest, here: mouse embryonic fibroblasts (see Note 1)
- 70% (v/v) ethanol (Chem Supply, catalog number: EA043 )
- Dulbecco’s modified Eagle medium (DMEM) or other suitable complete growth medium for culture of cell line of interest
- Fetal bovine serum (FBS) (Sigma-Aldrich, catalog number: F9423 )
- Phosphate-buffered saline (PBS) (sterile) (Thermo Fisher Scientific, GibcoTM, catalog number: 14190250 )
- Penicillin/streptomycin solution (Sigma-Aldrich, catalog number: P4333 )
- Poly(I:C)-fluorescein (InvivoGen, catalog number: tlrl-picf )
- Poly(I:C)-rhodamine (InvivoGen, catalog number: tlrl-picr )
- dsRNA specific monoclonal antibody (J2, SCICONS English and Scientific Consulting, catalog number: 10010200 )
- RNase A enzyme (Sigma-Aldrich, catalog number: R4875 )
- 1x trypsin-EDTA solution (Sigma-Aldrich, catalog number: 59430C )
- Paraformaldehyde (PFA) powder (Sigma-Aldrich, catalog number: 158127 )
- Tween 20 (Sigma-Aldrich, catalog number: P1379 )
- DAPI (4’,6-Diamidine-2’-phenylindole dihydrochloride) powder (Sigma-Aldrich, catalog number: D9542 )
- ImmersolTM Immersion Oil (Carl Zeiss, catalog number: 4449620000000 )
- Complete growth medium (see Recipes)
- 10% FBS/PBS (see Recipes)
- 4% paraformaldehyde (w/v) (see Recipes)
- Permeabilization buffer (see Recipes)
- DAPI solution (see Recipes)
Equipment
- Pipetting aid (Thermo Fisher Scientific, catalog number: 9531 )
- Micropipettes from 0.5 µl to 1 ml (Mettler-Toledo International, Rainin, model: Pipet-LiteTM XLS+ )
- Hemocytometer
- Class II biological safety cabinet/tissue culture hood
- Humidified CO2 incubator (95% air, 5% CO2, 37 °C)
- Inverted light microscope (phase contrast)
- 37 °C water bath
- Vacuum aspiration system with glass Pasteur pipettes
- Table top centrifuge equipped with a swing-out rotor for 10 ml conical tubes
- Microcentrifuge
- LSRFortessa X20 (BD, BD Biosciences, model: LSRFortessaTM X-20 ) or equivalent flow cytometer
- LSM 780 confocal laser scanning microscope (ZEISS, model: LSM 780 ) or equivalent microscope
Software
- FIJI/ImageJ
- Zeiss ZEN package
- Microsoft Excel
- FlowJo
- GraphPad Prism 7
Procedure
- Cell culture and maintenance
- Perform all cell culture-based work in a class II biological safety cabinet/tissue culture hood. Ensure work surface and materials are sterilized using 70% (v/v) ethanol.
- Grow mammalian cells of choice in complete growth medium using 10 cm cell culture dishes or cell culture flasks in a humidified CO2 incubator (95% air, 5% CO2, 37 °C). Here, we use mouse embryonic fibroblasts (MEFs) derived from C57BL/6 mice and MEFs lacking the dsRNA transporter, SIDT2, as our gene of interest (see Note 2).
- Maintain cells using standard cell culture procedures or as recommended by the supplier. Here, split MEFs approximately every 3-4 days or before cells reach 100% confluency using standard cell culture procedures (see Note 3).
- One day prior to stimulation, split the cells as above and determine cell number using a hemocytometer or other appropriate methods under a light microscope.
- Perform all cell culture-based work in a class II biological safety cabinet/tissue culture hood. Ensure work surface and materials are sterilized using 70% (v/v) ethanol.
