- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

High Dimensional Functionomic Analysis of Human Hematopoietic Stem and Progenitor Cells at a Single Cell Level

Published: Vol 8, Iss 10, May 20, 2018 DOI: 10.21769/BioProtoc.2851 Views: 8691
Reviewed by: Ruth A. FranklinLokesh KalekarChris Tibbitt
Original research article
The authors used this protocol in:

Jul 2017

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
The ability to conduct investigation of cellular transcription, signaling, and function at the single-cell level has opened opportunities to examine heterogeneous populations at unprecedented resolutions. Although methods have been developed to evaluate high-dimensional transcriptomic and proteomic data (relating to cellular mRNA and protein), there has not been a method to evaluate corresponding high-dimensional functionomic data (relating to cellular functions) from single cells. Here, we present a protocol to quantitatively measure the differentiation potentials of single human hematopoietic stem and progenitor cells, and then cluster the cells according to these measurements. High dimensional functionomic analysis of cell potential allows cell function to be linked to molecular mechanisms within the same progenitor population.
Keywords: CD34+ hematopoietic stem and progenitor cellsBackground
The development of techniques for single-cell measurements of cell transcription, signaling, and function at the single-cell level, alongside preexisting technologies such as flow cytometry, has allowed new lenses to examine complex, heterogeneous populations. Such methods generate large amounts of data, which can be interpreted with aid from dimensionality reduction algorithms, as illustrated on single-cell RNA-Seq using Mpath, Monocole, PCA, Wishbone, or diffusion map algorithms (Paul et al., 2016; See et al., 2017), and on CyTOF using tSNE or PhenoGraph (Amir el et al., 2013; Levine et al., 2015).
We developed this protocol to allow functional analysis and subsequent dimension reduction of large-scale culture of hematopoietic progenitors in single-cell environments. In this protocol, we describe a method to culture single cells of human CD34+ hematopoietic stem and progenitor cells (HSPCs) in a stromal cell culture with cytokines, to enumerate the clonal outcome (functionomics) of six different lineages (i.e., granulocyte, monocyte, lymphocyte, CD141+ dendritic cell (conventional type 1 DC (cDC1)), CD1c+ dendritic cell (conventional type 2 DC (cDC2)), and plasmacytoid dendritic cells (pDC)) from each progenitor, and to cluster the progenitors according to function with a dimension reduction method. In our previous paper, we showed that this is feasible for a population of 2,247 progenitors; each progenitor can be individually plotted to form a two-dimensional ‘map’ of 2,247 data points (Lee et al., 2017). Our protocol allows for the functional clustering of single cells. Such high dimensional functionomic analysis aids in linking cell function to molecular mechanism of any given cell population.
Materials and Reagents
- 15 cm culture plates (Corning, Falcon®, catalog number: 353025 )
- 15 ml Falcon tubes (Corning, Falcon®, catalog number: 352097 )
- 96-well V-bottom plates (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 249570 )
- Flat-bottom 96-well plates (Corning, Falcon®, catalog number: 353072 )
- 50 ml Falcon tubes (Corning, Falcon®, catalog number: 352098 )
- Serological pipettes (Fisher Scientific, catalog number: 13-678-11 )
- Opaque Eppendorf tubes (CELLTREAT Scientific Products, catalog number: 229437 )
- Eppendorf tubes (Fisher Scientific, catalog number: 05-408-129 )
- Pipette tips
- Pasteur pipette (Fisher Scientific, catalog number: 13-678-8B )
- 100 μm cell strainer (Corning, Falcon®, catalog number: 352360 )
- 0.20 μm filter (Corning, catalog number: 431229 )
- MS5 (Lee et al., 2015a and 2015b)
