- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Small Molecule-Based Retinal Differentiation of Human Embryonic Stem Cells and Induced Pluripotent Stem Cells
Published: Vol 8, Iss 12, Jun 20, 2018 DOI: 10.21769/BioProtoc.2882 Views: 8428
Reviewed by: Giusy TornilloDongsheng JiangAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Mar 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Retinal degeneration leads to loss of light-sensing photoreceptors eventually resulting in vision impairment and impose a heavy burden on both patients and the society. Currently available treatment options are very limited and mainly palliative. Ever since the discovery of human pluripotent stem cell technologies, cell replacement therapy has become a promising therapeutic strategy for these patients and may help restore visual function. Reproducibly generating enriched retinal cells including retinal progenitors and differentiated retinal neurons such as photoreceptors using human embryonic stem (ES) cells and induced pluripotent stem (iPS) cells in a dish is an essential first step for developing stem cell-based therapies. In addition, this will provide a reliable and sufficient supply of human retinal cells for studying the mechanisms of diseases. Here we describe a small molecule-based retinal induction protocol that has been used to generate retinal progenitors and differentiated retinal neurons including photoreceptors from several human ES and iPS cell lines. The retinal cells generated by this protocol can survive and functionally integrate into normal and diseased mouse retinas for several months following subretinal transplantation.
Keywords: HumanBackground
A number of groups around the world are developing methodologies to generate specific cell types from human pluripotent stem cells. These cells will likely play a critical role in the future of regenerative medicine as a source of replacement cells. These newly generated human cells will be very useful in developing better and more accurate human disease models that can then be used for discovery of novel drugs with better efficacy and safety profiles.
Our work focuses on retinal degenerative diseases such as macular degeneration and retinitis pigmentosa which affect millions of people worldwide. Death of light-sensing photoreceptors in the retina is commonly associated with those diseases and results in severe impairment or total loss of vision. There are no effective medical treatments available to cure those diseases.
Under specific conditions, human ES and iPS cells can be specifically used to generate retinal progenitor cells, and consequentially differentiate into specialized retinal neuronal subtypes (retinal ganglion cells, amacrine cells, bipolar cells, horizontal cells, and photoreceptors) (Osakada et al., 2008 and 2009; Hirami et al., 2009; Lamba et al., 2006, 2009 and 2010; Meyer et al., 2009; Hambright et al., 2012; Zhong et al., 2014). Establishment of effective and chemically-defined protocols to generate retinal progenitors as well as differentiated retinal cell types including photoreceptors from human ES cells and iPS cells is a critical step for developing cell replacement therapies for patients with a variety of incurable retinal degenerative diseases.
Here, we report a detailed small molecule-based retinal induction protocol that has been used to generate retinal cells in vitro and the derived retinal cells were used as donor cells in the transplantation studies carried out by Dr. Lamba’s research group. The derived retinal progenitors and retinal photoreceptors were tested in multiple host mouse lines with and without retinal degeneration conditions and showed the ability to survive and functionally integrate into the host mouse retina following transplantation (Zhu et al., 2017 and 2018).
Materials and Reagents
- 6-well plate (DNase and RNase free, treated) (Greiner Bio One International, catalog number: 657160 )
- Cryogenic vials (Corning, catalog number: 430659 )
- 2 ml serological pipette (VWR, catalog number: 76093-882 )
- 1,000 µl micropipette (VWR, catalog number: 89079-974 )
- 15 ml conical tube (VWR, catalog number: 490001-623 )
- Micropipette tip (VWR, catalog number: 490000-468 )
- Cell scraper (Corning, catalog number: 3008 )
