- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

The Long-lived Protein Degradation Assay: an Efficient Method for Quantitative Determination of the Autophagic Flux of Endogenous Proteins in Adherent Cell Lines


Published: Vol 8, Iss 9, May 5, 2018 DOI: 10.21769/BioProtoc.2836 Views: 11097
Reviewed by: Alessandro DidonnaPia GiovannelliManasi K. Mayekar
Original research article
The authors used this protocol in:

Oct 2013
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Autophagy is a key player in the maintenance of cellular homeostasis in eukaryotes, and numerous diseases, including cancer and neurodegenerative disorders, are associated with alterations in autophagy. The interest for studying autophagy has grown intensely in the last two decades, and so has the arsenal of methods utilised to study this highly dynamic and complex process. Changes in the expression and/or localisation of autophagy-related proteins are frequently assessed by Western blot and various microscopy techniques. Such analyses may be indicative of alterations in autophagy-related processes and informative about the specific marker being investigated. However, since these proteins are part of the autophagic machinery, and not autophagic cargo, they cannot be used to draw conclusions regarding autophagic cargo flux. Here, we provide a protocol to quantitatively assess bulk autophagic flux by employing the long-lived protein degradation assay. Our procedure, which traces the degradation of 14C valine-labelled proteins, is simple and quick, allows for processing of a relatively large number of samples in parallel, and can in principle be used with any adherent cell line. Most importantly, it enables quantitative measurements of endogenous cargo flux through the autophagic pathway. As such, it is one of the gold standards for studying autophagic activity.
Keywords: Long-lived protein degradationBackground
Pulse-chase labelling approaches have been used to study protein turnover for decades. In the long-lived protein degradation (LLPD) assay described here, the proteome of cells in culture is radiolabelled with 14C valine and chased in order to follow the decline in radioactive proteins as readout of protein degradation. After an initial chase period, the cells are washed to eliminate the degradation products of short-lived proteins, which predominantly result from proteasomal activity. Thereafter, a second chase is initiated, and proper controls are included in order to monitor the autophagic degradation of long-lived proteins. We recently used this method to discover that the calcium-modulating compounds thapsigargin and A23187, which based on results with autophagic markers were previously widely believed to activate autophagy, actually completely block bulk autophagic flux (Engedal et al., 2013). The starting material used in that and previous protein degradation protocols was derived from cells grown in 6-well plates (Bauvy et al., 2009; Engedal et al., 2013) or more (Ronning et al., 1979; Seglen et al., 1979), or involved relatively high amounts of radioactivity (Mizushima et al., 2001; Fuertes et al., 2003a), which is expensive. Recently, we have downscaled and simplified the LLPD protocol to the validated time- and cost-efficient version that we present here.