- OP9 (Lee et al., 2015a and 2017)
- dH2O (Thermo Fisher Scientific, InvitrogenTM, catalog number: 10977023 )
- Trypsin (Corning, catalog number: 25-052-CI )
- Trypan blue (Thermo Fisher Scientific, GibcoTM, catalog number: 15250061 )
- 70% ethanol
- Ficoll-Paque Plus (GE Healthcare, catalog number: 17144002 )
- Anti-human CD34 MACS microbeads (Miltenyi Biotec, catalog number: 130-046-702 )
- Recombinant human Flt3L (Celldex Therapeutics, catalog number: CDX-301 )
- Recombinant human SCF (PeproTech, catalog number: 300-07 )
- Recombinant human GM-CSF (PeproTech, catalog number: 300-03 )
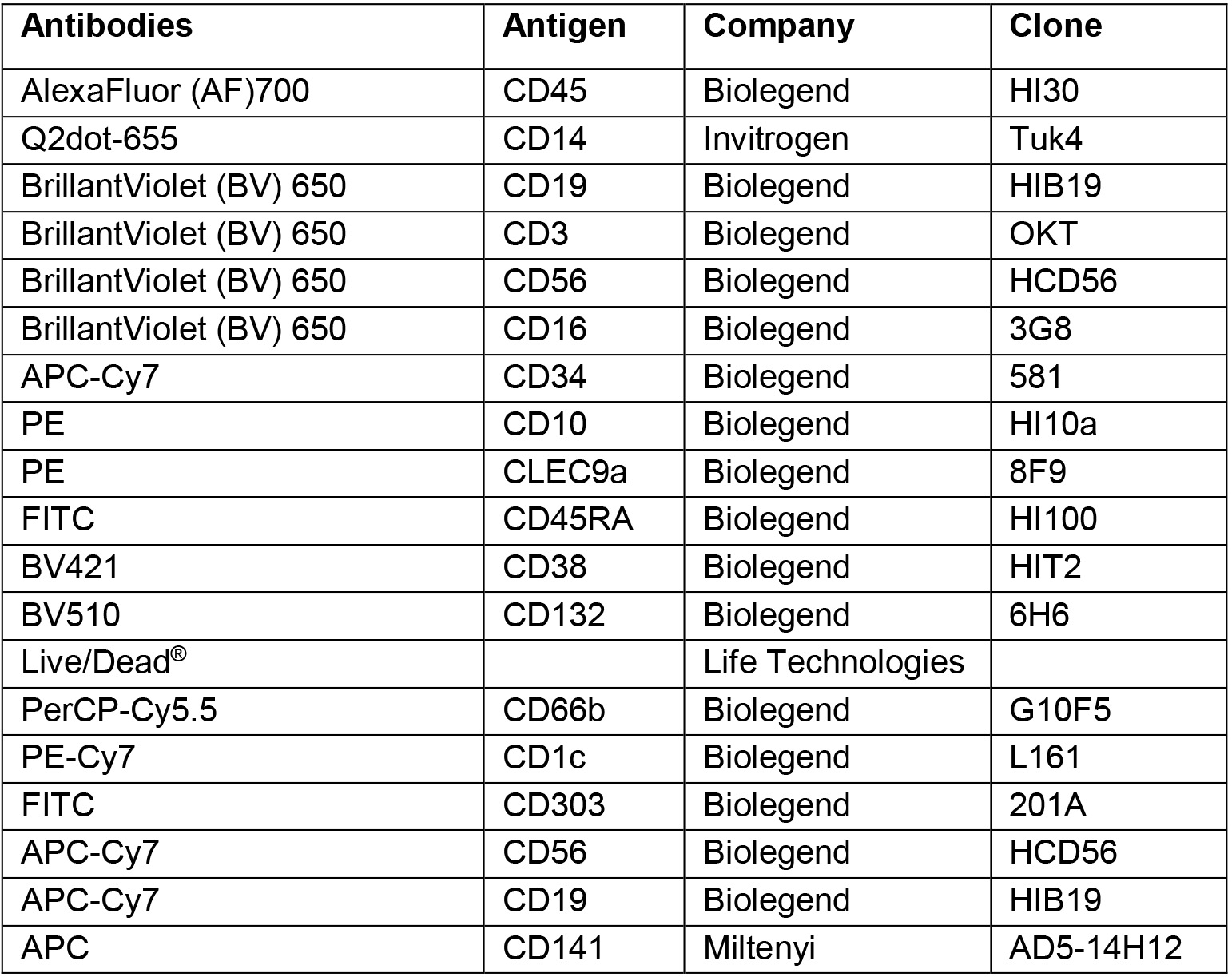
- Antibodies (details of clone name, fluorochrome, manufacturer and dilution are listed in Tables 1 and 3)
- MEMα with deoxyribonucleosides (Thermo Fisher Scientific, GibcoTM, catalog number: 12571071 )
- Fetal bovine serum (FBS) (Thermo Fisher Scientific, GibcoTM, catalog number: 10437028 )
- Pen-Strep (Thermo Fisher Scientific, GibcoTM, catalog number: 15140122 )
- Mitomycin C (Sigma-Aldrich, catalog number: M4287-5X2MG )
- Dulbecco’s phosphate-buffered saline (DPBS) (GE Healthcare, catalog number: SH30378.02 )
- Ethylenediaminetetraacetic acid (EDTA) (Corning, catalog number: 46-034-CI )
- Bovine serum albumin (BSA) (ThermoFisher, catalog number: BP1600-100 )
- FcR blocking buffer (Miltenyi Biotec, catalog number: 130-059-901 )
- CountBright Absolute Counting Beads (Thermo Fisher Scientific, InvitrogenTM, catalog number: C36950 )
- Complete MEMα medium (see Recipes)
- Mitomycin C stock (1 mg/ml) (see Recipes)
- FACS buffer (see Recipes)
- Anti-CD34 microbeads/FcR blocking mix (see Recipes)
Equipment
- Pipettes
- Incubator (Forma Scientific, model: 3354 )
- Centrifuge (Eppendorf, model: 5810 R , catalog number: 5811000320)
- Vortex with universal holder (VWR, catalog number: 97043-562 )
- Microscope
- Flow cytometer (BD, models: BD LSRII or LSRFortessaTM )
Procedure
- Culture of stromal cells (day -4 – day 0)
- Passaging stromal cells (maximum of 13 times)
- Grow MS5 and OP9 cells in complete MEMα medium. For 15 cm plates, seed 1.5 million cells in 20 ml medium.
- For normal passage, microscopically verify that the cells are 80-90% confluent.
- Remove medium from the culture plate by aspiration and wash cells with 10 ml sterile PBS to remove medium and FBS.
- Remove the PBS, and then add 3 ml trypsin pre-warmed to 37 °C.
- Incubate for 2-3 min at 37 °C, and microscopically verify that cells have started to detach.
- Add 10 ml complete MEMα medium pre-warmed to 37 °C to stop trypsin activity.
- Resuspend cells by pipetting up and down, and then transfer cells to 15 ml collecting tubes.
- Centrifuge at 450 x g (1,500 rpm) for 5 min at 4 °C.
- Remove supernatant, and then resuspend the cells in 1 ml complete MEMα medium (Cell Concentrate).
- In a new 15 cm plate, add 20 ml complete MEMα medium. Transfer 100 μl cell concentrate to the new plate. This new plate will become confluent after 3-4 days.
- Incubate at 37 °C, 5% CO2 for 3-4 days, or until cells are 80-90% confluent.
- Passage the cells again, to a maximum of 13 passages.
- For MS5 and OP9 cells, cell yield from a 15 cm plate with 80-90% confluency is 15-20 million cells.
- Grow MS5 and OP9 cells in complete MEMα medium. For 15 cm plates, seed 1.5 million cells in 20 ml medium.
- Treating stromal cells with Mitomycin-C
- Verify that MS5/OP9 cells are 80-90% confluent.
- Label collecting tubes (‘MS5 + mitC’, ‘OP9 + mitC’).
- Add Mitomycin C stock (1 mg/ml, 100x) directly into the medium to achieve a final concentration of 10 μg/ml. For example, aspirate medium and add back exactly 10 ml, and add 100 μl Mitomycin C stock.
- Mix by swirling gently for 3-5 sec.
- Incubate at 37 °C for 3 h.
- Verify that MS5/OP9 cells are 80-90% confluent.
- Plating stromal cells
- Remove medium, and then add 10 ml PBS. Gently swirl the plate for 3-5 sec.
- Remove the PBS, and then add 3 ml trypsin.