- Sterile Disposable Filter System with PES Membrane (0.22 µm pore size) (Thermo Fisher Scientific, catalog number: 566-0020 )
- Cover glasses (VWR, catalog number: 89015-725 )
- 70% ethanol (Sigma-Aldrich, catalog number: 459836 )
- Matrigel (BD, BD Biosciences, catalog number: 354234 )
- DMEM/F-12 basal medium (GE Healthcare, catalog number: SH30023.02 )
- Essential 8 medium (Thermo Fisher Scientific, catalog number: A2858301 )
- Penicillin Streptomycin Amphotericin B (Lonza, catalog number: 17-745E )
- Lyophilized Y27632 (Stemgent, catalog number: 04-0012 )
- 1x Dulbecco’s phosphate buffered saline (DPBS) without calcium and magnesium (Thermo Fisher Scientific, catalog number: 14190144 )
- 0.1% EDTA (Diluted 1x EDTA in DPBS at 1:1,000) (Corning, catalog number: 46-034-CI )
- Sodium Pyruvate (Corning, catalog number: 25-000-Cl )
- HEPES Buffer (Corning, catalog number: 25-060-CI )
- Sodium Bicarbonate (Corning, catalog number: 25-035-CI )
- Non-essential Amino Acid (Corning, catalog number: 25-025-CI )
- N1 Supplement (Sigma-Aldrich, catalog number: N6530 )
- Fetal Bovine Serum (FBS) (Atlanta Biologicals, catalog number: S11150 )
- KnockOutTM Serum Replacer (KSR) (Thermo Fisher Scientific, catalog number: 10828028 )
- IWR1-endo (Stemgent, catalog number: 04-0010 )
- LDN 193189 (Stemgent, catalog number: 04-0074 )
- Insulin-like growth factor 1 (IGF1) (R&D Systems, catalog number: 291-G1 )
- TrypLE Express dissociation reagent (Thermo Fisher Scientific, catalog number: 12604021 )
- 1x HBSS (Corning, catalog number: 21-022-CM )
- Dimethyl Sulfoxide (DMSO) (Fisher Scientific, catalog number: BP231-1 )
- Poly-D-lysine (Sigma-Aldrich, catalog number: P6407 )
- 4% Paraformaldehyde solution (Electron Microscopy Sciences, catalog number: 157-4 )
- 10% Normal Donkey Serum Blocking Solution (Jackson Immuno Research Laboratories, catalog number: 017-000-001 )
- Triton X-100 (Sigma-Aldrich, catalog number: T8787 )
- Anti-fading Fluoromount G mounting medium (Electron Microscopy Sciences, catalog number: 17984-25 )
- 4',6-diamidino-2-phenylindole (DAPI, Enzo Life Scienes, catalog number: ENZ-52404 )
- Monoclonal mouse anti PAX6 antibody (DHSB, catalog number: PAX6 )
- Polyclonal goat anti LHX2 antibody (Santa Cruz Biotechnology, catalog number: sc-19344 or sc-517243 )
- Biotinylated polyclonal goat anti human OTX2 antibody (R&D Systems, catalog number: BAF1979 )
- Polyclonal Rabbit anti CRX antibody (GeneTex, catalog number: GTX124188 )
- Anti-Recoverin Antibody (EMD Millipore, catalog number: AB5585 )
- Freezing Medium (NSC + 0.5% FBS Medium + 10% DMSO)
- Essential 8 human ESC/iPSC culture medium (see Recipes)
- Rock inhibitor Y-27632 (10 mM, 1,000x, Stemgent, catalog number: 04-0012 ) (see Recipes)
- Neuronal Stem Cell Culture Medium (NSC) (see Recipes)
- NSC + 0.5% FBS Medium (see Recipes)
- ISLI + KSR Retinal Induction Medium (see Recipes)
Equipment
- Vertical laminar flow hood certified for Level II handling of biological materials
- Water bath
- Incubator with humidity and gas control to maintain 37 °C and 95% humidity in an atmosphere of 5% CO2 in air
Note: The incubator needs to be able to adjust the O2 level, as required for Step A4i. - Low-speed centrifuge with a swinging bucket rotor (e.g., Beckman Coulter, model: GS-6 ) with an adaptor for plate holders
- Pipette-aid with appropriate serological pipettes
- Hemocytometer
- Micropipette with appropriate tips
- Mr. Frosty freezing container at room temperature (Thermo Fisher Scientific, catalog number: 5100-0001 )
- -150 °C freezer or liquid nitrogen (LN2) vapor tank
- -80 °C freezer
- -20 °C freezer
- Refrigerator (2-8 °C)
- Inverted microscope
Procedure
- Thawing and passaging human ES and iPS cells
- Sterilization before handling the cells
Expose the Biological Safety Cabinet to UV light for 1 h prior use, spray 70% ethanol and wipe the working area inside the cabinet, the tools and supplies that will be used inside the cabinet. - Preparation of Matrigel-coated plates
- Take 1 aliquot of frozen Matrigel (110 µl) (BD Biosciences) and thaw on ice.
Note: The Matrigel aliquots are prepared ahead of time and stored in -80 °C freezer. - Dissolve 110 µl Matrigel in 12 ml chilled (4 °C) DMEM/F-12 basal medium (GE Healthcare), pipette up and down to mix the Matrigel and medium very well.
