- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Extraction of Small Molecules from Fecal Samples and Testing of Their Activity on Microbial Physiology

Published: Vol 8, Iss 8, Apr 20, 2018 DOI: 10.21769/BioProtoc.2808 Views: 8019
Reviewed by: David CisnerosParul MehrotraAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jul 2017
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols



Abstract
The human body is colonized by vast communities of microbes, collectively known as microbiota, or microbiome. Although microbes colonize every surface of our bodies that is exposed to the external environment, the biggest collection of microbes colonizing humans and other mammals can be found in the gastrointestinal tract. Given the fact that the human gut is colonized by several hundred microbial species, our group hypothesized that the chemical diversity of this environment should be significant, and that many of the molecules present in that environment would have important signaling roles. Therefore, we devised a protocol to extract these molecules from human feces and test their signaling properties. Potentially bioactive extracts can be tested through addition to culture medium and analyses of bacterial growth and gene expression, among other properties. The protocol described herein provides an easy and rapid method for the extraction and testing of metabolites from fecal samples using Salmonella enterica as a model organism. This protocol can also be adapted to the extraction of small molecules from other matrices, such as cultured mammalian cells, tissues, body fluids, and axenic microbial cultures, and the resulting extracts can be tested against various microbial species.
Keywords: MetabolomeBackground
Complex assemblages of microbes live in and on humans, colonizing every surface exposed to the external environment. These communities have received several denominations over the decades, including normal flora, microbiota, and, more recently, microbiome (Sekirov et al., 2010; Kashyap et al., 2017). In humans, these vast microbial communities colonize our skin, respiratory tract, genitals, gastrointestinal tract, and many other sites. By far, the most heavily colonized site is the gastrointestinal tract, where trillions of microbes, encompassing several hundred species, coexist peacefully with their hosts. Some of these species knowingly live in symbiotic associations with the human organism, where both parts benefit from the interactions. For others, the relationship may be purely commensal, where the parts coexist without causing any harm to each other, but without providing or obtaining any benefit (Sekirov et al., 2010; Kundu et al., 2017).
The human gastrointestinal microbiota presents significant diversity in their composition, and stands out as a complex environment where interactions between different microbes as well as between microbes and host cells constantly occur. Bacteria are known to produce a plethora of bioactive small molecules, such as antibiotics, bacteriocins, pigments, secondary metabolites, quorum sensing signals, and many others (Antunes and Ferreira, 2009; Antunes et al., 2010; Antunes et al., 2011a). In such a complex environment as the intestinal microbiome, it is almost imperative to consider the production and accumulation of such molecules. These small molecules may represent by-products of metabolic activities or signals with specific roles, and can be produced both by the host itself as well as the microbes living in that environment; in many cases these small molecules are the tools used by these organisms to interact. Using high-throughput mass spectrometry-based metabolomics, we have previously shown that thousands of small molecules can be found in the lumen of the mammalian intestinal tract, and that the gut microbiome is involved in the production of many of them (Antunes et al., 2011b). In order to ascertain the signaling potential of small molecules from the gut metabolome, we have