An overview of the method is shown in Figure 1. To label proteins with radioactive valine, cells are seeded in 24-well plates in complete medium supplemented with 14C valine. As proteins are synthesised, they incorporate amino acids, including the 14C valine present in the medium, and therefore the amount of long-lived 14C valine-labelled proteins increases with time (Figure 1, first part of the curve). Valine is an optimal amino acid to use in the LLPD method, since it is a poorly metabolised amino acid, is well tolerated at high doses, and does not influence autophagy or protein degradation rates (Seglen et al., 1979). Moreover, free valine is readily exchanged over the plasma membrane (Seglen and Solheim, 1978), enabling efficient wash-out of released 14C valine. After 2-3 days of labelling, unincorporated 14C valine is removed by a simple wash procedure, and the cells receive new medium supplemented with a high concentration of non-radioactive (‘cold’) valine (‘chase medium’). The large surplus of cold valine prevents reincorporation of released 14C valine. Thus, from this point on the presence of free 14C valine is a result of endogenous protein degradation. After a chase period of 18 h, the free 14C valine that has been produced by degradation of short-lived proteins (predominantly due to proteasomal degradation) is washed out. Next, a second chase period, which we call the ‘sampling period’, is initiated along with experimental treatments and proper controls. Generally, we use a sampling period of 2-6 h to monitor the degradation of long-lived proteins. At the end of the sampling period, trichloroacetic acid (TCA) is added to precipitate intact proteins. The TCA-soluble fraction of degraded proteins (containing free amino acids and small peptides) is separated from the TCA-insoluble fraction (containing intact proteins) by centrifugation, and the radioactivity in each fraction is measured by liquid scintillation counting. This allows calculation of the rate of long-lived protein degradation in the sampling period, expressed as the percentage of radioactivity in the TCA-soluble fraction versus the total amount of radioactivity in the TCA-soluble and–insoluble fractions, divided by the duration of the sampling period (Figure 1).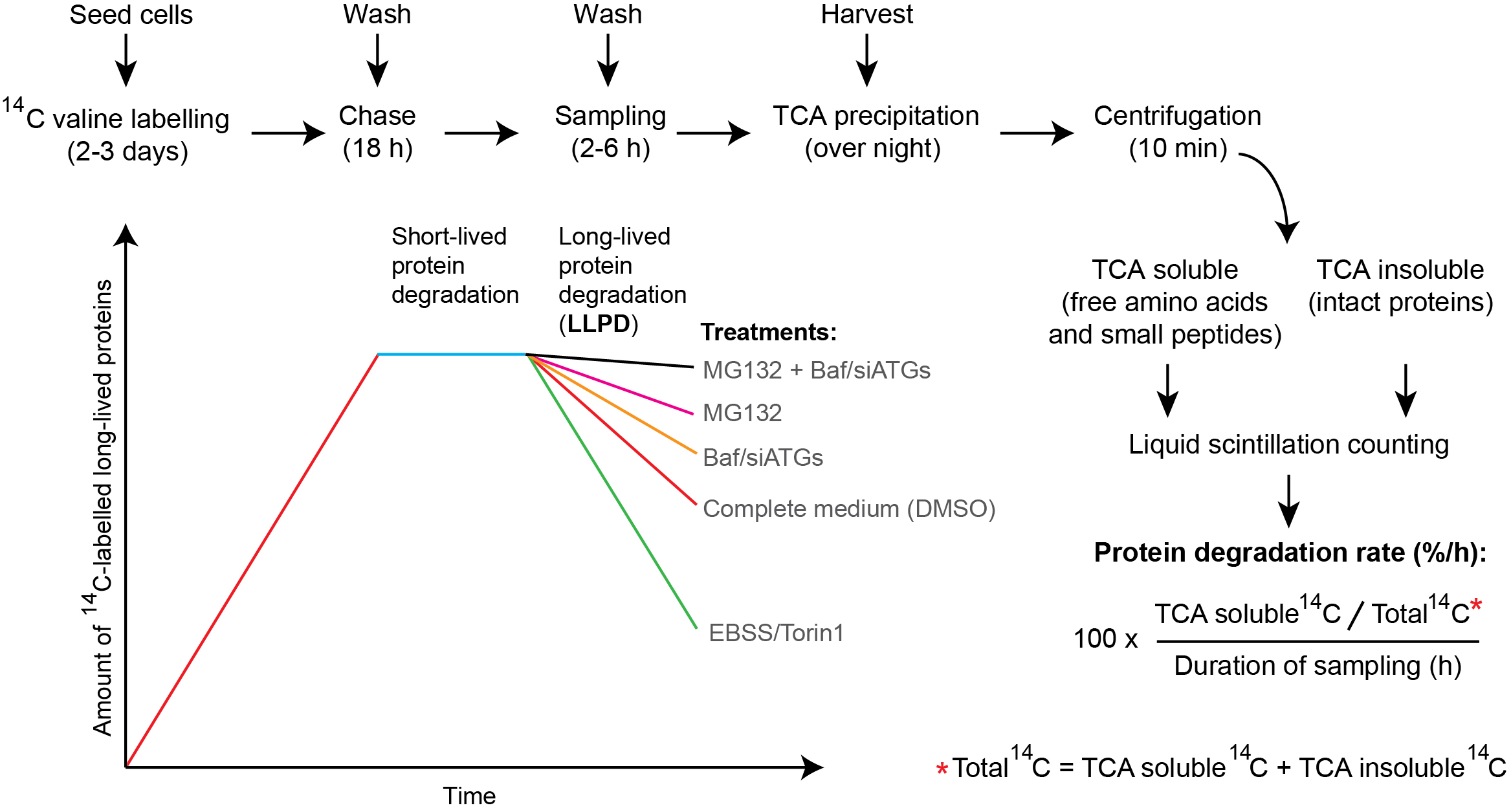
Figure 1. Overview of the long-lived protein degradation (LLPD) assay. During the labelling period (2-3 days), the amount of radioactive long-lived proteins increases with time. Thereafter, an 18 h chase period allows for degradation of the short-lived proteins and subsequent elimination of the released 14C valine by a washing step. Consequently, only the degradation of long-lived proteins is followed in the 2-6 h sampling period. Compared to cells kept in complete, nutrient-rich medium (red line), incubating cells with EBSS starvation medium or the mTOR-inhibitor Torin1 produces a very strong degradation of long-lived proteins in the sampling period, due to enhanced bulk autophagy (green line). Autophagic-lysosomal LLPD can be revealed by treatment with the lysosomal inhibitor Bafilomycin A1 (Baf) or RNAi-mediated silencing of key ATGs (siATGs) (yellow line), whereas the contribution to LLPD from the proteasome can be assessed by treatment with proteasomal inhibitors like MG132 (purple line). Blocking both autophagic-lysosomal and proteasomal activity simultaneously will abrogate both main sources of LLPD, thus resulting in minimal loss of 14C-labelled intact protein (black line). Note that the rise and fall in the curves are schematic and purely intended for illustrative purposes–they are not intended to indicate exact details in the kinetics of long-lived protein labelling and/or degradation. See text for a more detailed description of each of the protocol steps and for representative examples of data.