- Add 2 ml of the mixture of Matrigel and medium to each well of a 6-well plate (Greiner Bio One, DNase and RNase free, treated), and let the plate sit in an incubator (37 °C) for 2-12 h before seeding the cells into the plate.
- Take 1 aliquot of frozen Matrigel (110 µl) (BD Biosciences) and thaw on ice.
- Preparation of Essential 8 human ESC/iPSC culture medium (see Recipe 1)
- Thawing and seeding cells
In general, 1 well of the cells in a 6-well plate that is at 60-70% of confluence is prepared to be frozen into 2 cryovials. One vial of cryopreserved cells can be successfully recovered into 1-2 wells of a Matrigel-coated 6-well plate.
Note: The number of wells plated may need to be adjusted depending on how many cell aggregates were cryopreserved. Typically, twice as many cell aggregates will need to be plated per well upon thawing to account for lower post-thaw viability than during routine passaging.- Label the Matrigel-coated plate, warm Essential 8 culture medium (with supplement of Rock Inhibitor [Recipe 2]) to room temperature (~25 °C), and prepare all other cell culture supplies before starting the protocol to ensure that the thawing procedure is done as quickly as possible.
- Quickly thaw the cells in a 37 °C water bath by gently shaking the cryovial continuously until only a small frozen cell pellet remains.
- Remove the cryovial from the water bath and wipe it with 70% ethanol.
- Use a 2 ml serological pipette to transfer the contents of the cryovial to a 15 ml conical tube.
Note: Using a 2 ml serological pipette instead of a 1 ml micropipette will minimize breakage of cell aggregates. - Add 5-7 ml of warm Essential 8 medium (supplied with Rock Inhibitor) dropwise to the 15 ml tube, gently mixing as the medium is added.
- Centrifuge cells at 270 x g for 3 min at room temperature.
- Aspirate the medium, leaving the cell pellet intact, gently resuspend the cell pellet in 1 ml of Essential 8 medium (with Rock Inhibitor) using a 2 ml serological pipette, maintain the cells as aggregates.
- Transfer 0.5 ml of the cell mixture onto a well of a Matrigel-coated 6-well plate containing Essential 8 with Rock Inhibitor (i.e., two wells can be plated from each cryovial).
- Place the plate in a 37 °C hypoxia-capable incubator (5% CO2, 5% O2), move the plate in several quick, short, back-and-forth and side-to-side motions to evenly distribute the cell aggregates. Do not disturb the plate for 24 h.
Note: Uneven distribution of aggregates may result in increased differentiation of human ES and iPS cells (Figure 2). - Perform daily medium changes using Essential 8 medium and visually assess cultures to monitor the growth until the next passaging time. Check for undifferentiated colonies that are ready to be passaged (dense-centered) approximately 6-7 days after thawing (Figure 1 and Figure 2A).
Note: If only a few undifferentiated colonies are observed after thawing, it may be necessary to select only these colonies for passaging and replate them in the same size well (i.e., without splitting) on a newly-coated plate.
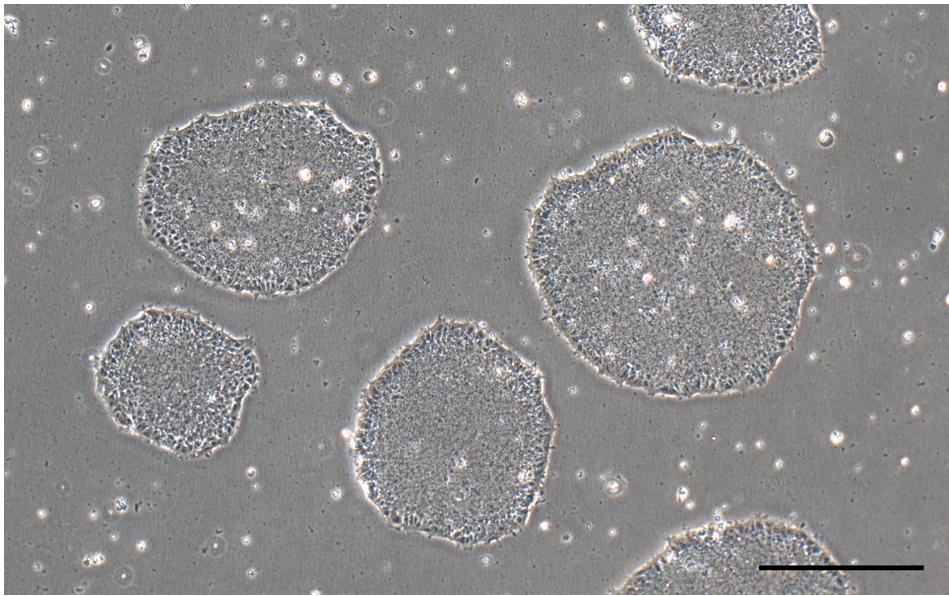
Figure 1. Undifferentiated iPSC colonies in Essential 8 media (scale bar = 400 μm) - Label the Matrigel-coated plate, warm Essential 8 culture medium (with supplement of Rock Inhibitor [Recipe 2]) to room temperature (~25 °C), and prepare all other cell culture supplies before starting the protocol to ensure that the thawing procedure is done as quickly as possible.