- Quantification of poly(I:C) uptake by flow cytometry
- Seed 1 x 104 cells in a 24 well plate in triplicate for the following conditions in a total volume of 500 µl of complete growth medium (cells may be seeded for additional conditions as required):
- Unstimulated
- Stimulated with poly(I:C) (see Note 4)
- Unstimulated
- Let cells adhere and rest overnight in a humidified CO2 incubator (95% air, 5% CO2, 37 °C).
- The next morning, remove cells from the incubator and add 1 µg/ml of fluorescein-poly(I:C) to the cell culture medium (see Note 5).
- Stimulate cells in an incubator at 37 °C, 5% CO2 for 24 h.
- After 24 h of stimulation, carefully aspirate complete growth medium from each well with a glass Pasteur pipette using a vacuum aspiration system.
- Wash cells with 1 ml of cold 1x PBS using a P1000 pipette. Carefully add PBS on the side of the well to avoid dislodging cells.
- Repeat Step B6 for a total of 3 washes.
- Harvest cells by adding 500 µl of Trypsin to each well and incubate cells at 37 °C, 5% CO2 for 5 min.
- Add 500 µl of complete growth medium to each well.
- Gently resuspend cells by pipetting up and down 3-5 times.
- Transfer resuspended cells to new individual 1.5 ml microcentrifuge tubes.
- Centrifuge cells in a microcentrifuge for 3 min at 500 x g and resuspend the pellet in 500 µl PBS supplemented with 10% FBS.
- Add 10 µl of RNase A enzyme (5 mg/ml stock) to each well at a final concentration of 100 µg/ml (see Note 6).
- Incubate cells at 37 °C and 5% CO2 for 30 min.
- Centrifuge cells in a microcentrifuge for 3 min at 500 x g.
- Carefully aspirate RNase A solution from each well.
- Resuspend cells in 1 ml cold PBS.
- Repeat Step B15 for a total of 3-5 washes.
- Resuspend cells in 200 µl of 10% FBS/PBS.
- Transfer cells to individual 1.2 ml Micro Titertubes and leave on ice.
- Analyze the incorporated fluorescence of cells of both genotypes and using flow cytometry. Compare the histograms and corresponding mean fluorescence intensities (MFI) between fluorescein-poly(I:C)-stimulated cells and unstimulated cells (Figure 1).
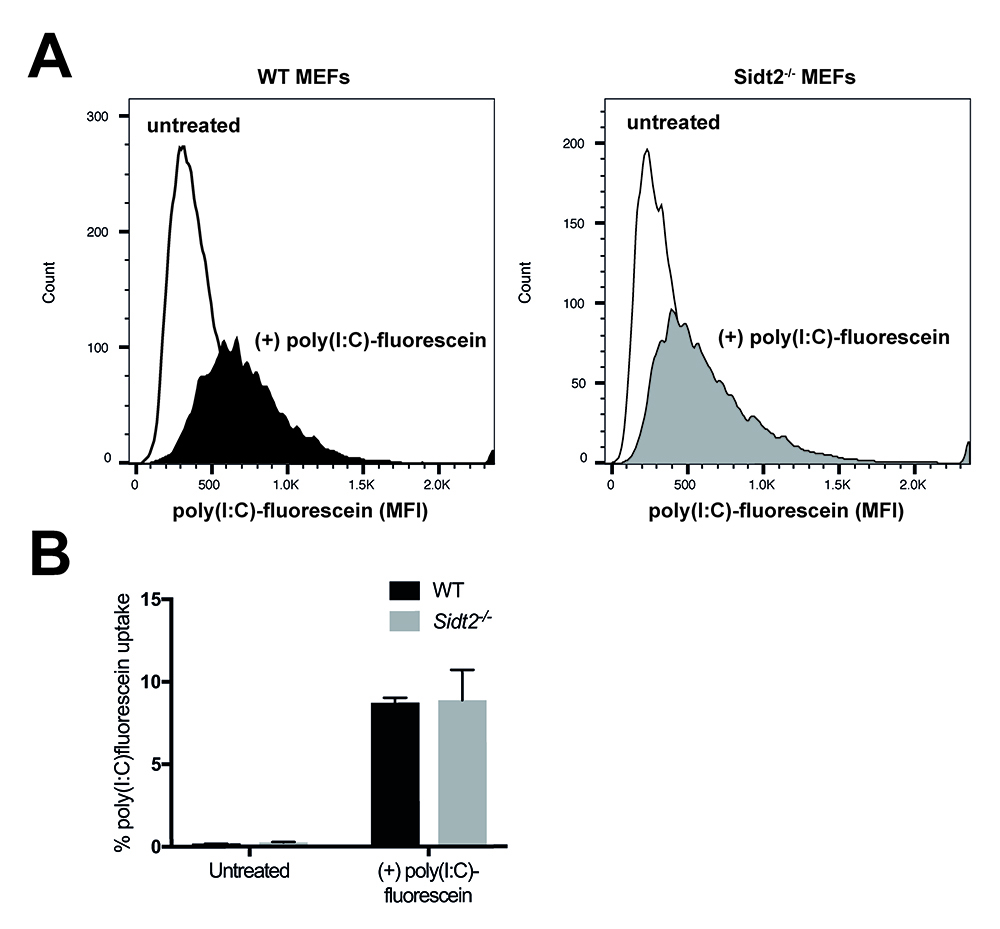