Materials and Reagents
In the current protocol we use:
- Pipette tips (Thermo Fisher ART Barrier tips) (VWR, catalog numbers: 732-2223 (0.5-20 µl), 732-2207 (1-200 µl), and 732-2355 (100-1,000 µl))
- 75 cm2 tissue culture flask (Corning, Falcon®, catalog number: 353136 )
- 24-well tissue culture plates (Corning, Falcon®, catalog number: 353047 )
- Microcentrifuge tubes (VWR, catalog number: 211-2130 )
- Scintillation vials (PerkinElmer, catalog number: 6000292 )
- Nitrile gloves (VWR, catalog number: 112-2372 )
- 50 ml tube (VWR, catalog number: 525-0402 )
- 0.45 µm filter (VWR, catalog number: 514-0075 )
- LNCaP cells (ATCC, catalog number: CRL-1740 )
- U2OS cells (ATCC, catalog number: HTB-96 )
- VCaP cells (ATCC, catalog number: CRL-2876 )
- Huh7 cells (Nakabayashi et al., 1982) (Kindly provided by Dr. Line M. Grønning-Wang, Oslo, Norway)
- PBS (Thermo Fisher Scientific, GibcoTM, catalog number: 20012019 )
- 0.25% Trypsin-EDTA (Thermo Fisher Scientific, GibcoTM, catalog number: 25200056 )
- RPMI 1640 (Thermo Fisher Scientific, GibcoTM, catalog number: 21875091 )
- Fetal bovine serum (FBS) (Sigma-Aldrich, catalog number: F7524 )
- [1-14C] L-valine, 45 mCi/mmol, 0.1 mCi/ml (Vitrax, catalog number: VC 308 )
- Poly-D-lysine (Sigma-Aldrich, catalog number: P6407-10X5MG )
- For RNAi reverse transfection:
Lipofectamine RNAiMAX (Thermo Fisher Scientific, InvitrogenTM, catalog number: 13778150 )
Ambion SilencerTM select siRNAs (negative control ‘siCtrl’, Thermo Fisher Scientific, InvitrogenTM, catalog number: 4390843 ; siULK1, s15964; siULK2, s18706)
Opti-MEM reduced serum medium (Thermo Fisher Scientific, GibcoTM, catalog number: 11058021 ) - Earle’s balanced salt solution (EBSS) (Thermo Fisher Scientific, GibcoTM, catalog number: 24010043 )
- Scintillation liquid (PerkinElmer, catalog number: 6013199 )
- DMSO (Sigma-Aldrich, catalog number: D2650 )
- Bafilomycin A1 (Enzo Life Sciences, catalog number: BML-CM110-0100 )
- Ammonium chloride (NH4Cl) (Sigma-Aldrich, catalog number: A4514 )
- SAR-405 (Magento, ApexBio, catalog number: A8883 )
- Torin1 (Tocris Bioscience, catalog number: 4247 )
- Non-radioactive L-valine (Sigma-Aldrich, catalog number: V0513 )
- Bovine serum albumin (BSA) (VWR, catalog number: 422361V )
- Trichloroacetic acid (TCA) (Sigma-Aldrich, catalog number: T0699 )
- Potassium hydroxide (KOH) (Sigma-Aldrich, catalog number: 60377 )
- 200 mM cold L-valine (see Recipes)
- RPMI 1640/10%FBS (see Recipes)
- 1 mg/ml poly-D-lysine (see Recipes)
- 1 mM Torin1 (see Recipes)
- PBS/2%BSA (see Recipes)
- 25% TCA (see Recipes)
- 0.2 M KOH (see Recipes)
- 0.2 mM Bafilomycin A1 (see Recipes)
- 160 mM NH4Cl (see Recipes)
Equipment
- Pipettes (FinnpipetteTM F2 GLP Kit) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 4701070 )
- Humidified incubator
- Tube rotator (VWR, catalog number: 444-0502 )
- Plate shaker (Grant Instruments, model: PMS-1000i )
- Magnetic stirrer (IKA, catalog number: 0003810001 )
- Autoflow IR Direct Heat CO2 incubator (NuAire, model: NU-5510E )
- Vortexer (Denville Scientific, catalog number: S7030 )
- Benchtop centrifuge with cooling (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 75002430 )
- Scintillation counter (Liquid Scintillation Analyzer) (Packard, model: 1600 TR )
Procedure
- Radioactive labelling (standard 2-3 days)
- Culture adherent cells in a 75 cm2 tissue culture flask in a humidified incubator with 5% CO2 at 37 °C until a near confluent cell layer has been obtained. Wash once with 3 ml 37 °C PBS and incubate the cells with 3 ml 0.25% trypsin-EDTA at 37 °C until the cells have detached (typically 3-5 min).
- Collect the detached cells in 10 ml 37 °C RPMI 1640 supplemented with FBS to 10% final concentration (= Complete Medium, CM).
Note: The concentrations of radioactive and cold valine in this protocol have been adapted specifically for use in RPMI 1640, based on the concentration of valine in this medium, and based on cost-efficiency with respect to the amount of radioactive valine used. When following this protocol, cells should therefore be resuspended in RPMI 1640 after trypsinization, even though they are not normally cultured in RPMI 1640. For the same reason, RPMI 1640 should be used for the remainder of the protocol. We have validated this assay in > 20 cell lines and have never observed signs of toxicity or altered phenotypes in cells that are normally not cultured RPMI 1640, as long as the cells do not require any special supplements. Other media can be used instead of RPMI 1640, but users should adjust valine concentrations throughout the protocol to account for differences in the valine concentration of RPMI 1640 versus the alternative medium used. - Seed cells in a 24-well plate in 0.5 ml CM supplemented with 14C-valine to a final concentration 0.1 µCi/ml. Seed a cell number that will lead to the desired monolayer confluency at the time of experimental treatment. In general, we seed cells so that they reach 50-80% confluency after 2-3 days incubation in a humidified incubator with 5% CO2 at 37 °C.
Notes:- The protocol presented here is compatible with a 24-well plate format. Because of the ease of the procedure and its high reproducibility, we routinely process two 24-well plates (48 wells) per experiment with duplicate or triplicate wells per treatment condition. This makes it possible to test a relatively large number of conditions in a single experiment.
- For LNCaP cells, we seed 1 x 105 cells per well. For U2OS, VCaP and Huh7, we seed 3 x 104, 2.5 x 105, and 1.2 x 104 cells per well, respectively.
- When working with cell types that adhere poorly (e.g., LNCaP cells, HEK293 cells, and others), it is recommended to use tissue culture plates that have been coated to improve cell adherence (due to multiple wash steps in the protocol). We prefer to use poly-D-lysine (PDL), as it is relatively inexpensive and easy to use, and does not interfere with amino acid starvation (since it, unlike, e.g. poly-L-lysine, is not metabolised by cells). Per well in a 24-well tissue culture plate, add 300 µl sterile PDL (2.5 µg/ml). Incubate the plates in a sterile environment for 30 min at room temperature. Afterwards, remove the PDL using suction, and wash each well briefly with 0.5 ml sterile H2O. The plates are ready for use when dry. PDL-coated plates are stable for up to several weeks when stored at room temperature. Of note, for most cell types coating is not necessary.
