- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Generation of microRNA Sponge Library

Published: Vol 8, Iss 8, Apr 20, 2018 DOI: 10.21769/BioProtoc.2820 Views: 8348
Reviewed by: Alka MehraAntoine de MorreeSmita Nair
Original research article
The authors used this protocol in:

Sep 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
This protocol describes the generation and functional validation of microRNA (miRNA) sponge or decoy constructs. When expressed from a strong promoter, these transcripts can sequester specific miRNA:RISC complexes, thereby resulting in a derepression of endogenous target mRNA. Hence, cells expressing such sponges display a partial or full miRNA loss-of-function phenotype.
Depending on the sponge sequence, the activity of any miRNA of choice can be inhibited by sponge sequestration, but it should be noted that these constructs do not seem to be specific for one particular miRNA. Rather, all miRNAs of the same family as defined by the seed sequence will be affected, albeit to a different degree.
Background
microRNAs (miRNAs) are short, non-coding RNAs with a size of about 21-24 nucleotides that post-transcriptionally silence the majority of all protein-coding genes in mammals. Since their discovery, more and more studies have clearly identified this regulatory layer as a crucial element for almost all physiological processes. Not surprising in this context, aberrant expression of miRNAs has also been causally linked to several human malignancies including cancer.
Using classic gain-of-function approaches, early studies have often utilized overexpression to evaluate the function of a particular miRNA. However, this can reach miRNA levels up to 100-fold or higher compared to the physiological context and may generate a phenotype that is not necessarily linked to the miRNA’s normal function.
To avoid this obvious problem with possible overexpression artifacts, in the last years several techniques for miRNA inhibition have been developed both for in vitro and in vivo use, thereby allowing the analysis of a specific miRNA or a group of miRNAs in a loss-of-function approach. These include e.g., the expression of miRNA decoys or sponges, long transcripts that interfere with miRNA function by sequestering miRNA:RISC complexes in a sequence-specific manner (Ebert et al., 2007; Gentner et al., 2009). Another frequently used strategy to interfere with miRNA function are antagomirs, small antisense RNAs complementary to specific miRNAs that can be brought into cells by different means such as by transfection or by viral transduction (Krützfeldt et al., 2005; Scherr et al., 2007). Notably, antagomirs have also been modified in a way that facilitates their cellular uptake, making them an attractive therapeutic tool. More recently, CRISPR/Cas9-mediated genome editing has been shown to be able to abolish miRNA expression on the level of its gene (Chang et al., 2016).
Here I provide a detailed protocol for the generation and functional validation of microRNA sponges as recently described (Lindner et al., 2017). Compared to synthesized small RNAs such as antagomirs, miRNA sponges are cheap and can be stably expressed by retroviral integration. This allows the long-term miRNA knockdown in almost every type of cell and tissue, even in specimen that are difficult to transfect. Moreover, the expression of the sponge RNA can be linked to a fluorescent and/or genetic marker, allowing the easy tracking of sponge-positive cells e.g., by microscopy or flow cytometry. Last, miRNA sponges have been shown to sequester not single miRNAs, but rather all miRNAs of the same family as defined by their seed sequence, i.e., sponges may uncover redundant roles of miRNAs expressed in a particular tissue.
Materials and Reagents
- 1.5 ml microcentrifuge tubes (SARSTEDT, catalog number: 72.690.001 )
- PCR tubes (SARSTEDT, catalog numbers: 72.985.002 and 65.986.002 )
- Chemically competent E. coli cells (home-made or New England Biolabs, catalog number: C2987H )
- DNA oligonucleotides, reverse phase cartridge or HPLC-purified (Sigma-Aldrich, catalog number: OLIGO )
- PHUSION polymerase (New England Biolabs, catalog number: M0530 )
- Deoxynucleotide (dNTP) solution mix (Carl Roth, catalog number: K039.1 )
- TOPO PCR Cloning Kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: 451245 )
- Plasmid Mini Kit (Carl Roth, catalog number: HP29.2 )
- Restriction enzymes (New England Biolabs)
- CloneJET PCR Cloning Kit (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: K1231 )
- Agarose Gel Extraction Kit (Jena Bioscience, catalog number: PP-202L )
- DNA ladder (Promega, catalog number: G5711 )
- LE Agarose (Biozym Scientific, catalog number: 840004 )
- Tris base (Sigma-Aldrich, catalog number: T1503 )
- Glacial acetic acid (Sigma-Aldrich, EMD-Millipore, catalog number: 27225-M )
- EDTA, disodium salt (Sigma-Aldrich, catalog number: E5134 )
- Deionized water
- 10x TAE for DNA electrophoresis (see Recipes)
Equipment
- Pipettes (Eppendorf, model: Research® Plus, catalog numbers: 3123000918 , 3123000020 )
- PCR machine TProfessional Basic (Biometra, catalog number: 070-701 )
- Electrophoresis equipment (Bio-Rad Laboratories)
Procedure
Here, I exemplarily describe the generation of a miRNA sponge sequestering miR-1839, however, by altering the sequence accordingly, any miRNA family can be targeted. The sponge construct itself is generated by a concatemeric PCR reaction (Figure 1), the design of the corresponding primers is explained below.