- Incubate for 2-3 min at 37 °C, and use a microscope to verify that cells have started to detach.
- Add 10 ml complete MEMα medium to stop trypsin.
- Resuspend cells by pipetting up and down, and then transfer cells to labeled harvesting tubes.
- Centrifuge at 450 x g for 5 min at 4 °C.
- Remove medium, and then resuspend in 1 ml complete MEMα medium.
- Count cells, and calculate the number and volume of cells needed for the final plating. Each well in a flat-bottom 96-well plate, should contain 6,250 OP9 cells and 37,500 MS5 cells in 50 μl complete MEMα medium. To prepare for one 96-well plate, resuspend 0.625 x 106 OP9 cells and 3.75 x 106 MS5 cells in 5 ml complete MEMα medium, mix well, and then distribute 50 μl into each well.
- Incubate cells at 37 °C overnight.
- Verify that cells are securely attached to plate by shaking gently while viewing under a light microscope. With secure attachment, the cells should not drift around with the fluid movement.
- Within 1-3 days, sort and plate progenitors on these cells.
- Remove medium, and then add 10 ml PBS. Gently swirl the plate for 3-5 sec.
- Passaging stromal cells (maximum of 13 times)
- Purification of CD34+ hematopoietic stem and progenitor cells from human cord blood (day 0)
- Isolation of cord blood mononuclear cells using Ficoll-Paque
Note: This step should be handled at room temperature.- Add 25 ml PBS into multiple 50 ml tubes.
- Sterilize the outside of the cord blood sample container with ethanol. Samples usually contain about 100 ml of blood.
- In 15 ml portions, evenly distribute the cord blood sample into the prepared 50 ml Falcon tubes containing PBS.
- Rinse the blood sample container with some additional room temperature PBS, and use the rinse to ‘top off’ the last Falcon tube (such that each tube has a fluid volume of 40 ml). On average, use 7 Falcon tubes per bag of cord blood.
- Tightly seal each of the tubes, and gently invert them 3-5 times to resuspend any sedimented cells.
- Underlay the cell suspension with 10.5 ml of Ficoll-Paque with a 10 ml Serological pipette, in the following manner:
- Load 10.5 ml into the pipette.
- Without releasing any fluid, gently bring the pipette to the bottom of the tube.
- For the first 2 ml, very slowly release Ficoll-Paque into the bottom of the tube. The goal is to have a pure layer of Ficoll-Paque at the bottom of the tube.
- For the next 8 ml, the Ficoll-Paque release can be slightly sped up, but use caution to avoid turbulence that may disrupt the Ficoll-Paque-blood boundary.
- For the last 0.5 ml, slow down the Ficoll-Paque release, and observe the meniscus in the pipette. Use caution to prevent introducing air bubbles into Ficoll-Paque.
- Load 10.5 ml into the pipette.
- Centrifuge at 450 x g (1,500 rpm) for 25 min at room temperature. Use lower acceleration (level 3 of 9) and no brakes for deceleration. Orient the opaque sections of the test tube face towards the central motor of the centrifuge, so that cell deposits will collect on a transparent section of the test tube, away from the motor, and so can be easily visualized.
- After centrifugation, there should be four layers:
- The topmost layer (yellow/clear, ~35 ml) is a PBS/serum mix.
- The second layer from top (cloudy, ~1 ml) is a leukocyte-containing buffy coat.
- The third layer from top (clear, ~10 ml) contains Ficoll-Paque.
- The bottommost layer (red, ~5 ml) contains RBCs.
- Additionally, at the buffy coat layer, a smeared pellet of cells (cloudy, ~1 mm) should be deposited along the test tube wall.
- The topmost layer (yellow/clear, ~35 ml) is a PBS/serum mix.
- Transfer the collected cells into buffy collection tube:
- Prepare one 15 ml buffy collection tube for each 50 ml tube from the previous steps. Add 2 ml PBS in each tube.
- Remove the PBS/serum layer until only 10 ml of this layer remains, using caution to avoid turbulence by aspirating from the top of the layer. (i.e., if the buffy coat line is at the 30 ml line, then retain 40 ml total fluid remains.)
- With a 10 ml Serological pipette, transfer the buffy coat layer to a buffy collection tube, and use caution to minimize fluid transfer. To do so, hold the pipette tip directly above the buffy coat, and slowly aspirate, then eject fluid to the buffy collection tube. Leave ~5 ml of PBS/serum for the next step.
- Resuspend the smeared pellet of cells in the Ficoll-Paque by gently rocking the tube, using caution to avoid mixing the fluid layers. The rocking motion allows the liquid-liquid interface between the Ficoll-Paque and the PBS/serum mix to rub the pellet off the wall.
- Transfer as much Ficoll-Paque layer to the buffy collection tube as possible, while completely avoiding the RBC layer. Avoiding the PBS/serum layer is ideal, but not critical.
- Prepare one 15 ml buffy collection tube for each 50 ml tube from the previous steps. Add 2 ml PBS in each tube.
- Centrifuge at 450 x g (1,500 rpm) for 10 min at 4 °C.
- Remove supernatant, and resuspend cells in 1 ml FACS buffer.
- Collect cell suspensions from all collection tubes into a single 50 ml Falcon tube.
- Wash the cells by adding FACS buffer until the total fluid volume is 50 ml.
- Count the cells. Each bag of cord blood yields ~300-1,000 million cells.
- Centrifuge at 450 x g for 5 min at 4 °C.
- Remove supernatant, deriving a mononuclear cell pellet.
- Add 25 ml PBS into multiple 50 ml tubes.
- Enrichment of CD34+ cells
- Prepare anti-CD34 microbeads/FcR blocking mix.
- In a 15 ml conical tube, dilute the cells in 2 μl anti-CD34 microbeads/FcR blocking mix per 1 million nucleated cells.
- Incubate at 4 °C for 30-40 min.
- Add FACS buffer up to the 15 ml line.
- Centrifuge at 450 x g (1,500 rpm) for 5 min at 4 °C.
- Remove supernatant.
- Resuspend cells in FACS buffer (1 ml per 100 million cord blood mononuclear cells).