Figure 1. Analysis of poly(I:C)-fluorescein uptake by flow cytometry. The endocytic activity of WT and Sidt2-/- MEFs was assessed by measuring the uptake of fluorescein-conjugated poly(I:C) following 24 h stimulation. A. Representative histograms for the mean fluorescence intensities (MFI) for each genotype are shown. B. Mean percentage of poly(I:C)-fluorescein positive cells. n = 3 technical replicates and errors bars represent ± SEM. Loss of SIDT2 did not affect poly(I:C) uptake.
- Seed 1 x 104 cells in a 24 well plate in triplicate for the following conditions in a total volume of 500 µl of complete growth medium (cells may be seeded for additional conditions as required):
- Confocal analysis of poly(I:C) subcellular localization
- Seed 5 x 103 cells in an 8 well chamber slide for the following conditions in a total volume of 200 µl of complete growth medium (cells may be seeded for additional conditions as required):
- Unstimulated
- Stimulated with poly(I:C)
- Unstimulated
- The next morning, remove cells from the incubator and add 1 µg/ml of rhodamine-poly(I:C) to the cell culture medium (see Note 5).
- Incubate cells at 37 °C, 5% CO2 for 24 h.
- Carefully aspirate and discard complete growth medium from each well with a P200 pipette.
- Wash cells with 200 ml of cold 1x PBS using a P1000 pipette.
- Repeat Step C5 for a total of 3 washes.
- After washes, add 200 µl PBS supplemented with 10% FBS to each well.
- Add 10 µl of RNase A enzyme (5 mg/ml stock) to each well at a final concentration of 100 µg/ml (see Note 6).
- Incubate cells at 37 °C and 5% CO2 for 30 min.
- Carefully aspirate RNase A solution from each well.
- Wash cells with 200 µl cold PBS.
- Repeat Step C11 for a total of 3 washes.
- Fix cells with 200 µl of 4% PFA for 10 min on ice.
- Wash cells with 200 µl cold PBS.
- Repeat Step C14 for a total of 3 washes.
- Stain cells with 100 ng/ml DAPI solution in a final volume of 200 µl per well for 10 min at room temperature (see Note 7).
- Wash cells with 200 µl cold PBS.
- Repeat Step C17 for a total of 3 washes.
- After the final wash, add 200 µl of PBS to each well.
- Slides are now ready for imaging or can be stored at 4 °C for up to 2 weeks.
- Acquire images using a ZEISS LSM 780 confocal microscope (an equivalent microscope can be used) with a 25x oil immersion objective lens.
- Acquire Z-stack of 0.25 μm slices from the top to the bottom of the cells (Figure 2A).
- Save images as .lsm files and separate each condition into individual folders (e.g., WT untreated, WT pIC treated, KO untreated, KO pIC treated).