Figure 1. Schematic illustration of the concatemeric chain reaction that generates concatemeric repeats of the miRNA binding sites (exemplified for mmu-miRNA-1839). In the first PCR cycle, forward and reverse primers anneal with a small overlap that is extended during the elongation. In the subsequent PCR cycle, these extended oligonucleotides can anneal in different ways, i.e., with a full or a partial overlap (Step 3), eventually giving rise to longer and longer dsDNA fragments.
- Primer design
- Retrieve the sequence of the mature miRNA you want to target, e.g., at mirbase.org.
mmu-miR-1839 5’-AAGGUAGAUAGAACAGGUCUUG-3’ - Mark the four nucleotides (pos. 9-12) following the seed region (position 2-8).
mmu-miR-1839 5’-AAGGUAGAUAGAACAGGUCUUG-3’ - Form the reverse complement of the sequence and transform RNA into DNA.
mmu-miR-1839rc 5’-CAAGACCTGTTCTATCTACCTT-3’ - Generate a mismatch between your sponge sequence and the miRNA to be targeted by deletion of one nucleotide at the marked positions and by alteration of the three remaining nucleotides (in this example, TCTA is changed to GAT). This mismatch interferes with the slicing activity of the RISC complex and improves the miRNA sequestration by the sponge.
mmu-miR-1839rc_mismatch 5’-CAAGACCTGTGATTCTACCTT-3’ - Add a linker to the oligonucleotide, e.g., CCGT.
Sponge mmu-miR-1839F 5’-CAAGACCTGTGATTCTACCTTCCGT-3’
This is the first primer for the sponge PCR (see below).
Note: While we have used the CCGT linker in all our sponges, there is no apparent restriction with respect to its size, sequence or GC content. However, one should keep in mind that the linker sequence itself may accidentally function as a sponge if it corresponds to a microRNA seed region together with its surrounding sequences. We therefore recommend that the concatemeric sequence including the linker be checked for any unexpected miRNA binding, e.g., using the RegRNA 2.0 tool (Chang et al., 2013; http://regrna2.mbc.nctu.edu.tw/detection.html). - Form the reverse complement of the primer sponge mmu-miR-1839F.
5’-ACGGAAGGTAGAATCACAGGTCTTG-3’ - Split the sequence close to the middle and copy the sequence of the second half in front of the first half.
5’-ACGGAAGGTAGA ATCACAGGTCTTG-3’
Sponge mmu-miR-1839R 5’-ATCACAGGTCTTGACGGAAGGTAGA-3’
This is the second primer for the sponge PCR. - Order both primers from the company of your choice (e.g., Sigma-Aldrich), at least in reverse phase cartridge-purified quality.
- Retrieve the sequence of the mature miRNA you want to target, e.g., at mirbase.org.
- Sponge cloning
- Use the designed primers to perform the first round of PCR with the following conditions (see White et al., 1991 for details).
0.2 µl dNTPs (25 mM each)
0.5 µl PHUSION polymerase
6 µl GC buffer (supplied with the polymerase)
0.75 µl sponge forward oligo (10 µM)
0.75 µl sponge reverse oligo (10 µM)
H2O ad 30 µl
PCR program: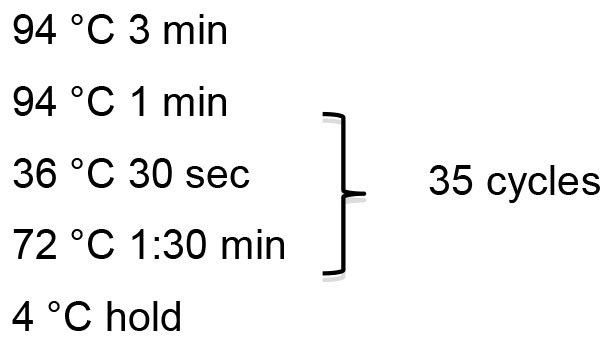
- Use an aliquot of the first PCR as a template for the second round of PCR.