- When doing siRNA transfection, we recommend using reverse transfection so that the cells can be transfected and radiolabelled simultaneously. This is both practical and gives a very high transfection efficiency. Combine Opti-MEM, siRNA, and Lipofectamine RNAiMAX according to the manufacturer’s protocol, and aliquot the transfection mix into a 24-well plate (50 µl per well). Incubate at room temperature for 20-45 min, and seed 0.5 ml cell suspension in CM containing 14C-valine in the wells containing the siRNA-Lipofectamine mixture. Carefully agitate the plate and place it in the incubator.
- Follow the rules of your institution with regard to work with radioactivity. For own protection, wear gloves with a thickness of ≥ 0.25 mm.
- The protocol presented here is compatible with a 24-well plate format. Because of the ease of the procedure and its high reproducibility, we routinely process two 24-well plates (48 wells) per experiment with duplicate or triplicate wells per treatment condition. This makes it possible to test a relatively large number of conditions in a single experiment.
- Incubate the cells for 2-3 days in a humidified incubator at 37 °C and 5% CO2 to allow ample incorporation of 14C valine into long-lived proteins.
- Culture adherent cells in a 75 cm2 tissue culture flask in a humidified incubator with 5% CO2 at 37 °C until a near confluent cell layer has been obtained. Wash once with 3 ml 37 °C PBS and incubate the cells with 3 ml 0.25% trypsin-EDTA at 37 °C until the cells have detached (typically 3-5 min).
- Chase (standard 18 h)
- Aspirate the medium to remove unincorporated radioactivity.
- Wash cells once with 0.5 ml 37 °C CM supplemented with 10 mM cold valine (CM-V).
Note: Since RPMI 1640 contains 0.17 mM L-valine, the final concentration of L-valine in CM-V will be 10.17 mM. The use of 10 mM cold L-valine to block re-incorporation of released 14C-valine is standard for this type of assay (Bauvy et al., 2009; Mizushima et al., 2001; Fuertes et al., 2003a). It is not known whether there is an upper limit of how high concentration of cold L-valine can be used, but in isolated rat hepatocytes it has been shown that up to 20 mM cold L-valine does not affect protein degradation activity (Seglen et al., 1979). - Aspirate the medium and replace with 0.5 ml 37 °C CM-V.
- Incubate the cells for 18 h in a humidified incubator at 37 °C and 5% CO2.
Notes:- The chase does not need to be 18 h. However, altering the duration of the chase will change the protein degradation rate during the sampling (Figure 2A), as well as the signal intensity during liquid scintillation counting (see Note 2 in the Notes section for comments on scintillation counting and the variability of assay measurements), and will also change the pool of proteins analysed (see Note 3 in the Notes section). One should keep this in mind when comparing results obtained with different chase durations.
- If doing experimental treatments longer than 6 h, add your treatments prior to or during the chase. This additionally gives an opportunity to sample treatment effects on LLPD at various time periods after initiation of the treatment.
- The chase does not need to be 18 h. However, altering the duration of the chase will change the protein degradation rate during the sampling (Figure 2A), as well as the signal intensity during liquid scintillation counting (see Note 2 in the Notes section for comments on scintillation counting and the variability of assay measurements), and will also change the pool of proteins analysed (see Note 3 in the Notes section). One should keep this in mind when comparing results obtained with different chase durations.
- Aspirate the medium to remove unincorporated radioactivity.
- Sampling (standard 2-6 h)
- Remove released 14C valine (resulting from the degradation of short-lived proteins) by aspirating the medium.
- Wash cells once with 0.5 ml 37 °C of appropriate medium. If the subsequent treatment will be in complete medium (e.g., Torin1), wash with CM-V. If the subsequent treatment will be in amino acid starvation medium (e.g., EBSS), wash with EBSS supplemented with 10 mM cold valine (EBSS-V).
- Aspirate the medium and add your treatments in 0.25 ml 37 °C CM-V or EBSS-V.
- Incubate the cells for 2-6 h in a humidified incubator at 37 °C and 5% CO2.
- Remove released 14C valine (resulting from the degradation of short-lived proteins) by aspirating the medium.
- Harvest
Day 1- Remove the plate with cells from the incubator and cool it on ice for ~2 min.
- Add 50 µl ice-cold PBS/2%BSA per well.
Note: BSA is added as a protein carrier to facilitate protein precipitation with TCA. - Add 200 µl ice-cold 25% TCA per well to precipitate protein.
- Place the plate on a shaker at 4 °C and let it shake over night at 600 rpm.
Note: Whereas overnight TCA precipitation is sufficient, the precipitation may be extended for several days without affecting the results.
- Transfer the precipitated protein (~500 µl) from each well to 1.5 ml microcentrifuge tubes.
Note: Transfer the solution to microcentrifuge tubes without scraping. The protein precipitate remaining in the wells will be solubilised and collected in the protocol Steps D10-D12. - Sediment precipitates by centrifugation at 5,000 x g for 10 min at 4 °C. After the centrifugation, the supernatant (TCA soluble fraction) contains amino acids and small peptides (including 14C valine released from proteins due to degradation during the sampling period) and the pellet (TCA insoluble fraction) contains intact proteins.
- After the centrifugation, place the tubes on ice.
- Transfer the supernatant (~500 µl) from each tube into 6 ml scintillation vials.
- Solubilise the pellet by adding 250 µl 0.2 M KOH per tube and rotating the tubes for at least 1 h at room temperature.
Note: It is not necessary to wash the pellet prior to solubilising it in KOH. - Solubilise remaining protein precipitate in the 24-well plate by adding 250 µl 0.2 M KOH per well.