- Drag and drop all folders into a single folder (see Note 8).
- Images are now ready for analysis using FIJI/ImageJ software (see below).
- Seed 5 x 103 cells in an 8 well chamber slide for the following conditions in a total volume of 200 µl of complete growth medium (cells may be seeded for additional conditions as required):
Data analysis
- Determine appropriate threshold value for images
- Open an image file that contains cells with representative poly(I:C)-rhodamine or alternative dsRNA signal intensity in FIJI/ImageJ.
- Acquire maximum projection image (Navigate to toolbar on the top left corner of the screen → Image → Stacks → Z Project → Projection type: Max intensity → Okay).
- Select channel corresponding to poly(I:C)-rhodamine by adjusting the C slider on the bottom of the maximum projection image window.
- From the toolbar, select Image → Adjust → Threshold…
- Set bottom slider to 255.
- Adjust top slider until threshold (red) is able to distinguish between each individual punctate endosome (Figure 2B). Here, we used a threshold of 60.
- Note down threshold value.
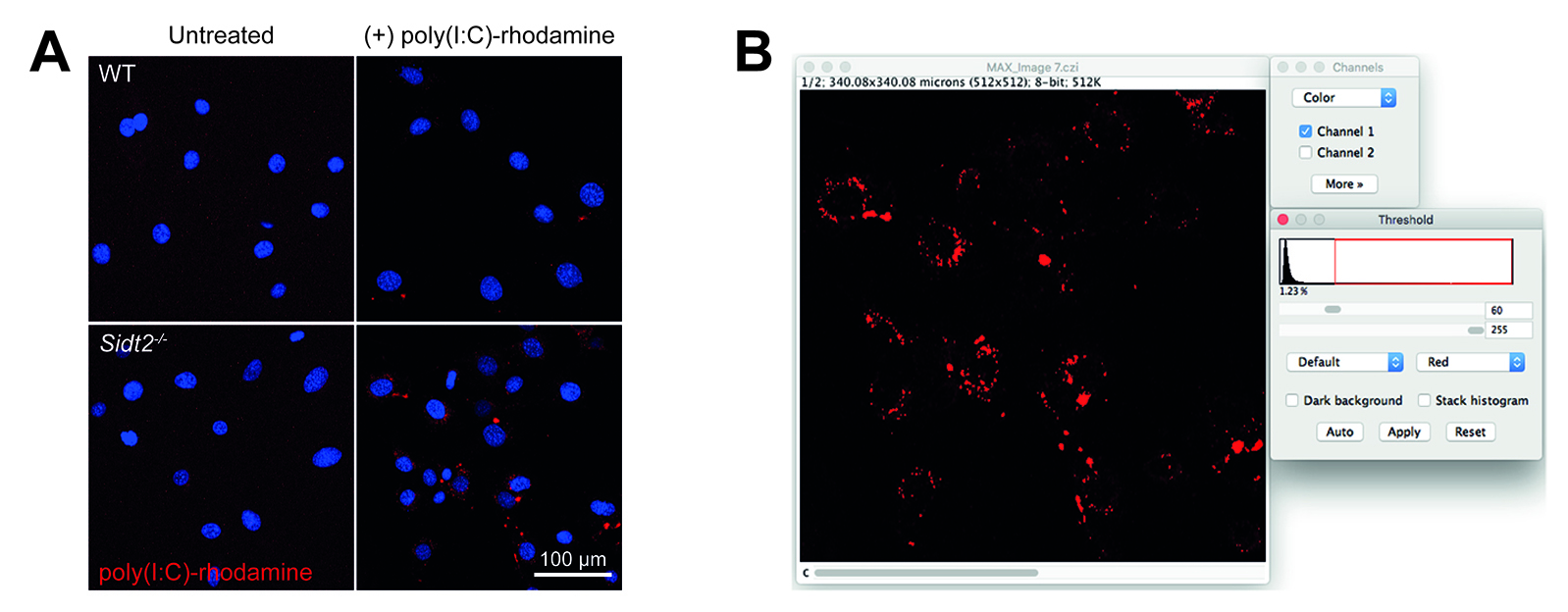
Figure 2. Analysis of poly(I:C)-rhodamine subcellular localization by confocal microscopy. A. WT and Sidt2-/- MEFs were treated with rhodamine-conjugated poly(I:C) for 24 h and assessed via confocal microscopy. Representative maximum projection images are shown, red = poly(I:C), blue = DAPI. B. Snapshot from FIJI/ImageJ showing selection of threshold value step.