0.2 µl dNTPs (25 mM each)
0.5 µl PHUSION polymerase
10 µl GC buffer (supplied with the polymerase)
5 µl template (unpurified PCR from the first round)
0.75 µl sponge forward oligo (10 µM)
0.75 µl sponge reverse oligo (10 µM)
H2O ad 50 µl
PCR program: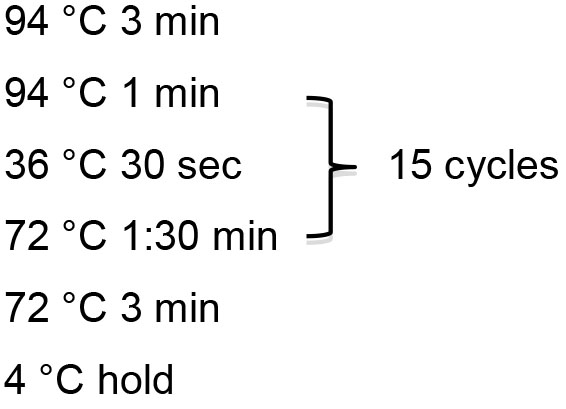
Anticipated result: In the concatemeric chain reaction (Figure 1), each PCR cycle extends the existing DNA strands and thereby adds more concatemer units. However, this extension is not ‘synchronized’, i.e., the second PCR will not generate a DNA fragment of a defined size, but rather a mixture of dsDNA fragments with different numbers of concatemeric repeats. In consequence, you should see a smear of DNA on an agarose gel that actually represents a DNA ladder of fragments differing in size by the length of one concatemeric repeat (Figure 2). We typically aim for a band size of about 750 bp, which encodes about 30 miRNA binding sites (the concatemeric repeat for the miR-1839 sponge e.g., has a length of 21 bp + 4 bp of linker; 25 bp x 30 repeats results in a fragment of 750 bp).
Of note, the efficiency of the concatemeric chain reaction is not the same for every set of oligonucleotides. Therefore, it may be that 15 cycles in the second PCR is too low (no or only very small PCR products on a gel) or too high (Figure 2, lanes 3 and 4). If this is the case, adjust the number of rounds for the second PCR accordingly, i.e., increase or reduce the number of cycles for the second PCR to 20 and 10 or even 5, respectively.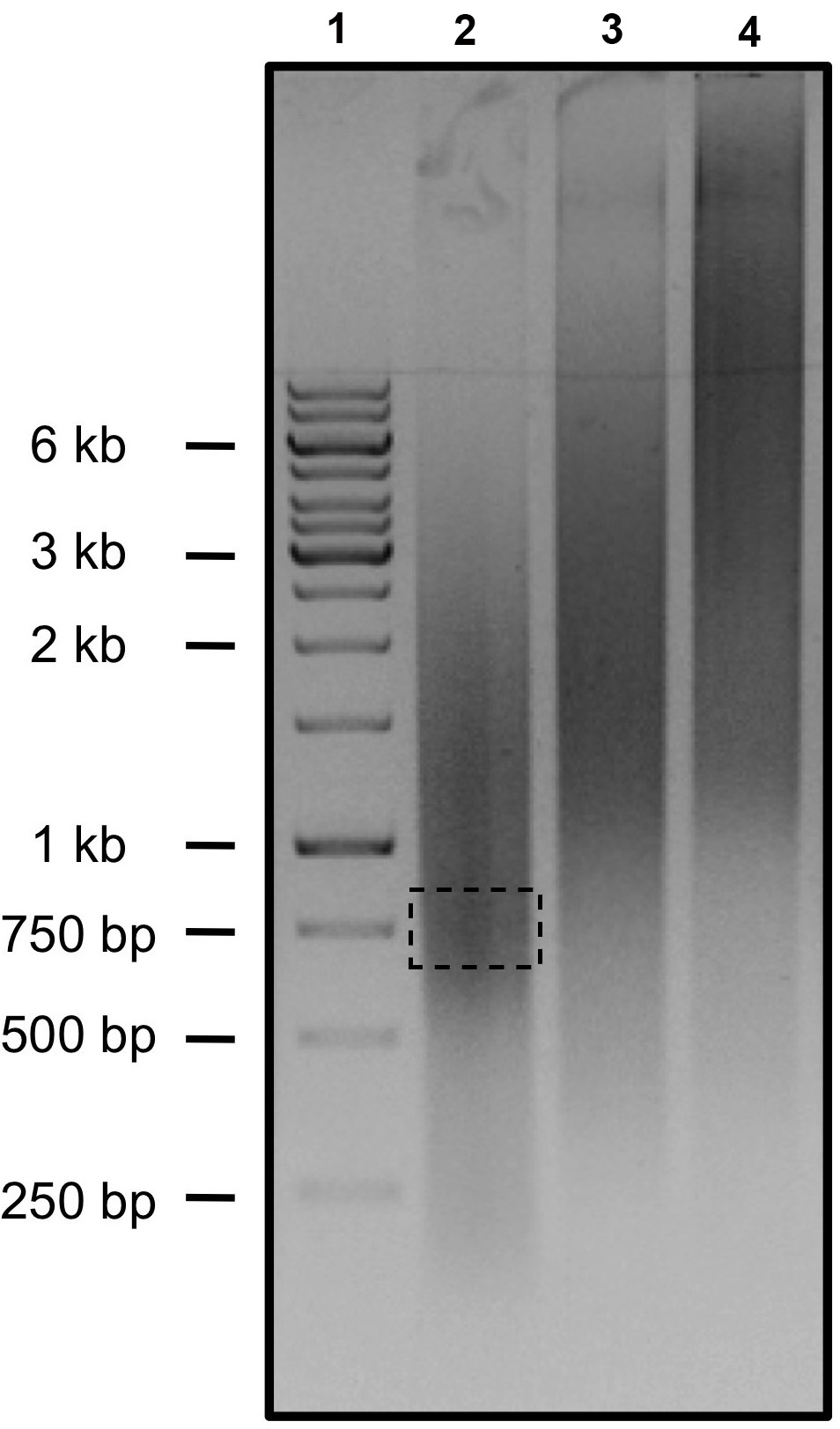