- Rotate on a shaker at 600 rpm for 1 h at room temperature.
- Merge the solubilised protein from the respective wells and tubes (~500 µl) and transfer to 6 ml scintillation vials.
- Fill each vial from Steps D8 and D12 with 4 ml scintillation liquid. Cap the tubes, and vortex hard.
Note: The scintillation liquid is an organic and toxic substance. For your own protection, add scintillation liquid, cap tubes, and vortex in a flow-cabinet, wearing protective gloves and lab coat. - Measure the amount of radioactivity in all of the tubes by liquid scintillation counting (see Note 2 in the separate Notes section for comments on scintillation counting and the variability of assay measurements).
- Remove the plate with cells from the incubator and cool it on ice for ~2 min.
Data analysis
- Representative examples of data
As detailed in the protocol above, we in general label proteins for 2-3 days followed by a chase for 18 h to enrich for radioactively labelled long-lived proteins. The chase can be reduced or extended to suit other experimental setups than shown here. It is, however, important to be aware that this will enrich for proteins with a different half-life, which in turn may alter the overall protein degradation rate during the sampling period, as well as the outcome of your experimental treatments (see further explanation in Note 3 in the Notes section). We therefore recommend keeping the labelling and chase periods as consistent as possible. As an example, decreasing the chase period from 18 h to 1 h increased the overall protein degradation rate from ≈ 1.05%/h to ≈ 1.3 %/h in LNCaP cells (Figure 2A).
To identify the proportion of lysosomal protein degradation taking place under the experimental conditions examined, we recommend including an inhibitor of lysosomal degradation such as the vacuolar-type H+-ATPase-inhibitor Bafilomycin A1 (Baf). Baf deacidifies lysosomes and resultantly inhibits lysosomal degradation activity in a dose-dependent manner. In LNCaP cells, the Baf-mediated inhibition of long-lived protein degradation is saturated at 25-50 nM in complete medium (Figure 2B). The Baf-sensitive fraction represents lysosomal degradation, whereas the Baf-insensitive fraction represents non-lysosomal degradation, mainly proteasomal protein degradation. Confirming this, the lysosomotropic compound NH4Cl reduces LLPD to the same extent as Baf (Figure 2C). Lysosomal LLPD is caused by autophagy. In LNCaP cells, we find that treatment with the Phosphatidylinositol 3-Kinase Catalytic Subunit Type 3 (PIK3C3)-inhibitor SAR-405 or knockdown of the autophagy-related proteins Unc-51 like Autophagy Activating Kinase 1 (ULK1) and ULK2 inhibit LLPD to the same extent as Baf or NH4Cl (Figure 2C). Thus, in LNCaP cells, basal lysosomal LLPD is due to PIK3C3/ULK-dependent autophagy, i.e., canonical autophagosome-mediated autophagy (macroautophagy), whereas putative contributions from micro-autophagy, endosomal micro-autophagy, or chaperone-mediated autophagy (all of which are PIK3C3/ULK-independent) are negligible. The contribution of the proteasome to LLPD may be determined by applying proteasomal inhibitors such as MG132 (Fuertes et al., 2003a; Engedal et al., 2013). Lysosomal and proteasomal inhibitors can be used to infer whether treatment effects on LLPD are caused by alterations in either lysosomal and proteasomal activity. For example, when combining autophagy-inhibitory calcium modulators (A23187 and thapsigargin) with either Baf or MG132, we found additive reducing effects on LLPD with MG132, but not with Baf (Engedal et al., 2013), indicating that the calcium modulators inhibit autophagic-lysosomal LLPD rather than proteasomal LLPD. Of note, one should be aware that crosstalk between proteasomal and autophagic-lysosomal pathways may occur (Dikic, 2017). However, within the time frame (2-6 h) and conditions (basal conditions, or conditions of amino acid starvation, mammalian target of rapamycin (mTOR)-inhibition, or endoplasmic reticulum stress) that we have tested, we have not observed signs of such crosstalk at the level of LLPD. In LNCaP and U2OS cells we found the effects of Baf and MG132 to be perfectly additive (Engedal et al., 2013), thus indicating absence of any measurable effect of autophagy-proteasome crosstalk on LLPD.