also extracted these molecules and tested their ability to modulate growth and gene expression of an enteric pathogen. As shown by our previous results, Salmonella enterica serovar Typhimurium responds to these molecules, and modulates the expression of over one hundred genes in response to bioactive small molecules from human feces (Antunes et al., 2014). Interestingly, many of the genes regulated by the fecal extract are required for the pathogenesis of Salmonella, such as those involved in the invasion of non-phagocytic host cells. More recently, we were able to purify and identify small aromatic compounds as the culprits for the regulation of Salmonella genes by the human gut metabolome (Peixoto et al., 2017). Here, we describe in detail the methods used by our group to obtain small molecules from the human gut metabolome and test them against Salmonella for various biological activities. A workflow of the procedures described herein can be found in Figure 1. 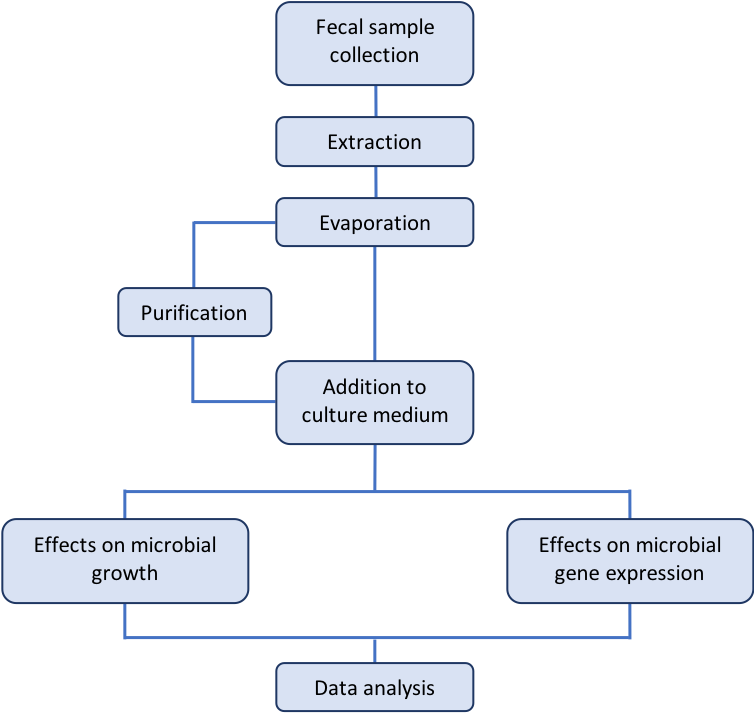
Figure 1. Workflow of the procedures described in this protocol
Materials and Reagents
- Polypropylene container (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 193A )
- Aluminum foil
- Tape
- Graduated glass pipettes (Fisher Scientific, catalog number: 13-678-25E )
- 2-ml Safe-Lock tubes (Eppendorf, catalog number: 0030120094 )
- Syringes (Descarpack, catalog number: 0324501 )
- Conical tubes (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 362694 )
- Axygen universal pipette tips (Corning, Axygen®, catalog number: T-200-C-L-R )
- Barrier tips (Fisher Scientific, catalog number: 02-707-430 )
- Syringe filter, 0.22-µm pore (KASVI, catalog number: K18-230 )
- Cuvettes (BRAND, catalog number: 759115 )
- Inoculation loop
- Borosilicate tubes, 16 x 100 mm (DWK Life Sciences, Kimble, catalog number: 73500-16100 )
- Bacterial culture (Salmonella enterica serovar Typhimurium SL1344)
- HPLC-grade ethyl acetate, ≥ 99.7% pure (Sigma-Aldrich, catalog number: 34858 )
- Nitrogen gas
- HPLC-grade methanol (Sigma-Aldrich, catalog number: 34860 )
- C18 cartridges containing 360 mg of sorbent (WATERS, catalog number: WAT051910 )
- Distilled water
- Phosphate-buffered saline (Sigma-Aldrich, catalog number: P5493-1L )
- RNeasy Mini Kit (QIAGEN, catalog number: 74106 )
- Agarose (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 17850 )
- QuantiTect Reverse Transcription Kit (QIAGEN, catalog number: 205311 )
- MinElute Reaction Cleanup Kit (QIAGEN, catalog number: 28204 )
- Primers (Integrated DNA Technologies)
- Power SYBR Green PCR Master Mix (Thermo Fisher Scientific, Applied BiosystemsTM, catalog number: 4367659 )
- Hydrochloric acid, 36.5-38% (Sigma-Aldrich, catalog number: H1758 )
- Sodium hydroxide, ≥ 98% pure (Sigma-Aldrich, catalog number: S8045 )
- Luria-Bertani (LB) Broth (BD, catalog number: 244620 )
- Bacteriological agar (Sigma-Aldrich, catalog number: A5306 )
- Streptomycin (Sigma-Aldrich, catalog number: S9137-25G )
- 1 N HCl (see Recipes)
- 1 N NaOH (see Recipes)
- LB broth or agar (see Recipes)
- LB with streptomycin (see Recipes)
Equipment
- Digital scale (Ohaus, model: SP202 )
- Glass bottle (Fisher Scientific, catalog number: FB800500 )
- Orbital shaker (BiomiXer, model: TS-2000A )
- Graduated glass cylinder (Laborglas, catalog number: 91376 )
- Glass boiling flask (Corning, PYREX®, catalog number: 4100-125 )
- Electronic pipette (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 9501 )
- Fume hood
- Rotary evaporator (Heidolph, catalog number: 560-01300-00 )
- 37 °C incubator
- Freezer, -20 °C
- P20 micropipette (Gilson, catalog number: F123600 )
- P200 micropipette (Gilson, catalog number: F123601 )
- P1000 micropipette (Gilson, catalog number: F123602 )
- Speed-Vac concentrator (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: SPD131DDA-115 )
- Vortex (Scientific Industries, model: Vortex-Genie 2, catalog number: SI-0236 )
- pH meter (Sigma-Aldrich, catalog number: MT 51302916 )