Figure 2. Agarose gel of the amplified DNA fragments after the second round of PCR for three different sponge constructs. Lane 1: Size marker. Lane 2: Product of a concatemeric PCR with the characteristic distribution of DNA fragment sizes. A fragment of about 750 bp (illustrated by a dashed rectangle), corresponding to about 30 miRNA binding sites, can be easily isolated from the gel. Lanes 3 and 4: Example of two sponges in which the second PCR with 15 cycles generated DNA fragments that exceeded the projected size. To isolate a fragment of about 750 bp, the second PCRs need to be repeated with only 5 or 10 cycles of amplification. - Cut a gel slice of the appropriate size, purify the DNA fragment using the Agarose Gel Extraction Kit (Jena Bioscience) according to the manufacturer’s protocol and blunt-end subclone it into a vector of choice, e.g., via TOPO cloning (Thermo Fisher Scientific) into pCR-BluntII-TOPO or by CloneJET PCR cloning (Thermo Fisher Scientific) into pJET1.2.
- Amplify selected clones and isolate the plasmid DNA using the Miniprep Kit.
- Verify clones by DNA digestion and sequencing, especially with respect to insert size.
- Clone the sponge fragment 3’ of a fluorescent and/or genetic marker into the expression vector of your choice. In line with the abovementioned example, the fragment should be oriented in the following way:
5’-[fluorescent/genetic marker]-[CAAGACCTGTGATTCTACCTTCCGT]n-3’
We typically use a pMIG-derived retroviral vector in which the retroviral LTR together with a pPGK promoter transcribes a PAC(confers puromycin resistance)-IRES-dsRed-sponge cassette, enabling the selection as well as identification of sponge expressing cells by flow cytometry or microscopy (Lindner et al., 2017). However, every expression vector should be fine as long as its promoter gives rise to a reasonably high sponge expression (see also Gentner et al., 2009).
- Use the designed primers to perform the first round of PCR with the following conditions (see White et al., 1991 for details).