- Passaging human ES and iPS cells
Human ES and iPS cells maintained in Essential 8 medium can be passaged using several different methods. The non-enzymatic EDTA dissociation method is described below:- Clean off any flat or differentiating cells (Figure 2B) with a 1,000 µl pipette tip under a microscope in a sterilized working area.
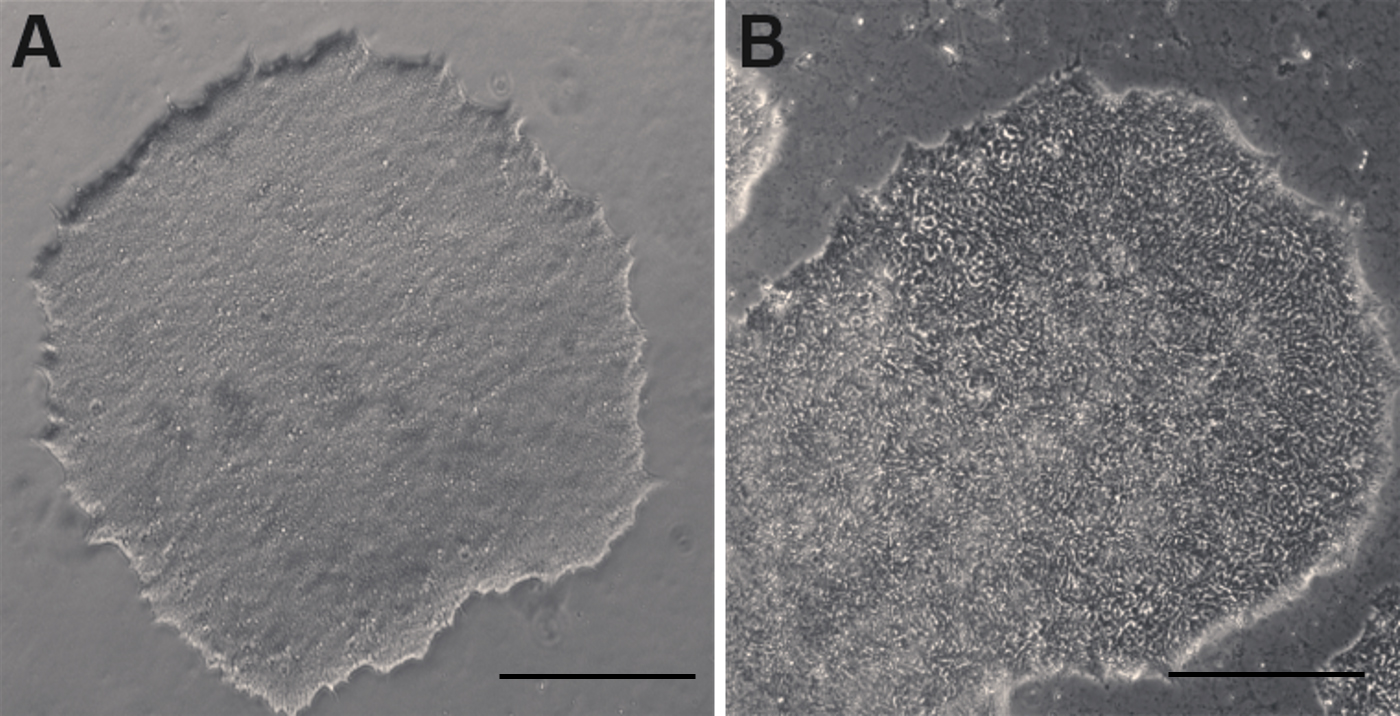
Figure 2. Undifferentiated and differentiating human ESC colonies in culture. A. An undifferentiated human ESC colony. B. A differentiating human ESC colony with whorled appearance that needs to be cleaned up prior to subculture. (scale bars = 400 µm)- Rinse cells with 2 ml of DPBS once or twice.