mTOR suppresses autophagy in a manner that is sensitive to the presence of amino acids and growth factors (Meijer et al., 2015). Thus, upon removal of amino acids and serum from the medium, mTOR activity is inhibited, and autophagy is induced. Figure 2D shows the typical response of LNCaP cells to treatment with the mTOR-inhibitor Torin1 or acute amino acid- and serum-starvation; total LLPD is increased by about two-fold, in a manner that in both cases is strongly sensitive to Baf, i.e., dependent on lysosomal activity. The increase in LLPD observed upon acute starvation differs from cell line to cell line. For example, the effect is weaker in VCaP and Huh7 cells compared to U2OS (Figure 2E) and LNCaP cells (Figure 2D). Importantly, the starvation-induced LLPD is sensitive to Baf in all cell lines, demonstrating that it mainly induces lysosomal LLPD. Of note, however, Baf does not completely abolish the effects of mTOR-inhibition or starvation (Figures 2D-2E), likely because these conditions slightly elevate proteasomal activity as well as autophagy (Fuertes et al., 2003a; Zhao et al., 2015 and our unpublished results). Also note that the degradation rate, and the degree of Baf-sensitive degradation, varies substantially between cell types in both complete medium and starvation medium (Figures 2D-2E). This reflects different degrees of basal autophagy (Baf-sensitive degradation in complete medium) and autophagic capacity (Baf-sensitive degradation in EBSS) in different cell lines.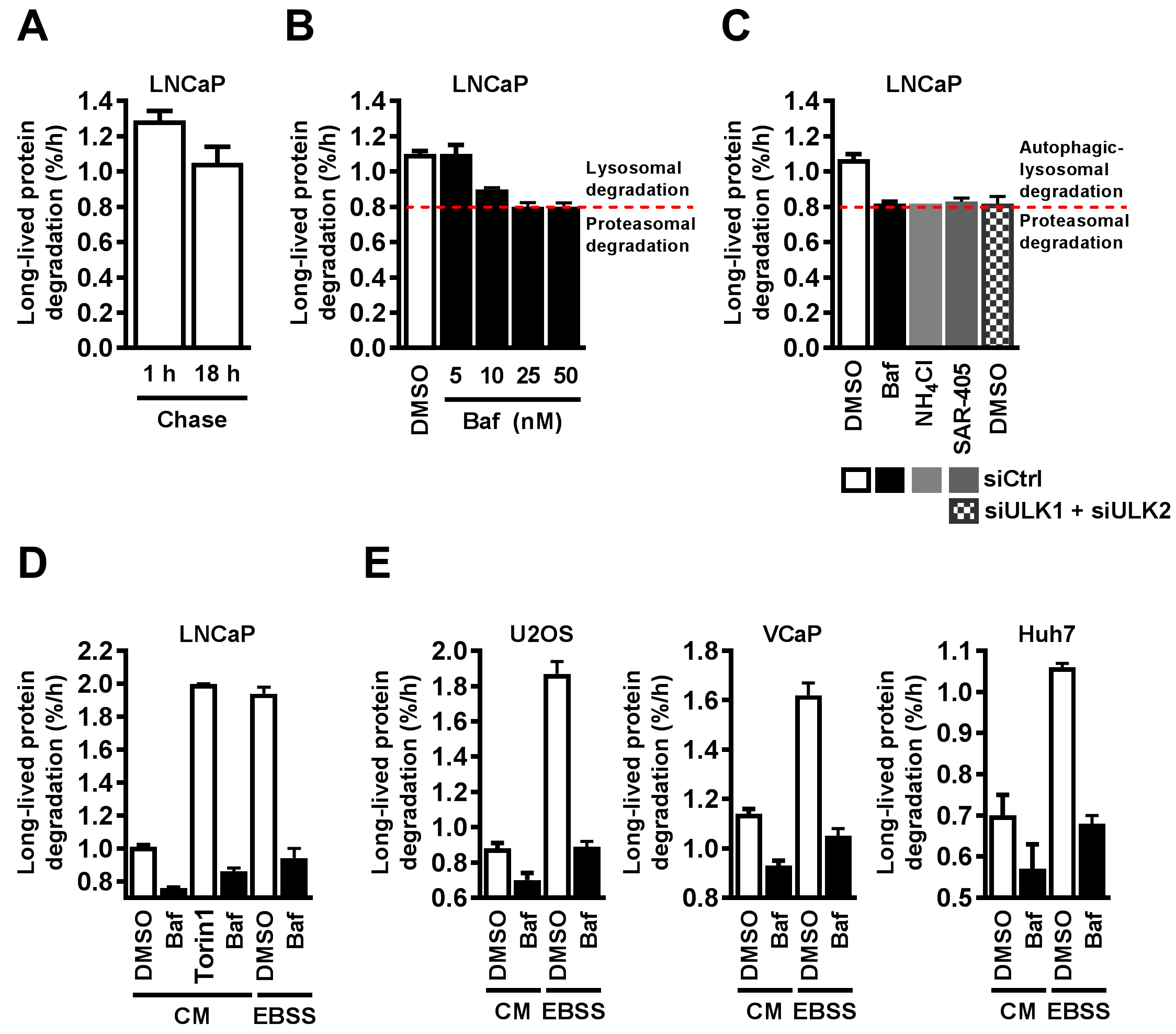
Figure 2. Quantitatively measuring autophagic flux of endogenous proteins using the long-lived protein degradation assay. A-D. LNCaP and (E) U2OS, VCaP, and Huh7 cell lines were labelled with 14C valine for 2-3 days, chased for (A, left bar) 1 h or (A, right bar, B-E) 18 h, and subjected to the indicated treatments in chase medium (the ‘sampling period’). Long-lived protein degradation (LLPD) was subsequently measured. A. LLPD was measured during a 6 h sampling period, subsequent to either 1 h or 18 h chase. B. LLPD is inhibited by Baf in a dose-dependent manner. The red, dotted line shows where Baf-mediated inhibition of LLPD is saturated. The remaining level of protein degradation is predominantly caused by the proteasome. C. Cells were reverse transfected with 10 nM non-targeting siRNA (‘siCtrl’) or 5 nM siULK1 in combination with 5 nM siULK2, and treated with DMSO (0.1%), Baf (50 nM), NH4Cl (10 mM), or 3 µM SAR-405 for 4 h. The red line indicates where LLPD inhibition is saturated. The remaining level of protein degradation is predominantly caused by the proteasome. D. Cells were subjected to the indicated treatments for 4 h. Baf and Torin1 were used at 50 nM. E. U2OS (left), VCaP (centre), and Huh7 (right) were subjected to the indicated treatments for 4 h. Baf was used at 50 nM in all cell lines. A-E. One representative of two independent experiments is shown (mean ± SD of 3 biological replicates). Baf, Bafilomycin A1; CM, complete medium; EBSS, Earle’s balanced salt solution. - Data analysis
The scintillation counter generates a text file where the sample numbers are listed with corresponding ‘counts per minute’ (CPM). The CPM values reflect the quantity of 14C in the individual samples. The degradation rate is calculated by dividing the amount of radioactivity in the TCA-soluble fraction by the total amount of radioactivity (the sum of radioactivity in the TCA-soluble and –insoluble fractions), divided by the duration of the sampling period. The degradation rate is expressed as ‘long-lived protein degradation (%/h)’.