- Open an image file that contains cells with representative poly(I:C)-rhodamine or alternative dsRNA signal intensity in FIJI/ImageJ.
- Subcellular localization quantification
- Download FIJI macro file from: https://bitbucket.org/DrLachie/rna_subcell.
- Open macro file in FIJI/ImageJ.
- Change ‘GreenThreshold’ to the threshold value determined in the above section (e.g., ‘60’).
- Click ‘Run’.
- Open output\ folder and check the accuracy of cell segmentation (Figure 3A) and individual cell by cell quantification of the punctate area, intensity and number of puncta determined by macro.
- Open results.xls file located in top folder using Microsoft Excel.
- Calculate and Graph results for the average punctate area, intensity and number of puncta in Prism GraphPad or equivalent software (Figure 3B) (see Note 9).
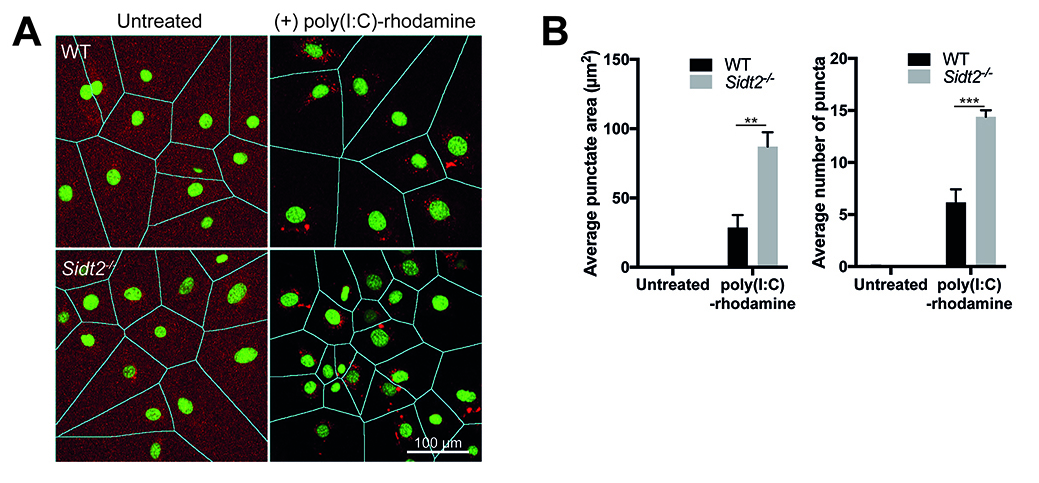
Figure 3. Quantification of poly(I:C)-rhodamine subcellular localization using FIJI/ImageJ macro. A. Representative output image following macro quantification showing segmentation of cells using DAPI to delineate between individual cells. Note that poly(I:C) localizes in the cytoplasm and not in the nucleus. B. Comparison of the average percentage punctate area, average intensity and average number of puncta between WT and Sidt2-/- MEFs. Data are plotted as mean ± SEM and at least 5 representative fields of view each containing 5-22 cells were analyzed per condition. **P < 0.01, ***P < 0.001.
- Download FIJI macro file from: https://bitbucket.org/DrLachie/rna_subcell.
Notes
- Other adherent cell lines of interest can also be used such as bone marrow-derived macrophages, HEK293T and NIH3T3, etc. We have also used non-adherent cells such as DC2.4 and bone marrow-derived dendritic cells by treating suspension cells in 1.5 ml microcentrifuge tubes and subsequently mounting fixed cells onto a microscope slide via a cytospin. However, we find that this method leads to a loss of cell morphology and therefore recommend the use of adherent cells if possible.