- Add 1 ml of 0.1% EDTA dissociation reagent per well on a 6-well plate, incubate at room temperature for 3-5 min.
- Check under a microscope for signs of dissociation.
- Aspirate EDTA dissociation reagent without disturbing the cells.
- Add 1 ml of Essential 8 medium (with Rock Inhibitor supplement).
- Use a cell scraper to lift the cells from the bottom of the well, triturate with a 1,000 µl pipette gently to break down into cell clusters containing 20-40 cells.
- Add 2 ml of Essential 8 medium (with Rock Inhibitor added) to a new well.
- Add cells from original well to the new well to reach 20% of confluence. Usually 1:5 ratio for a confluent plate.
Note: If there are too few cells, cells don't grow as well; if there are too many cells, cells grow too quickly and should be split soon. Small clumps are ideal, if cells remain as big chunks, gently triturate the large size cell aggregates with a 1,000 µl pipette for a few times to break them down. - Place the plate back into a hypoxia 37 °C incubator (5% CO2 and 5% O2) and evenly distribute cells by shaking plate front to back and side to side as above.
- Perform daily medium changes, 2 ml Essential 8 medium per well, and visually assess cultures to monitor growth until the next passaging time.
- Clean off any flat or differentiating cells (Figure 2B) with a 1,000 µl pipette tip under a microscope in a sterilized working area.
- Sterilization before handling the cells
- Induction of neuro-retinal fate
- Medium preparation
- Preparation of Neuronal Stem Cell Culture Medium (NSC) (500 ml) (see Recipe 3).
- Preparation of NSC medium with 0.5% FBS (100 ml) (see Recipe 4).
- Preparation of ISLI + KSR Retinal Induction Medium (50 ml) (see Recipe 5).
- Retinal induction in human ES and iPS cells
When the cells in wells reach 60-70% confluence, start treating cells with ISLI + KSR retinal induction medium (2 ml per well of a 6-well plate), daily medium change for 5-6 days. Once the cells are in ISLI + KSR medium, culture the cells in a normoxic 37 °C incubator with 5% CO2 level.
Procedure of splitting differentiating cells (Days 6-7)- Change 2 ml fresh ISLI + KSR retinal induction medium for cells at least 1 h before splitting.
- At the time of splitting, aspirate the medium.
- Add 1 ml TrypLE Express dissociation reagent to the well; incubate 6-8 min at room temperature until the cells loose contact with each other and the plate.
- Scrape the cells gently from the bottom of the well and transfer the cell suspension into a 15 ml centrifuge tube. Take care not to make single cell suspension.
- Rinse the well with 2 ml 1x HBSS once to collect the remaining cells from the well.
- Centrifuge at 270 x g for 3 min.
- Aspirate the supernatant gently without disturbing the cell pellet.
- Add 3-4 ml 1x HBSS to resuspend the cell pellet, centrifuge again at 270 x g for 3 min.
- Aspirate 1x HBSS gently without disturbing the cell pellet.
- Resuspend the cell pellet in fresh ISLI + KSR retinal induction medium. The splitting ratio is 1:3.
- Evenly distribute the cells as above by shaking the plate and return to the incubator under normoxic conditions.
- Next day, check the cell survival by looking the percentage of cells attached to the bottom of the plate, dead cells do not attach and float in culture medium. If there is too much cell death, rinse cells with 2 ml 1x HBSS once or twice to clean up the dead cells.
- Wean cells into NSC culture medium supplemented with 0.5% FBS gradually by adding 1 ml ISLI + KSR medium and 1 ml NSC + 0.5% FBS medium on Day 1 post-split; 0.5 ml ILSI + KSR medium + 1.5 ml NSC + 0.5% FBS medium on Day 2; from Day 3 and onward, the differentiating cells will be cultured in NSC + 0.5% FBS medium to let them undergo further differentiation. When the cells reach confluence, the cells need to be split into new Matrigel-coated plates with the TrypLE dissociation method. This is usually done every 7 days with a split ratio of 1:3 to 1:5 depending on the cell line.
Note: Rock Inhibitor can be added to the NSC medium at the step to help cell survive after dissociation if the cells are old or stressed.
- Change 2 ml fresh ISLI + KSR retinal induction medium for cells at least 1 h before splitting.