- Prepare an elution tube: 15 ml Falcon tube + 2 ml FACS buffer.
- Perform positive selection for CD34+ cells with an LS MACS column:
- Place a 100 μm cell strainer on top of the column.
- Equilibrate the column by filtering 5 ml FACS buffer through the strainer; discard the flow-through.
- Gently vortex the cells to ensure suspension.
- Filter the cells through the LS MACS column; retain the flow-through.
- Optional: wash the column once by passing 4 ml FACS buffer through it; discard this wash. Then, re-load the flow-through from the previous step on the column to increase cell yield.
- Wash the column by passing 4 ml FACS buffer through it; discard the wash. Repeat the wash a second time, and discard the wash again.
- Remove the MACS column from its magnetic holder, and place it on top of the elution tube.
- Add 4 ml of FACS buffer to the column, and allow gravity elution to occur.
- Add 4 ml of FACS buffer, taking caution to prevent the column from drying out, and use a plunger to push the FACS buffer through.
- Place a 100 μm cell strainer on top of the column.
- Count the cells. Each bag of cord blood yields 300-1,000 x 106 CD34+ cells.
- Centrifuge at 450 x g for 5 min at 4 °C.
- Remove supernatant.
- (Optional) If necessary, cells can be stored overnight on ice in 4 °C, or frozen in 10% DMSO + FBS media; however, proceeding with fresh cells without an intermediate freezing step is recommended to avoid affecting clonal output and cell viability.
- Prepare anti-CD34 microbeads/FcR blocking mix.
- Isolation of cord blood mononuclear cells using Ficoll-Paque
- Culture of purified cells in single-cell environment (day 0-7)
- Single-cell sorting and plating
- Stain CD34+ HSPCs with antibody mix. Use 10 μl antibody mix for 1 x 106 cells according to the following dilution (Table 1):
Table 1. Antibody preparation for purification of progenitors from human cord blood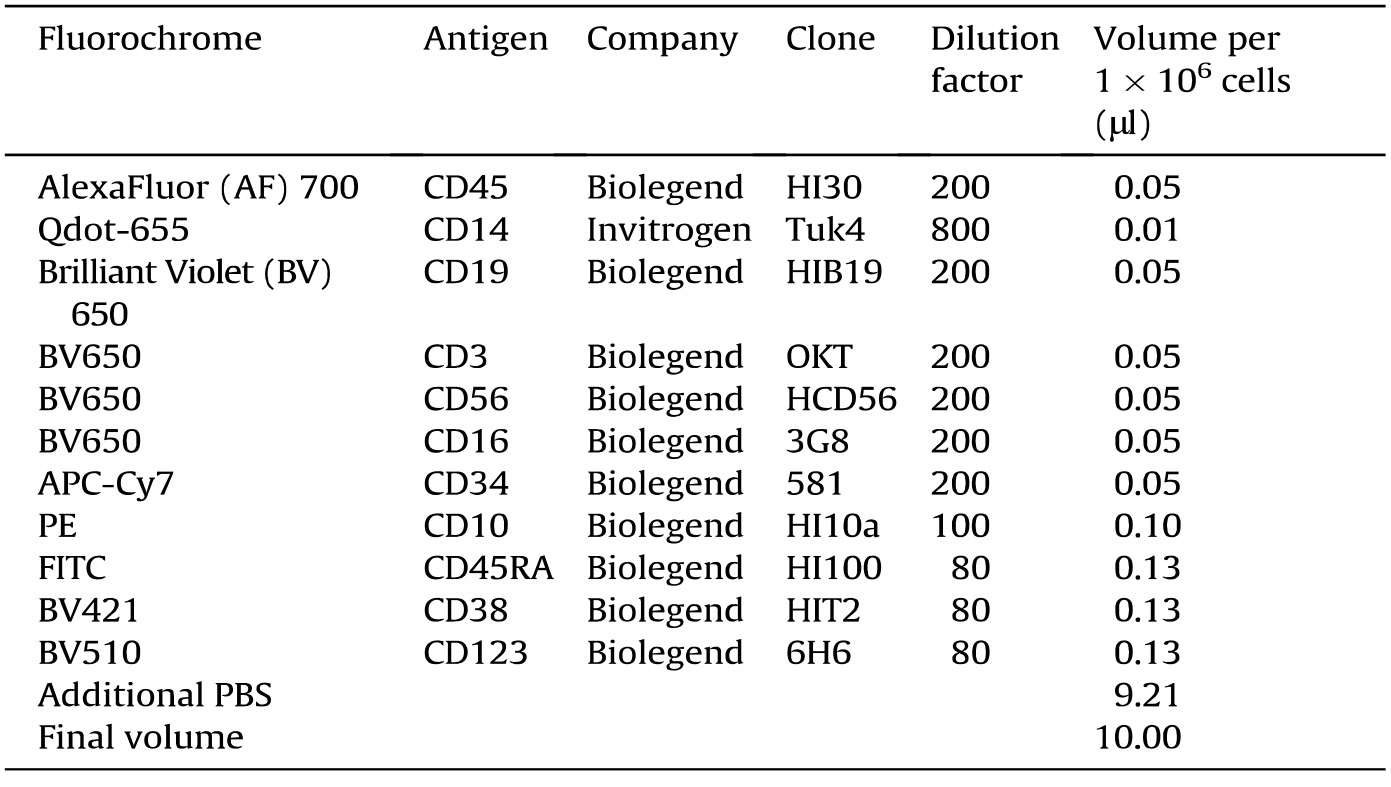
- Incubate cells on ice for 40 min, and using flow cytometry, sort cells according to following gating (Table 2, Figure 1):
Table 2. Characterization of progenitor populations in human cord blood