- Functional validation of the miRNA sponges
In our experience, the expression of a miRNA sponge does not necessarily result in reduced miRNA levels, i.e., the binding of the miRNA:RISC complex to the sponge does not always induce miRNA decay. Thus, mature miRNA levels, e.g., quantified by Northern blot or qPCR, are a poor indicator of sponge function. If a validated target gene of the miRNA that is sponged is known, its expression level can be quantified by PCR. If the sponge successfully sequesters the specific miRNA:RISC complexes, the endogenous target mRNA should be stabilized. This may even result in higher protein expression as determined by Western blotting.
In case that no target gene is known, or if it is unclear to what extent this gene is regulated by the miRNA to be analyzed, we regularly validate sponge function by fluorescent reporters. These reporters contain a perfect complementary miRNA binding site in the 3’-UTR of the cDNA encoding the fluorescent protein (Lindner et al., 2017).
Anticipated result: When expressed in cells that have been selected for expression of different GFP-based reporters (Figure 3), dsRed-based sponge constructs should have no effect on a control reporter lacking a miRNA binding site or a reporter for another miRNA. However, in cells in which the sponge sequesters the miRNA:RISC-complex from the corresponding reporter, the GFP expression should be significantly increased, indicating a derepression of endogenous miRNA targets. Of note, this type of experiment has always been highly reproducible in our hands, resulting in little biological variation. In consequence, for most miRNA sponges only a few biological replicates are needed to reach statistically significant reporter derepression, e.g., when evaluated by a Student’s t-test.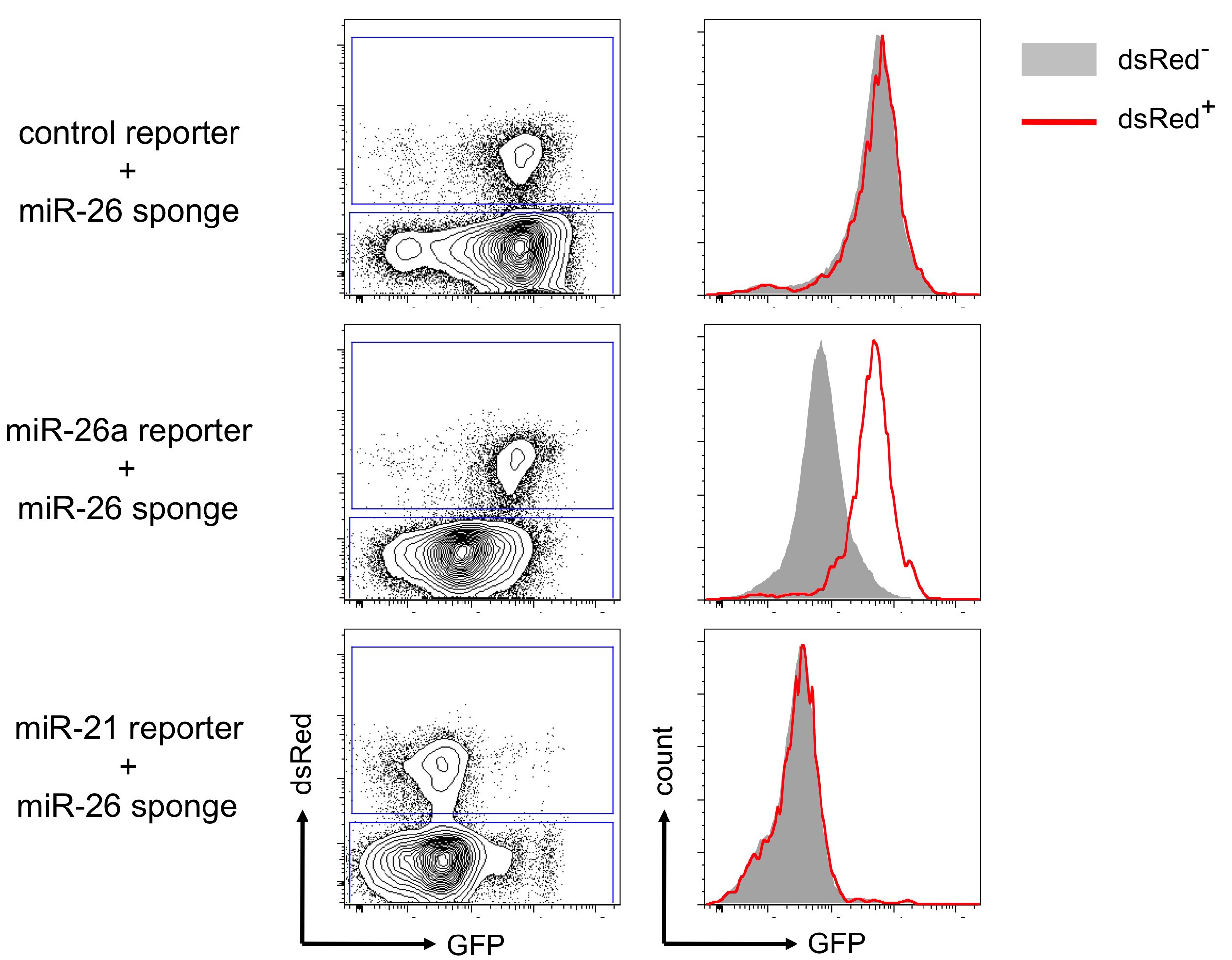
Figure 3. Precursor B cells selected for expression of a GFP-based control reporter, a reporter for miR-26a and a reporter for miR-21 were retrovirally transduced with a dsRed-based sponge construct targeting the miR-26 family. Density plots show the gating strategy, discriminating the transduced, dsRed-positive cells (upper gate) and the non-transduced, dsRed-negative (lower gate) population. Histogram overlays show a shift indicating reduced miRNA activity only in the cells expressing the miR-26a reporter together with the miR-26 sponge.
Recipes
- 10x TAE for DNA electrophoresis
48.4 g Tris base