Manufacturer: Mettler-Toledo, catalog number: 51302916 . - Autoclave
- Visible range spectrophotometer
- Nucleic acid (UV) spectrophotometer
- Shaker (NOVATECNICA, model number: NT145 )
- Centrifuge for 96-well plates (Eppendorf, model: 5810 , catalog number: 5810000017)
- Real Time PCR Machine (Thermo Fisher Scientific, Applied BiosystemsTM, catalog number: 4376357 )
Software
- GraphPad Prism (GraphPad Software)
- StepOne Software v2.3 (Thermo Fisher Scientific)
Procedure
- Extraction of small molecules from fecal samples
- The donor should collect a fresh fecal sample using a clean–not necessarily sterile–polypropylene container, store it in the fridge, and bring it to the laboratory within the next 24 h.
- Once the fecal sample is in the laboratory, weight the sample using a digital scale.
Note: Extracts can be prepared from samples of any weight. However, in order to achieve the desired concentration for testing, we recommend a minimum sample weight of 50 g. - Transfer the sample to a clean glass bottle, not sterile.
- Based on the weight of the sample, add 1 volume of ethyl acetate into the glass bottle containing the sample.
- Close the bottle tightly, wrap it with aluminum foil, and place it horizontally in an orbital shaker.
- Fix the bottle to the shaker using tape.
- Incubate it overnight at room temperature, setting the orbital shaker to 50 rotations per minute.
- After the incubation, remove the bottle from the shaker, place it vertically on the bench and let it stand for 5 min, so that the solid material can precipitate.
- Carefully remove the solvent from the bottle using a glass pipette and transfer it to a new bottle.
- Measure and record the volume of solvent recovered using a graduated glass cylinder.
Note: It is not possible to recover the entire volume of ethyl acetate used to perform this extraction. This is due to the fact that some of the solvent will be absorbed by the fecal sample. - The extract can then be stored in a -20 °C freezer (Note 1).
- The donor should collect a fresh fecal sample using a clean–not necessarily sterile–polypropylene container, store it in the fridge, and bring it to the laboratory within the next 24 h.
- Drying the extract
- Transfer the desired volume of extract to a glass boiling flask or 2-ml Safe-Lock tubes using a glass pipette or Axygen pipette tips and a micropipettor.
- Completely evaporate the solvent using a rotary evaporator or a Speed-Vac concentrator (Note 2).
Note: Because ethyl acetate has a low boiling point and some of the bioactive molecules may be temperature-sensitive, it is not recommended to increase the temperature on the evaporator or Speed-Vac above 37 °C. - Store the dried extract in the original vial (a glass boiling flask or 2-ml plastic tubes) at -20 °C (Note 1).
- Transfer the desired volume of extract to a glass boiling flask or 2-ml Safe-Lock tubes using a glass pipette or Axygen pipette tips and a micropipettor.
- Purification of small molecules with potential bioactivity
- Thaw the dried extracts at room temperature.
Note: Thawing the dried extract may produce some water precipitation. If this is the case, the precipitate can be dried by applying a gentle stream of nitrogen gas into the flask or tube. - Add 1 volume of 25% methanol (in water) to the vial(s) containing the dried extract.
- Scrape the walls of the vial with a glass pipette or a pipette tip, pipetting up and down and thoroughly vortexing the vials periodically.
- Take the desired number of C18 cartridges and, using a syringe, apply 2 ml of methanol through each cartridge.
- Using a syringe, apply 2 ml of water through each cartridge.
- Apply a maximum of 5 ml of extract through each column.
- Collect the flow-through using a conical tube.
- Wash each column with 4 ml of distilled water.
- Elute the molecules bound to the resin using 2 ml of solutions of increasing methanol concentrations–concentrations should be determined by the user, we suggest using 30, 40, 50, 60, 70, 80, 90, and 100% methanol (in water).