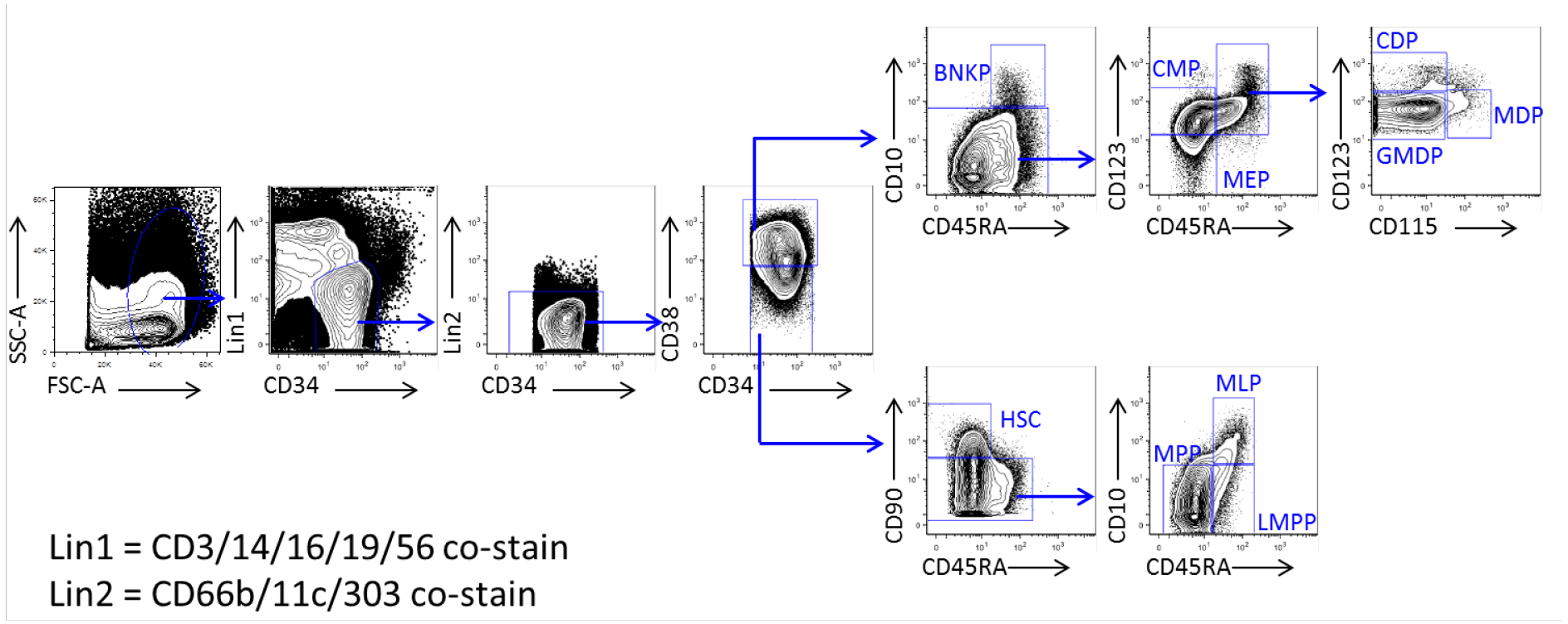
Figure 1. Visual gating strategy of progenitor populations in human cord blood. Flow cytometry plots show separation of CD34+ cord blood cells into ten populations, HSC, MPP, LMPP, MLP, BNKP, CMP, MEP, GMDP, MDP and CDP. - Sort single cells into each well of stromal-cell-containing 96-well plates with 50 μl complete MEMα medium per well.
- Stain CD34+ HSPCs with antibody mix. Use 10 μl antibody mix for 1 x 106 cells according to the following dilution (Table 1):
- Cell culture over time
- Prepare a 2x mixture of cytokines in complete medium:
- 100 ng/ml Flt3L (final 1x concentration 50 ng/ml).
- 20 ng/ml SCF (final 1x concentration 10 ng/ml).
- 10 ng/ml GM-CSF (final 1x concentration 5 ng/ml) (not required to culture cells; stimulates and inhibits proliferation of various cell subtypes).
- 100 ng/ml Flt3L (final 1x concentration 50 ng/ml).
- Add the 2x mixture of cytokines to seeded stromal cell wells. (Add 50 μl for 96-well plate)
- Replenish media with cytokine every 7 days.
- On day 7, for each well, prepare 50 μl medium containing cytokines at 3x concentration. Gently add 50 μl fresh media with 3x cytokines into the well by pipetting onto the wall.
Note: A higher concentration of cytokines is prepared because the final media volume will be increased, while the volume of media-cytokines mixture remains the same. Media is not removed from the wells yet, due to the possibility of disturbing cell attachment. - On day 14, prepare 50 μl medium containing cytokine at 3x concentration as in Step C2c i. Gently remove 50 μl of the old medium from each well, and then add 50 μl fresh media into the well by pipetting onto the wall.
- On day 7, for each well, prepare 50 μl medium containing cytokines at 3x concentration. Gently add 50 μl fresh media with 3x cytokines into the well by pipetting onto the wall.
- Prepare a 2x mixture of cytokines in complete medium:
- Single-cell sorting and plating
- Harvest and analyze cells (day 7~21)
- Read cells via flow cytometry
- Prepare antibody mixture in FACS buffer using the following dilution (Table 3).