- Medium preparation
- Isolation of Neuroretinal Rosettes (Days 18-21)
The differentiating cells are mixed populations that are composed of retinal progenitor cell population and retinal pigmented epithelial progenitor cells (RPE) (Figure 3A) as they both arise from the same optic vesicle. The retinal stem/progenitors form clusters (neuroretinal rosettes) and can be manually separated and expanded (Figure 3B).
Figure 3. Differentiated Early Retinal Rosettes in Culture. A. Retinal Rosettes prior to picking and sorting at 3 weeks; B. Purified neuro-retinal cultures following picking and replating. Scale bars = 100 μm.
Procedure of manually separation of the retinal stem/progenitor cells and retinal pigmented epithelial cells- Sterilize the bench surface and any areas on and around the microscope and tools that need to be in direct contact with the cells with 70% ethanol.
- Change to fresh NSC + 0.5% FBS medium for the cells before picking.
- Under a microscope, gently scrape the areas that have densely-packed neuronal cells with a sterile micropipette tip. If too large, scrape regions containing 100-200 cell clusters.
- After most of neuronal areas are lifted, collect the medium that contains the floating neuronal rosettes.
- Transfer them into a new Matrigel-coated well with a 1,000 µl pipette to let the cells attach and grow in NSC medium in the incubator (37 °C, 5% CO2).
- Sterilize the bench surface and any areas on and around the microscope and tools that need to be in direct contact with the cells with 70% ethanol.
- Retinal Stem/Progenitor cell expansion
The manually purified retinal stem/progenitors need to be cultured in NSC + 0.5% FBS medium for several months to allow the cells undergo further differentiation to give rise to all types of differentiated retinal neurons (Figure 4). The dissociation method is described below:
TrypLE Dissociation Method of Differentiating Retinal Cells- Aspirate the old medium.
- Rinse cells with 1-2 ml 1x HBSS (or DPBS) once or twice.
- Aspirate 1x HBSS.
- Add 1 ml of TrypLE Express dissociation reagent to the cells, incubate for 6-8 min at room temperature, and check under the microscope for signs of dissociation (The cell-cell connections loosen up and empty spaces show up among densely packed cells). Do not wait until the cells are lifted (Figure 4).

Figure 4. TrypLE dissociation of differentiating retinal cells for expansion. A. Morphology of retinal cells before dissociation. B. Morphology of retinal cells after 6 min of TrypLE dissociation. Scale bars = 400 µm. - Scrape cells using a cell scraper.
- Transfer cells into a 15 ml centrifuge tube.
- Spin at 270 x g for 3 min.
- Aspirate the supernatant and add 2 ml 1x HBSS and resuspend the cells.
- Spin at 270 x g for 3 min.
- Aspirate the supernatant and add NSC + 0.5% FBS medium to resuspend the cells (2 ml per well), usually the splitting ratio is 1:3.
Note: Rock Inhibitor can be added to the medium at the step to help cell survive after dissociation if the cells appear stressed. - Add 2 ml of cells in NSC + 0.5% FBS medium into the wells.
- Put the plate into an incubator (37 °C, 5% CO2) and shake left to right and back and forth to evenly distribute the cells.
- Cells will be cultured in NSC + 0.5% FBS medium.
- Change media every 2-3 days as needed.
- Aspirate the old medium.
- Retinal Stem/Progenitor cell cryopreservation
Freezing Neuro-retinal Stem Cells- Rinse cells with 1-2 ml of 1x HBSS twice.
- Add 1 ml of TrypLE per well on a 6-well plate and incubate for 6-8 min.
- During this time, start labeling cryovials with date, cell line, info, etc.
- Check under a microscope for signs of dissociation as in Step D4.
- Scrape cells using a cell scraper.
- Transfer everything into a 15 ml tube.
- Spin for 3 min at 270 x g.
- Dispose of the supernatant.
- Add 2 ml of 1x HBSS and resuspend the cells.
- Spin for 3 min at 270 x g.
- Dispose of the supernatant.
- Add the calculated amount of freezing medium to resuspend the cells.
- Add 1 ml of cells and freezing medium to each cryovial.
- Immediately place all cryovials into a freezing chamber.
- Transfer chamber into a -80 °C freezer.
- Next day, remove vials and place into an appropriate box in the -80 °C freezer.
- Transfer vials to liquid nitrogen next day.
- Rinse cells with 1-2 ml of 1x HBSS twice.