- We have previously reported that the loss of SIDT2 leads to accumulation of dsRNA within endosomes (Nguyen et al., 2017). Here, Sidt2-/- MEFs were used to assess endosomal subcellular localization compared to WT MEFs. Cells lacking or overexpressing gene of interest can be used in place of Sidt2-/- MEFs as desired.
- Here, we pre-warm 1x PBS, 1x Trypsin solution and complete growth medium to 37 °C using a water bath. Old medium is removed from the cell culture dish using a Pasteur pipette by vacuum aspiration. Cells are subsequently washed with 10 ml of 1x PBS before the addition of 3 ml (per 10 cm dish) of Trypsin solution and incubate for 3-5 min at 37 °C. Detached cells are resuspended in 10 ml/10 cm dish by pipetting 2-3 times up and down and transfer to a 10 ml centrifuge tube. Cells are pelleted in a tabletop centrifuge at 500 x g, 5 min at RT and resuspended in 10 ml of fresh complete growth medium. For cell maintenance, add 1 ml of cell suspension to a new 10 cm cell culture dish with 10 ml of complete growth medium and incubate in a humidified CO2 incubator (95% air, 5% CO2, 37 °C).
- Here, rhodamine or fluorescein conjugated poly(I:C) (InvivoGen) was used to stimulate cells as it is commercially available and readily accessible. Alternate fluorescently-tagged ligands can also be used as required. We have also successfully detected viral dsRNA via immunofluorescence staining using a dsRNA specific monoclonal antibody (J2, English and Scientific Consulting).
- The concentration of poly(I:C) and length of stimulation should be optimized according to cell line of interest. We found that MEFs do not efficiently internalize poly(I:C) and therefore require a 24 h stimulation time. However, bone marrow-derived dendritic cells and macrophages require much shorter stimulation times (1 to 3 h) for sufficient uptake.
- Treating cells with RNase will degrade any surface-bound dsRNA while retaining internalized dsRNA. Here, we used RNase A which is able to cleave both ssRNA and dsRNA at low salt concentrations. Alternatively, RNase III can be used to specifically cleave dsRNA.
- It is important to stain cells with DAPI in the presence of detergent to permeabilize the cell membrane and allow DAPI access to nuclear DNA. We use 0.1% Tween to permeabilize cells; however, alternative detergents can be used such as Saponin or Triton-X.
- The FIJI macro used for image analysis requires two layers of folders in order to proceed with analysis.
- Here, we demonstrate that loss of SIDT2 results in endosomal accumulation of poly(I:C), consistent with our previous findings (Nguyen et al., 2017). In that study, we also performed transient transfection and immunofluorescence staining of various endosomal markers – EEA-1 (early endosomes), RAB-7 (late endosomes) and LAMP-1 (lysosomes) – in order to precisely determine the subcellular localization of poly(I:C) within Sidt2-/- cells.
Recipes
- Complete growth medium
Dulbecco’s modified Eagle medium supplemented with:
10% fetal bovine serum
100 U/ml penicillin
100 µg/ml streptomycin
Filter sterilize, store at 4 °C - 10% FBS/PBS
450 ml Sterile Phosphate Buffered Saline
50 ml fetal bovine serum
Store at 4 °C - 4% paraformaldehyde (w/v)
- Dissolve 4 g of PFA powder in 90 ml PBS and heat to 65 °C while stirring. If PFA does not dissolve, add drops of 1 M NaOH until the solution becomes clear.
- Bring to 100 ml with PBS. Cool and filter. Aliquot and store at -20 °C. Thaw aliquots as needed and use immediately.
- Dissolve 4 g of PFA powder in 90 ml PBS and heat to 65 °C while stirring. If PFA does not dissolve, add drops of 1 M NaOH until the solution becomes clear.