- Collect each fraction using a conical or Safe-Lock tube.
- Completely evaporate the solvent using a Speed-Vac concentrator.
- Save fractions in a -20 °C freezer until testing (Note 1).
- Thaw the dried extracts at room temperature.
- Preparing culture medium containing the desired extract
- The desired extracts or fractions obtained above should be removed from the freezer and LB broth should be added to the tubes–the volume added will vary, but we recommend starting with the same volume of culture medium as the volume of solvent evaporated (1x concentration).
- Scrape the walls of the tubes with a glass pipette or a pipette tip, pipetting up and down and thoroughly vortexing the tubes periodically.
Note: Some samples will not solubilize completely. Due to the nature of the solvents used, some extracts will have a high fat content. In some cases, a significant amount of the material will either stick to the tube wall or form insoluble flakes in the culture medium. - Filter the culture medium using a syringe and a 0.22-µm-pore filter to remove debris and any insolubilized material.
- Measure the pH of the solution.
- Adjust the pH to approximately 7.3 using HCl or NaOH.
- Add streptomycin to achieve a final concentration of 100 µg/ml.
- Filter-sterilize the solution using a syringe and a 0.22-µm-pore filter.
- Save the medium at room temperature until used (for storage times longer than 48 h we recommend storing the medium in a 4 °C fridge) (Note 1).
- The desired extracts or fractions obtained above should be removed from the freezer and LB broth should be added to the tubes–the volume added will vary, but we recommend starting with the same volume of culture medium as the volume of solvent evaporated (1x concentration).
- Testing the effect of small molecules from the fecal metabolome on bacterial growth
- Using an inoculation loop, take a small amount of bacterial culture from a frozen stock of Salmonella enterica serovar Typhimurium SL1344 and streak a plate of LB containing 100 µg/ml of streptomycin.
Note: SL1344 can be obtained from the National Collection of Type Cultures, Public Health England, at https://www.phe-culturecollections.org.uk/collections/nctc.aspx. - Incubate overnight at 37 °C.
- Using an inoculation loop, take a single colony of Salmonella and transfer it to approximately 2 ml of LB broth containing 100 µg/ml of streptomycin, in 16 x 100 mm borosilicate tubes.
- Incubate it overnight at 37 °C with shaking (225 RPM).
- Dilute the culture 1:10 using phosphate-buffered saline.
- Measure the optical density of the suspension and multiply the value obtained by 10 to achieve the optical density of the original suspension.
- Calculate the volume of culture required to inoculate 8 ml of culture medium and achieve a starting optical density at 600 nm (OD600) of 0.05.
- Inoculate the desired number of 16 x 100 mm borosilicate tubes containing 8 ml of either LB broth or LB broth to which the fecal extract had been added, and 100 µg/ml of streptomycin.
Note: If previously stored at 4 °C, culture medium should be equilibrated to room temperature before inoculation with Salmonella. - Incubate at 37 °C with shaking (225 RPM).
- Every 30 min, remove a small aliquot from each of the cultures and use it to measure and record the OD600 of the cultures.
Note: After the initial time points, the cultures will need to be diluted in phosphate-buffered saline before measurements can be made. This should be done to make sure that the optical density values obtained fall within the linear range of the equipment. Please be mindful of the linear range of your specific spectrophotometer. - Using GraphPad Prism, plot the values obtained in a curve where the y-axis represents the optical density values and the x-axis represents the time of incubation.
- Compare the curves to evaluate if the extract has any effect on bacterial growth–statistical analyses can be performed within GraphPad Prism.
- Using an inoculation loop, take a small amount of bacterial culture from a frozen stock of Salmonella enterica serovar Typhimurium SL1344 and streak a plate of LB containing 100 µg/ml of streptomycin.
- Testing the effect of small molecules present in the extract on bacterial gene expression
- Prepare a growth curve experiment exactly as described above, using either LB or LB containing a fecal extract–the number of replicates per condition can vary, but we recommend a minimum of 4 replicates per condition.
- Based on the growth curve previously performed using the same fecal extract, grow the cultures to the desired growth stage–we suggest that the cultures used for RNA extraction be in the exponential growth phase.
- Collect between 1-2 ml of each culture and transfer to a conical tube containing 2-4 ml (2 volumes) of RNAprotect Bacteria Reagent.