11.4 ml acetic acid, glacial
3.7 g EDTA, disodium salt
Dissolve all components in 800 ml of deionized water and fill up to 1 L. Dilute 1:10 for electrophoresis
Acknowledgments
This protocol was previously used in Lindner et al. (2017), which was funded by the Deutsche-Forschungsgemeinschaft through Grant EXC294 (the Centre for Biological Signalling Studies, BIOSS), the Tiroler Zukunftsstiftung and the elite postdoctoral program of the Baden-Württemberg foundation. The author declares no conflict of interest.
References
- Chang, H., Yi, B., Ma, R., Zhang, X., Zhao, H. and Xi, Y. (2016). CRISPR/cas9, a novel genomic tool to knock down microRNA in vitro and in vivo. Sci Rep 6: 22312.
- Chang, T. H., Huang, H. Y., Hsu, J. B., Weng, S. L., Horng, J. T. and Huang, H. D. (2013). An enhanced computational platform for investigating the roles of regulatory RNA and for identifying functional RNA motifs. BMC Bioinformatics 14(Suppl 2): S4
- Ebert, M. S., Neilson, J. R. and Sharp, P. A. (2007). MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods 4(9): 721-726.
- Gentner, B., Schira, G., Giustacchini, A., Amendola, M., Brown, B. D., Ponzoni, M. and Naldini, L. (2009). Stable knockdown of microRNA in vivo by lentiviral vectors. Nat Methods 6(1): 63-66.
- Krützfeldt, J., Rajewsky, N., Braich, R., Rajeev, K. G., Tuschl, T., Manoharan, M. and Stoffel, M. (2005). Silencing of microRNAs in vivo with 'antagomirs'. Nature 438(7068): 685-689.
- Lindner, S. E., Lohmuller, M., Kotkamp, B., Schuler, F., Knust, Z., Villunger, A. and Herzog, S. (2017). The miR-15 family reinforces the transition from proliferation to differentiation in pre-B cells. EMBO Rep 18(9): 1604-1617.
- Scherr, M., Venturini, L., Battmer, K., Schaller-Schoenitz, M., Schaefer, D., Dallmann, I., Ganser, A. and Eder, M. (2007). Lentivirus-mediated antagomir expression for specific inhibition of miRNA function. Nucleic Acids Res 35(22): e149.
- White, M. J., Fristensky, B. W. and Thompson, W. F. (1991). Concatemer chain reaction: a Taq DNA polymerase-mediated mechanism for generating long tandemly repetitive DNA sequences. Anal Biochem 199(2): 184-190.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Herzog, S. (2018). Generation of microRNA Sponge Library. Bio-protocol 8(8): e2820. DOI: 10.21769/BioProtoc.2820.
Category
Molecular Biology > RNA > miRNA interference
Molecular Biology > RNA > miRNA-mRNA interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.