Notes
- In general, we perform the experiment with duplicate or triplicate wells per treatment condition (i.e., two or three biological replicates in each experiment), whereas the scintillation counting is performed on the whole samples (i.e., no technical replicates for the measurement of radioactivity). The deviation between biological replicates is generally very low when following this protocol. From 23 independent experiments across 6 different cell lines and covering a total of 292 experimental conditions performed in triplicate, we have calculated the coefficient of variation (CV) of the LLPD rates of treatment replicates to be 2.3% ± 1.5% (mean CV ± standard deviation). More specifically, the CV values ranged from 0.1-8.9%, with 5%, 25%, 50%, 75%, and 95% percentile values of 0.4%, 1.2%, 2.0%, 3.2%, and 5.1%, respectively.
- Liquid scintillation counters can be set to record counts either for a specific time period, or until a specific number of counts has been accumulated. The uncertainty in the measurements is inversely correlated with the total number of counts. We recommend setting up the counter to accumulate at least 10,000 counts, whereby the standard deviation will be ≤ 1.0%. Following the current protocol with 2-3 days labelling with 0.1 µCi/ml 14C valine, these settings typically require less than a minute of counting for the TCA-insoluble fractions and a few minutes of counting for the TCA-soluble fractions. The background level is set during standard 14C calibration, and is very low (≤ 22 CPM). In the standard measurement protocol, this background is automatically subtracted from each sample. There is therefore no need to include additional background sample measurements (which would require many hours of sampling time for accurate measurement). As a general note, we recommend contacting your local service engineer for maintenance, calibration, and service questions.
- Different durations of the labelling, chase, and sampling periods than those presented here can be used. For example, some previous protocols used an 18-24 h labelling period, followed by a 1 h chase period (Mizushima et al., 2001; Bauvy et al., 2009). The advantage of increasing the labelling period to 2-3 days (as here) is that it can be conveniently combined with 2-3 days of transfection, e.g., with the reverse siRNA transfection described here. Moreover, the longer the labelling period, the more long-lived proteins (which are slowly synthesized and slowly degraded) will be labelled. It should be noted that in human fibroblasts, a pool of short-lived proteins that is predominantly degraded by the proteasome and not by macroautophagy has been described (Fuertes et al., 2003b). This pool of proteins showed a half-life of ~1.1-1.3 h, and an ~8 h chase period was required to fully eliminate them (Fuertes et al., 2003b). It was found that during 1-4 h of chase, 5-7.5% of the initial TCA-insoluble radioactivity of short-labelled proteins reflected the cellular release of non-degraded TCA-insoluble proteins to the medium. This release decreased to about 1/10th of the above values after 8 h of chase (Fuertes et al., 2003b). In summary, we advise a chase period of > 8 h, to minimize the influence of degradation and/or secretion of short-lived proteins. To be on the safe side, we routinely use an 18 h chase period. Such a long chase period has the additional advantage that when wishing to analyse effects of long-term experimental treatments on LLPD, the treatments can be initiated during the chase period (see Step B4b). Moreover, since the chase period is usually followed by a sampling period of 2-6 h, it is in relation to working hours more convenient with an 18 h chase period than an 8 h chase period. The sampling period can be longer than 6 h if desired, but if combining the assay with control treatments such as lysosomal and proteasomal inhibitors (as we recommend), it is advisable to keep the period shorter than 6 h to minimize secondary effects of the inhibitors. If wishing to analyse effects of experimental treatments on LLPD after more than 18 h of treatment, one may either increase the duration of the chase period, or even start the treatments during the labelling period, as alternatives to increasing the duration of the sampling period. Finally, the assay can also be done with shorter sampling periods than 2 h, but then great care must be taken to assure even time points for each experimental sample, and one may have to increase the amount of radioactive 14C-valine to achieve a higher degree of labelling and thus obtain an adequate level of counts in the scintillation counter (see Note 2 above).