Table 3. Antibody preparation for analysis of progeny from human cord blood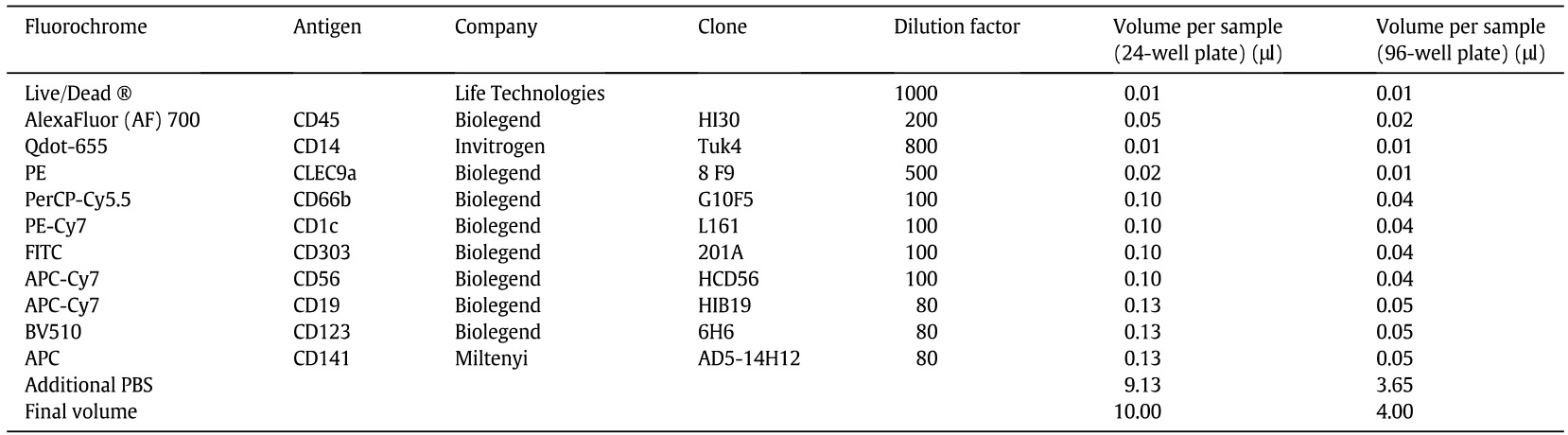
- Add PBS such that each well is filled, using a multichannel pipet. (250 μl on a 96-well plate)
- Pipet up and down 10-20 times to resuspend cells, and transfer 150 μl to 96-well V-bottom plates for staining.
- Centrifuge at 450 x g for 5 min at 4 °C.
- Discard the supernatant by microtip aspiration, using caution to keep the fluid level above the slanted portion of the well.
- Transfer the remaining 100 μl of cells to the 96-well V-bottom plates.
- Centrifuge at 450 x g for 4 min at 4 °C.
- Discard the supernatant, as before.
- Gently vortex the plate to resuspend the cells, using caution to avoid any fluid spillage. To do so, reduce the vortex speed to minimum, grasp the plate firmly, and firmly place it on the vortex mixer. Slowly increase the vortex speed, and gently reduce pressure on the vortex mixer so that the vortex mixer can shake. Then, decrease the vortex speed and remove the plate. As needed, this process may be repeated in each of the plate’s four quadrants. It is advised to practice this technique with a test plate and tap water to ensure no sample loss. (An alternative resuspension method is to gently pipet 10-20 times.)
- Add 4 μl of antibodies.
- Gently vortex the plate to mix cells with the antibody, as before.
- Centrifuge with a quick spin (50 x g, 5 sec) to bring down the cells and staining mix to the bottom of the well.
- Incubate in the dark for 40 min at 4 °C.
- Add 200 μl PBS to wash.
- Centrifuge at 450 x g for 5 min at 4 °C. Discard the supernatant.
- Resuspend in 50 μl FACS buffer.
- Vortex the counting beads stock solution for 30 sec, and make a 1:10 dilution of counting beads in FACS buffer.
- Add 10 μl of dilute counting beads to each well (~1,000 beads per well).
- Acquire cells on a flow cytometer (BD-LSRII or LSR Fortessa), and gate cells according to the following gating (Table 4) (Figure 2 and Lee et al., 2017).
Table 4. Characterization of progeny populations in human cord blood
- Prepare antibody mixture in FACS buffer using the following dilution (Table 3).
- Proceed to data analysis
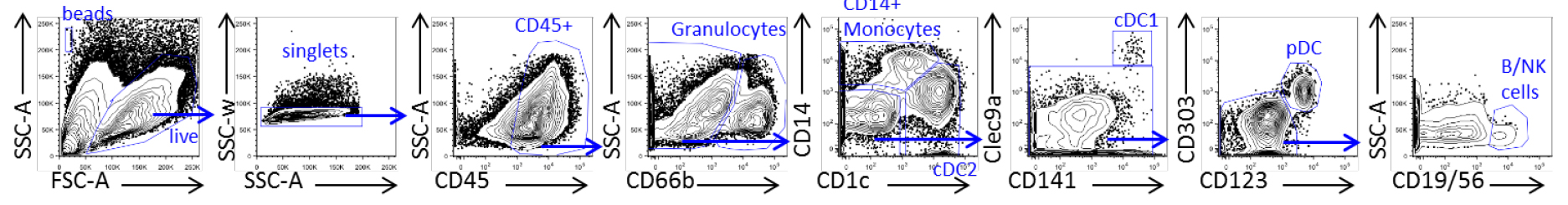
Figure 2. Visual gating strategy for analysis of progeny populations in human cord blood. Flow cytometry plots show phenotype of six cell types differentiated from CD34+ cells cultured in MP+FSG, granulocytes, monocytes, cDC1, cDC2, pDC and B/NK (lymphoid) cell.
- Read cells via flow cytometry
Data analysis
- Data preparation
- Export flow cytometry data and save in a .csv file, such that:
- Each data column contains one ‘dimension’ of data.
- Each row contains one sample’s data.
- One column ‘Batch’ exists (case sensitive); all samples from one patient should have identical batch IDs.
- One column ‘Progenitor’ exists (case sensitive).
- All data columns are consecutive, each containing the number of relevant reads in each sample-progeny data point. (Figure 3)
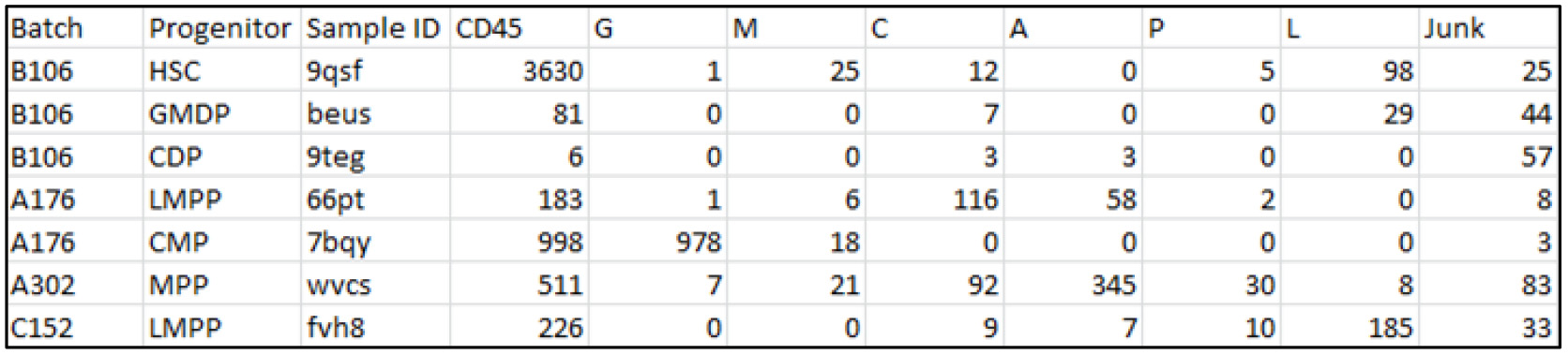
Figure 3. Example of first few rows of a valid .csv file viewed in Excel
- Each data column contains one ‘dimension’ of data.