- Thawing Retinal Stem/Progenitor cells
One vial of cryopreserved retinal stem/progenitor cells can be successfully thawed into 1 well of a Matrigel-coated 6-well plate.- Label the Matrigel-coated plate, warm NSC + 0.5% FBS medium (with supplement of Rock Inhibitor) to room temperature (~25 °C) and prepare all other cell culture supplies before starting the protocol to ensure that the thawing procedure is done as quickly as possible.
- Quickly thaw the cells in a 37 °C water bath by gently shaking the cryovial continuously until only a small frozen cell pellet remains.
- Remove the cryovial from the water bath and wipe it with 70% ethanol or isopropanol to sterilize.
- Use a 1 ml micropipette to transfer the contents of the cryovial to a 15 ml conical tube.
- Add 7 ml of 1x HBSS dropwise to the 15 ml tube, gently mixing as the medium is added.
- Centrifuge at 270 x g for 3 min.
- Aspirate the supernatant and add 2 ml of warm NSC medium (supplied with Rock Inhibitor) to the 15 ml tube to resuspend the cells.
- Transfer the cell mixture onto an empty well of a Matrigel-coated 6-well plate.
- Place the plate in a 37 °C normoxic incubator (5% CO2), move the plate in several quick, short, back-and-forth and side-to-side motions to distribute the cell aggregates. Do not disturb the plate for 24 h.
- Change media next day. If excessive cell death occurs, rinse cells with 1x HBSS before medium change.
- Perform medium changes every 2-3 days as needed using NSC medium and visually assess cultures to monitor the growth until the next passaging time.

Figure 5. Confluent Neuro-retinal culture following three months of culture (scale bar = 400 μm)
Figure 6. Immunocytochemistry analysis of the retinal cells generated from human iPSCs at 6 weeks of retinal differentiation. Cells expressed retinal stem cell markers PAX6 and LHX2 as well as photoreceptor markers, OTX2 and RCVRN (scale bars = 10 µm). - Label the Matrigel-coated plate, warm NSC + 0.5% FBS medium (with supplement of Rock Inhibitor) to room temperature (~25 °C) and prepare all other cell culture supplies before starting the protocol to ensure that the thawing procedure is done as quickly as possible.
- Immunocytochemistry protocol
- Aspirate the old culture media.
- Rinse cells in 1x HBSS 2 times gently without lifting the cells.
- Fix cells in 4% Paraformaldehyde solution (Electron Microscopy Sciences) for 30 min at room temperature.
- Rinse cells in 1x PBS for 3 times, 5 min for each rinse.
- Block the cells in 10% Normal Donkey Serum Blocking Solution (Jackson Immuno Research Laboratories, the blocking solution is prepared by diluting the stock in 1x PBS that contains 0.5% Triton X-100) for 1 h at room temperature.
- Add the primary antibodies with pre-determined concentrations (Table 1) (prepared in serum blocking solution) to the cells and incubate overnight in a cool room in a humidified chamber.
Table 1. Primary Antibodies for retinal analysis
- Next day, rinse off the non-binding primary antibodies with 1x PBS, 3 times, 5 min for each rinse.
- Add the secondary Antibodies and incubate at room temperature for 1 h in the dark in a humidified chamber.
- Rinse off the non-binding secondary antibodies with 1x PBS 3 times, 5 min for each rinse.
- DAPI (working concentration is 0.5 µg/ml, prepared in PBS) stains the nuclei of cells for 3-5 min.
- Mount the cells on cover glasses onto histology slides with anti-fading Fluoromount G mounting medium (Electron Microscopy Sciences).
- Aspirate the old culture media.
Data analysis
Data analysis for the differentiated retinal cells is carried out by t-test. Total cells expressing a given antibody as well as total cells in a field (marked by DAPI) were counted from 3-5 fields and at least 3 independent experiments. The results were compared to the marker expression in either undifferentiated cells or alternate differentiation media. Detailed analysis of induced retinal cell differentiation of human ES cells and iPS cells via this protocol can be found in a recently published open access paper by the lab (Zhu et al., 2018).
Notes
While the protocol works reliably in multiple cell lines tested by our lab. There is some inherent variability due to different culture conditions experienced by various undifferentiated cells or methods of derivation. Authors also advise all labs to routinely test lines for mycoplasma contamination using commercial kits as they significantly affect differentiation capacity.