Recipes
- 200 mM cold L-valine (Mw 117.15)
- Dissolve 1.172 g cold (i.e., non-radioactive) L-valine in 45 ml RPMI 1640 (without serum) or EBSS using a magnetic stirrer
- Adjust the volume to 50 ml with RPMI 1640 or EBSS, respectively
- Filter through a 0.45 µm filter into a sterile 50 ml tube
- This solution is stable for up to several months when stored at 4 °C. Do not preheat before use
- Dissolve 1.172 g cold (i.e., non-radioactive) L-valine in 45 ml RPMI 1640 (without serum) or EBSS using a magnetic stirrer
- RPMI 1640/10% FBS (Complete medium; CM) supplemented with 10 mM cold L-valine (CM-V)
- Add 1 part of the 200 mM cold L-valine solution (Recipe 1 above) to 19 parts of CM
- Make a new solution for each experiment
- Add 1 part of the 200 mM cold L-valine solution (Recipe 1 above) to 19 parts of CM
- 1 mg/ml poly-D-lysine
- Dissolve 5 mg poly-D-lysine (PDL) in 5 ml sterile H2O
- Aliquot in Eppendorf tubes and store at -20 °C
- The stock solution is stable for years when stored at -20 °C, and can be thawed and re-frozen several times
- Dissolve 5 mg poly-D-lysine (PDL) in 5 ml sterile H2O
- 1 mM Torin1 (Mw 607.62)
- Dissolve 10 mg Torin1 in 16.46 ml DMSO
- Aliquot in microcentrifuge tubes and store at -20 °C
- The stock solution is stable for years when stored at -20 °C, and can be thawed and re-frozen several times
- Dissolve 10 mg Torin1 in 16.46 ml DMSO
- PBS/2% BSA
- Dissolve 5 g BSA + in 200 ml PBS
- Add PBS to a total volume of 250 ml
- Filter through a 0.45 µm filter into a sterile glass flask
- The stock solution is stable for up to several months when stored at 4 °C
- Dissolve 5 g BSA + in 200 ml PBS
- 25% TCA
- For 10 ml, mix 2.5 ml 100% TCA with 7.5 ml Milli-Q (MQ) H2O
Caution: Add acid to the water, not vice-versa. - Make the solution fresh prior to harvesting
- For 10 ml, mix 2.5 ml 100% TCA with 7.5 ml Milli-Q (MQ) H2O
- 0.2 M KOH (Mw 56.11)
- Dissolve 1.12 g KOH in 90 ml MQ H2O
- Add MQ H2O to a total volume of 100 ml
- The solution is stable for up to several months when stored at room temperature
- Dissolve 1.12 g KOH in 90 ml MQ H2O
- 0.2 mM Bafilomycin A1 (Mw 622.84)
- Dissolve 0.1 mg Bafilomycin A1 in 803 µl DMSO
- Aliquot in microcentrifuge tubes and store at -20 °C
- The stock solution is stable for years when stored at -20 °C, and can be thawed and re-frozen several times
- Dissolve 0.1 mg Bafilomycin A1 in 803 µl DMSO
- 160 mM NH4Cl (Mw 53.49)
- Dissolve 256.8 mg NH4Cl in 25 ml MQ H2O
- Adjust to pH 7.74 at room temperature (= pH 7.4 at 37 °C)
- Adjust the final volume to 30 ml using MQ H2O
- Filter through a 0.45 µm filter into a sterile 50 ml tube
- This solution is stable for years when stored at -20 °C
- Dissolve 256.8 mg NH4Cl in 25 ml MQ H2O
Acknowledgments
This work was financially supported by the Research Council of Norway, the University of Oslo, the Anders Jahre Foundation, the Nansen Foundation, and the Legacy in the memory of Henrik Homan. This protocol is a modified version of that described in the methods section of Engedal et al., 2013, which in turn was adapted from Bauvy et al., 2009. We also would like to acknowledge the original work of Per O. Seglen, on which much of the method is based (Seglen and Solheim, 1978; Ronning et al., 1979; Seglen et al., 1979). The authors declare that they have no conflicts of interest.
References
- Bauvy, C., Meijer, A. J. and Codogno, P. (2009). Assaying of autophagic protein degradation. Methods Enzymol 452: 47-61.
- Dikic, I. (2017). Proteasomal and autophagic degradation systems. Annu Rev Biochem 86: 193-224.
- Engedal, N., Torgersen, M. L., Guldvik, I. J., Barfeld, S. J., Bakula, D., Sætre, F., Hagen, L. K., Patterson, J. B., Proikas-Cezanne, T., Seglen, P. O., Simonsen, A. and Mills, I. G. (2013). Modulation of intracellular calcium homeostasis blocks autophagosome formation. Autophagy 9(10): 1475-1490.
- Fuertes, G., Martin De Llano, J. J., Villarroya, A., Rivett, A. J. and Knecht, E. (2003a). Changes in the proteolytic activities of proteasomes and lysosomes in human fibroblasts produced by serum withdrawal, amino-acid deprivation and confluent conditions. Biochem J 375(Pt 1): 75-86.
- Fuertes, G., Villarroya, A. and Knecht. E. (2003b). Role of proteasomes in the degradation of short-lived proteins in human fibroblasts under various growth conditions. Int J Biochem Cell Biol 35: 651-664.
- Meijer, A. J., Lorin, S., Blommaart, E. F. and Codogno, P. (2015). Regulation of autophagy by amino acids and MTOR-dependent signal transduction. Amino Acids 47(10): 2037-2063.
- Mizushima, N., Yamamoto, A., Hatano, M., Kobayashi, Y., Kabeya, Y., Suzuki, K., Tokuhisa, T., Ohsumi, Y. and Yoshimori, T. (2001). Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol 152(4): 657-668.
- Nakabayashi, H., Taketa, K., Miyano, K., Yamane, T. and Sato, J. (1982). Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res 42(9): 3858-3863.
- Ronning, O. W., Pettersen, E. O. and Seglen, P. O. (1979). Protein synthesis and protein degradation through the cell cycle of human NHIK 3025 cells in vitro. Exp Cell Res 123(1): 63-72.
- Seglen, P. O., Grinde, B. and Solheim, A. E. (1979). Inhibition of the lysosomal pathway of protein degradation in isolated rat hepatocytes by ammonia, methylamine, chloroquine and leupeptin. Eur J Biochem 95(2): 215-225.
- Seglen P. O. and Solheim, A. E. (1978). Valine uptake and incorporation into protein in isolated rat hepatocytes. Nature of the precursor pool for protein synthesis. Eur J biochemi 85: 15-25
- Zhao, J., Zhai, B., Gygi, S. P. and Goldberg, A. L. (2015). mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc Natl Acad Sci U S A 112(52): 15790-15797.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Luhr, M., Sætre, F. and Engedal, N. (2018). The Long-lived Protein Degradation Assay: an Efficient Method for Quantitative Determination of the Autophagic Flux of Endogenous Proteins in Adherent Cell Lines. Bio-protocol 8(9): e2836. DOI: 10.21769/BioProtoc.2836.
Category
Biochemistry > Protein > Degradation
Cell Biology > Cell-based analysis > Autophagic activity
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.