- Permeabilization buffer
10 ml PBS
10 µl of Tween 20 - DAPI solution
- Dissolve stock solution in sterile dH2O at a final concentration of 1 mg/ml
- Dilute stock solution to a final concentration of 1 µg/ml in permeabilization buffer. Use immediately
- Dissolve stock solution in sterile dH2O at a final concentration of 1 mg/ml
Acknowledgments
We thank the members of the Wicks and Masters labs, WEHI for helpful discussions. This protocol was adapted from Nguyen et al. (2017) Immunity 47(3):498-509.e6. DOI: 10.1016/j.immuni.2017.08.007. This work was supported by Australian NHMRC (ID 520574 and 1064591), Royal Australasian College of Physicians, Menzies Foundation, CASS Foundation (SM13-4846 and SM14- 5566), and Reid Family Trust.
Competing interests
The authors declare no conflict of interest.
References
- Dansako, H., Yamane, D., Welsch, C., McGivern, D. R., Hu, F., Kato, N. and Lemon, S. M. (2013). Class A scavenger receptor 1 (MSR1) restricts hepatitis C virus replication by mediating toll-like receptor 3 recognition of viral RNAs produced in neighboring cells. PLoS Pathog 9(5): e1003345.
- DeWitte-Orr, S. J., Collins, S. E., Bauer, C. M. T., Bowdish, D. M., Mossman, K. L. (2010). An accessory to the ‘Trinity’: SR-As are essential pathogen sensors of extracellular dsRNA, mediating entry and leading to subsequent type I IFN responses. PLoS Pathog 6: e1000829.
- Itoh, K., Watanabe, A., Funami, K., Seya, T. and Matsumoto, M. (2008). The clathrin-mediated endocytic pathway participates in dsRNA-induced IFN-β production. J Immunol 181(8): 5522-5529.
- Kato, H., Takeuchi, O., Sato, S., Yoneyama, M., Yamamoto, M., Matsui, K., Uematsu, S., Jung, A., Kawai, T., Ishii, K. J., Yamaguchi, O., Otsu, K., Tsujimura, T., Koh, C. S., Reis e Sousa, C., Matsuura, Y., Fujita, T. and Akira, S. (2006). Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441(7089): 101-105.
- Matsumoto, M., Funami, K., Tanabe, M., Oshiumi, H., Shingai, M., Seto, Y., Yamamoto, A. and Seya, T. (2003). Subcellular localization of Toll-like receptor 3 in human dendritic cells. J Immunol 171(6): 3154-3162.
- Nellimarla, S. and Mossman, K. L. (2014). Extracellular dsRNA: its function and mechanism of cellular uptake. J Interferon Cytokine Res 34(6): 419-426.
- Nguyen, T. A., Smith, B. R. C., Tate, M. D., Belz, G. T., Barrios, M. H., Elgass, K. D., Weisman, A. S., Baker, P. J., Preston, S. P., Whitehead, L., Garnham, A., Lundie, R. J., Smyth, G. K., Pellegrini, M., O'Keeffe, M., Wicks, I. P., Masters, S. L., Hunter, C. P. and Pang, K. C. (2017). SIDT2 transports extracellular dsRNA into the cytoplasm for innate immune recognition. Immunity 47(3): 498-509 e496.
- Watanabe, A., Tatematsu, M., Saeki, K., Shibata, S., Shime, H., Yoshimura, A., Obuse, C., Seya, T. and Matsumoto, M. (2011). Raftlin is involved in the nucleocapture complex to induce poly(I:C)-mediated TLR3 activation. J Biol Chem 286(12): 10702-10711.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Nguyen, T. A., Whitehead, L. and Pang, K. C. (2018). Quantification of Extracellular Double-stranded RNA Uptake and Subcellular Localization Using Flow Cytometry and Confocal Microscopy. Bio-protocol 8(12): e2890. DOI: 10.21769/BioProtoc.2890.
Category
Immunology > Immune cell imaging > Confocal microscopy
Cell Biology > Cell imaging > Confocal microscopy
Molecular Biology > RNA > RNA detection
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.