Recipes
- Essential 8 human ESC/iPSC culture medium

For newly thawed cells, use Essential 8 medium with supplementation of Rock inhibitor (Y-27632, 10 µM) to promote cell survival. - Rock inhibitor (Y-27632) stock (10 mM, 1,000x)
Dissolve 2 mg lyophilized Y27632 (Stemgent) in 625 µl double distilled water to make 10 mM mother stock and make 10 µl aliquots and store in -20 °C freezer - Neuronal Stem Cell Culture Medium (NSC) (500 ml)

- NSC medium with 0.5% FBS (100 ml)

- ISLI + KSR Retinal Induction Medium (50 ml)
Note: In “ISLI + KSR”, “I” stands for IWR1, “S” stands for SB431542, “L” stands for LDN193189, the last “I” stands for IGF1, and KSR stands for KnockOutTM Serum Replacer.
Acknowledgments
We are grateful to Dr. Xianmin Zeng for the human iPS cell lines (NCL-1, NCL1-GFP). The human ES line was purchased from WiCell. The work was supported through funds from NEI grant EY025779, Buck Institute funds. This protocol is based on our previous work (Zhu et al., 2018).
References
- Hambright, D., Park, K. Y., Brooks, M., McKay, R., Swaroop, A. and Nasonkin, I. O. (2012). Long-term survival and differentiation of retinal neurons derived from human embryonic stem cell lines in un-immunosuppressed mouse retina. Mol Vis 18: 920-936.
- Hirami, Y., Osakada, F., Takahashi, K., Okita, K., Yamanaka, S., Ikeda, H., Yoshimura, N. and Takahashi, M. (2009). Generation of retinal cells from mouse and human induced pluripotent stem cells. Neurosci Lett 458(3): 126-131.
- Lamba, D. A., Karl, M. O., Ware, C. B. and Reh, T. A. (2006). Efficient generation of retinal progenitor cells from human embryonic stem cells. Proc Natl Acad Sci U S A 103(34): 12769-12774.
- Lamba, D. A., Gust, J. and Reh, T. A. (2009). Transplantation of human embryonic stem cell-derived photoreceptors restores some visual function in Crx-deficient mice. Cell Stem Cell 4(1): 73-79.
- Lamba, D. A., McUsic, A., Hirata, R. K., Wang, P. R., Russell, D. and Reh, T. A. (2010). Generation, purification and transplantation of photoreceptors derived from human induced pluripotent stem cells. PLoS One 5(1): e8763.
- Meyer, J. S., Shearer, R. L., Capowski, E. E., Wright, L. S., Wallace, K. A., McMillan, E. L., Zhang, S. C. and Gamm, D. M. (2009). Modeling early retinal development with human embryonic and induced pluripotent stem cells. Proc Natl Acad Sci U S A 106(39): 16698-16703.
- Osakada, F., Ikeda, H., Mandai, M., Wataya, T., Watanabe, K., Yoshimura, N., Akaike, A., Sasai, Y. and Takahashi, M. (2008). Toward the generation of rod and cone photoreceptors from mouse, monkey and human embryonic stem cells. Nat Biotechnol 26(2): 215-224.
- Osakada, F., Jin, Z. B., Hirami, Y., Ikeda, H., Danjyo, T., Watanabe, K., Sasai, Y. and Takahashi, M. (2009). In vitro differentiation of retinal cells from human pluripotent stem cells by small-molecule induction. J Cell Sci 122(Pt 17): 3169-3179.
- Zhong, X., Gutierrez, C., Xue, T., Hampton, C., Vergara, M. N., Cao, L. H., Peters, A., Park, T. S., Zambidis, E. T., Meyer, J. S., Gamm, D. M., Yau, K. W. and Canto-Soler, M. V. (2014). Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat Commun 5: 4047.
- Zhu, J., Cifuentes, H., Reynolds, J. and Lamba, D. A. (2017). Immunosuppression via loss of IL2rγ enhances long-term functional integration of hESC-derived photoreceptors in the mouse retina. Cell Stem Cell 20(3): 374-384 e375.
- Zhu, J., Reynolds, J., Garcia, T., Cifuentes, H., Chew, S., Zeng, X. and Lamba, D. A. (2018). Generation of transplantable retinal photoreceptors from a current good manufacture practice-manufactured human induced pluripotent stem cell line. Stem Cells Transl Med.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Zhu, J. and Lamba, D. A. (2018). Small Molecule-Based Retinal Differentiation of Human Embryonic Stem Cells and Induced Pluripotent Stem Cells. Bio-protocol 8(12): e2882. DOI: 10.21769/BioProtoc.2882.
Category
Stem Cell > Pluripotent stem cell > Cell differentiation
Stem Cell > Embryonic stem cell > Cell differentiation
Cell Biology > Cell isolation and culture > Cell differentiation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.