- Export flow cytometry data and save in a .csv file, such that:
- Data normalization in R
- Open cPlot.R in RStudio, then Source the file. (Code > Source) (The file is available online at https://github.com/kangliulab/cplot/blob/master/cPlot_v0.7.R)
- Change working directory to the location of data files. (Session > Set Working Directory > Choose Directory)
Note: Do not use a working directory in a synced folder (OneDrive, Dropbox, etc.), as this may cause some problems with file outputs. - Normalize data using one of the following functions. To use, enter the function into the console, replacing inFile with the file name, firstData with the first data column (leftmost column is #1), and dataCols with the number of data columns. Keyword may be omitted if desired; if used, it adds the keyword to the start of each row to help differentiate between files. When replacing words, keep any quotation marks that are denoted below.
- addCB (“inFile”, firstData, dataCols, “keyword”) is used for samples that must be normalized both by subject (i.e., batch) and by progeny (i.e., lineage).
- addPatient (“inFile”, firstData, dataCols, “keyword”) is used for samples that must be normalized only by subject (i.e., batch).
- addPreNorm (“inFile”, firstData, dataCols, “keyword”) is used for samples that have already been normalized.
- Examples:
- addCB (“data-file-v123_for-test-use.csv”, 5, 6).
- addCB (“data-file-v123_for-test-use.csv”, 5, 6, “v123”).
- addCB (“data-file-v123_for-test-use.csv”, 5, 6).
- addCB (“inFile”, firstData, dataCols, “keyword”) is used for samples that must be normalized both by subject (i.e., batch) and by progeny (i.e., lineage).
- Generate t-SNE maps using the below function. To use, enter it into the console. You may choose to omit all numbers, which causes the script to use the default numbers (denoted below). To replace numbers, indicate both the variable to be changed and its new value.
- generateManyMaps(dataCols = 6, perVector = c(50), thetaVector = c(0.10), iterVector = c(1000), etaVector = c(200), n = 1, seedSet = FALSE, randSeed = 6) allows multiple parameters to be simultaneously tested. It can also be used to generate multiple maps with a single set of parameters. Note that it will try every possible combination of parameters, and will try each combination n times. dataCols should be equal to your number of data columns, perVector selects perplexity values, thetaVector selects theta values, iterVector selects iterations to run, etaVector selects eta values, n selects number of repetitions per combination of parameters, seedSet forces each plot to have the same random seed, and randSeed selects the random seed of the first plot (or, the random seed of all plots if seedSet is TRUE).
- Examples:
- generateManyMaps().
- generateManyMaps(n = 10, seedSet = TRUE).
- generateManyMaps(perVector = c(40, 50, 60), etaVector = c(150, 200, 250), n = 3).
- generateManyMaps().
- generateManyMaps(dataCols = 6, perVector = c(50), thetaVector = c(0.10), iterVector = c(1000), etaVector = c(200), n = 1, seedSet = FALSE, randSeed = 6) allows multiple parameters to be simultaneously tested. It can also be used to generate multiple maps with a single set of parameters. Note that it will try every possible combination of parameters, and will try each combination n times. dataCols should be equal to your number of data columns, perVector selects perplexity values, thetaVector selects theta values, iterVector selects iterations to run, etaVector selects eta values, n selects number of repetitions per combination of parameters, seedSet forces each plot to have the same random seed, and randSeed selects the random seed of the first plot (or, the random seed of all plots if seedSet is TRUE).
- Assign cell lineages using each of the below functions in sequential order.
- pickMap(“inFile”) allows a particular t-SNE map to be imported for further analysis. inFile should represent a data file that was created by generateManyMaps().
- assignLineages() automatically calculates an appropriate lineage track for each data point. It can only be used after pickMap() has been used. It can take a long time (3-8 h) to run, especially for larger data files!
- compileAllData(“inFile”) generates a .csv with raw data, normalized data, tSNE coordinates, and assigned lineages. It assumes that data has already been imported into RStudio. If RStudio was closed sometime between importing the data (in step 3) and this step, then you should re-import the data before running compileAllData(). inFile should represent a data file that was created by assignLineages().
- Examples:
- pickMap(“data1_p50_t0.1_s6_i1000.csv”).
- assignLineages().
- compileAllData(“2assignedLineages_FC0.7.csv”).
- pickMap(“data1_p50_t0.1_s6_i1000.csv”).
- pickMap(“inFile”) allows a particular t-SNE map to be imported for further analysis. inFile should represent a data file that was created by generateManyMaps().
- Open cPlot.R in RStudio, then Source the file. (Code > Source) (The file is available online at https://github.com/kangliulab/cplot/blob/master/cPlot_v0.7.R)
Above, we describe an outlined approach to our data. As described in our previous paper, each progenitor’s output is imagined as a 6D vector. A preliminary data cleanup step involves neglecting low cell counts: below 2 cells for CDPs and below 6 cells for all other cell types; the neglect occurs by changing the reading to 0 cells. Progenitors with a total of 0 cell output after data cleanup should then be purged. Data normalization of cell type yield follows the DESeq procedure, assuming the geometric mean of total clonal output for a single progenitor phenotype across different donors should be similar. A second normalization of cell type yield was also done via DESeq, to ensure similar geometric mean of each progeny type yield across all progenitors.
Barnes-Hut t-SNE (Van Der Maaten, 2014) was performed to visualize the inferred pair-wise similarity data in 2 dimensions. Potency similarity between any pair of cells is inferred by their proximity in this space using Gaussian diffusion kernel.
To assign lineages to each progenitor, we begin by defining six ‘backbones’; one for each type of progeny output. The backbone of each progeny type consists of any progenitors with > 70% commitment degree to that progeny; commitment degree is defined as (yield of one progeny type)/(total yield of all progeny types). Then, distance of each progenitor to any given track is defined as the 6D Euclidean distance between a progenitor of interest and all backbone progenitors of that track. The progenitor is assigned to the lineage corresponding with the track that it has the lowest distance to.
Notes
Although this culture protocol only reports differentiation potential of granulocyte, monocyte, lymphocyte, CD141+ dendritic cell (cDC1), CD1c+ dendritic cell (cDC2), and plasmacytoid dendritic cells (pDC) of hematopoietic lineage, the analysis method can be applied to any culture system that nurtures different lineages as far as that the cellular output can be quantified.
Recipes
- Complete MEMα medium
MEMα
10% FBS
1x Pen/Strep - Mitomycin C stock (100x, 1 mg/ml)
- Suspend 2 mg Mitomycin C powder in 2 ml dH2O
- Vortex solution
- Filter solution with a 0.20 μm filter
- Keep stock for up to 3 months at -20 °C
- Suspend 2 mg Mitomycin C powder in 2 ml dH2O
- FACS buffer
DPBS
2 mM EDTA
0.5% BSA - Anti-CD34 microbeads/FcR blocking mix
Prepare 2 μl of the mix per 1 million total nucleated cells
15% FcR blocking buffer
25% anti-CD34 MACS beads
60% FACS buffer
Acknowledgments
This work was supported by the Empire State Stem Cell Fund through the New York State Department of Health (C029562 to K.L.) and The US National Institute of Health (AI101251 and OD023291 to K.L.). Research reported in this manuscript was performed partly by the Columbia Center for Translational Immunology Flow Cytometry Core, supported in part by the Office of the Director of the US National Institutes of Health (S10RR027050 and S10OD020056). This protocol was adapted from previous work Lee et al., 2017, Lee et al., 2015a. Authors declare no conflicts of interest or competing interests. The recombinant human Flt3L was a gift from Celldex Therapeutics.
References
- Amir el, A. D., Davis, K. L., Tadmor, M. D., Simonds, E. F., Levine, J. H., Bendall, S. C., Shenfeld, D. K., Krishnaswamy, S., Nolan, G. P. and Pe'er, D. (2013). viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol 31(6): 545-552.
- Levine, J. H., Simonds, E. F., Bendall, S. C., Davis, K. L., Amir el, A. D., Tadmor, M. D., Litvin, O., Fienberg, H. G., Jager, A., Zunder, E. R., Finck, R., Gedman, A. L., Radtke, I., Downing, J. R., Pe'er, D. and Nolan, G. P. (2015). Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell 162(1): 184-197.
- Lee, J., Zhou, Y. J., Ma, W., Zhang, W., Aljoufi, A., Luh, T., Lucero, K., Liang, D., Thomsen, M., Bhagat, G., Shen, Y. and Liu, K. (2017). Lineage specification of human dendritic cells is marked by IRF8 expression in hematopoietic stem cells and multipotent progenitors. Nat Immunol 18(8): 877-888.
- Lee, J., Breton, G., Aljoufi, A., Zhou, Y. J., Puhr, S., Nussenzweig, M. C. and Liu, K. (2015a). Clonal analysis of human dendritic cell progenitor using a stromal cell culture. J Immunol Methods 425: 21-26.
- Lee, J., Breton, G., Oliveira, T. Y., Zhou, Y. J., Aljoufi, A., Puhr, S., Cameron, M. J., Sekaly, R. P., Nussenzweig, M. C. and Liu, K. (2015b). Restricted dendritic cell and monocyte progenitors in human cord blood and bone marrow. J Exp Med 212(3): 385-399.
- Paul, F., Arkin, Y., Giladi, A., Jaitin, D. A., Kenigsberg, E., Keren-Shaul, H., Winter, D., Lara-Astiaso, D., Gury, M., Weiner, A., David, E., Cohen, N., Lauridsen, F. K. B., Haas, S., Schlitzer, A., Mildner, A., Ginhoux, F., Jung, S., Trumpp, A., Porse, B. T., Tanay, A. and Amit, I. (2016). Transcriptional heterogeneity and lineage commitment in myeloid progenitors. Cell 164(1-2): 325.
- See, P., Dutertre, C. A., Chen, J., Gunther, P., McGovern, N., Irac, S. E., Gunawan, M., Beyer, M., Handler, K., Duan, K., Sumatoh, H. R. B., Ruffin, N., Jouve, M., Gea-Mallorqui, E., Hennekam, R. C. M., Lim, T., Yip, C. C., Wen, M., Malleret, B., Low, I., Shadan, N. B., Fen, C. F. S., Tay, A., Lum, J., Zolezzi, F., Larbi, A., Poidinger, M., Chan, J. K. Y., Chen, Q., Renia, L., Haniffa, M., Benaroch, P., Schlitzer, A., Schultze, J. L., Newell, E. W. and Ginhoux, F. (2017). Mapping the human DC lineage through the integration of high-dimensional techniques. Science 356(6342).
- Van Der Maaten, L. (2014). Accelerating t-sne using tree-based algorithms. J Mach Learn Res 15: 3221-3245.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Luh, T., Lucero, K., Ma, W., Lee, J., Zhou, Y. J., Shen, Y. and Liu, K. (2018). High Dimensional Functionomic Analysis of Human Hematopoietic Stem and Progenitor Cells at a Single Cell Level. Bio-protocol 8(10): e2851. DOI: 10.21769/BioProtoc.2851.
Category
Immunology > Immune cell differentiation > HSPCs
Stem Cell > Adult stem cell > Hematopoietic stem cell
Cell Biology > Single cell analysis > Flow cytometry
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.