- Manually invert tubes 5 times to mix.
- Let it stand for 5 min at room temperature.
- Centrifuge at 5,000 x g for 5 min.
- Discard the supernatant.
Note: At this stage, the pellet can be stored at -80 °C and the extraction can continue in the next few days. - Extract RNA using the RNeasy Mini Kit, according to the manufacturer’s recommendations, using protocol 4 of the RNAprotect Bacteria Reagent Handbook (version 01/2015).
- Continue the extraction procedure using protocol 7 of the RNAprotect Bacteria Reagent Handbook (version 01/2015), including the on-column DNAse treatment.
- Determine the amount and concentration of the RNA obtained using a UV spectrophotometer.
- Test the samples for the presence of contaminant genomic DNA by polymerase chain reactions (PCR):
- Perform a regular PCR using Taq Polymerase and primers targeting the desired gene–we recommend using the housekeeping gene gapA (forward: GGCGCTAACTTTGACAAATACGAAGG, reverse: AGTCATCAGACCTTCGATGATGCCG; Peixoto et al., 2017).
- Use 1 ng of genomic DNA from Salmonella as a positive control and 100 ng of the RNA extracted for testing.
- Run the PCR products on a 1.5% agarose gel.
Note: Stain the gel using your preferred method of nucleic acid staining. - Observe the bands under UV light–the presence of a band of the expected size in the positive control and the absence of such band on the RNA samples tested indicates that no significant DNA contamination exists.
Note: RNA should be stored at -80 °C while the presence of DNA in the samples is being tested.
- Perform a regular PCR using Taq Polymerase and primers targeting the desired gene–we recommend using the housekeeping gene gapA (forward: GGCGCTAACTTTGACAAATACGAAGG, reverse: AGTCATCAGACCTTCGATGATGCCG; Peixoto et al., 2017).
- Synthesize complementary DNA (cDNA) using between 500-1,000 ng of RNA and the QuantiTect Reverse Transcription Kit, according to the manufacturer’s recommendations.
- Purify the cDNA using the MinElute Reaction Cleanup Kit, according to the manufacturer’s recommendations.
- Quantify the cDNA using a UV spectrophotometer.
- Dilute the cDNA so that all samples are at a concentration of 5-10 ng/µl.
- Perform Real-Time PCR using 1 ng of the cDNA as template, primers targeting the desired genes, and the Power SYBR Green PCR Master Mix.
- Determine the relative expression levels between samples grown in the absence or presence of the fecal extract using the ΔΔCt method, and the gapA gene as the housekeeping control.
- Prepare a growth curve experiment exactly as described above, using either LB or LB containing a fecal extract–the number of replicates per condition can vary, but we recommend a minimum of 4 replicates per condition.
Data analysis
- To analyze the effect of small-molecule extracts on bacterial growth, optical density values should be plotted in a curve where the y-axis represents the optical density values and the x-axis represents the time of incubation. This can be done using GraphPad Prism, and the average values should be plotted, together with the standard deviation or the standard error of means. The curves should be visually compared to evaluate if the small molecules have any effect on bacterial growth. For statistical purposes, we recommend a minimum of 4 replicates per condition and the use of two-tailed unpaired t-tests for the evaluation of statistical significance. In the unlikely event that values within one group of samples vary widely, outliers can be detected and removed using the Grubbs’ test (https://www.graphpad.com/quickcalcs/Grubbs1.cfm). Determining whether or not the extract has a remarkable effect on growth is somewhat subjective, but we suggest using a statistically-significant (P < 0.05), 25% (or higher) change on the optical density as a starting point.
- To analyze the effect of small-molecule extracts on bacterial gene expression, threshold cycle (Ct) values obtained from the Real-Time PCR equipment through the StepOne Software v2.3 should be processed using the ΔΔCt method. First, Ct values obtained with the housekeeping control gene (gapA is used here as an example) should be subtracted from the Ct values obtained with each the genes being tested. This should be done in a sample-specific manner, i.e., the gapA Ct from one sample should be subtracted from the test gene Ct of that same sample and no other. The resulting ΔCt should be transformed from its logarithmic form by applying the formula 2-ΔCt. The resulting values should then be normalized to the average of the values obtained in the control group. As a result, the average of the control group should be normalized to 1 and every single value from the control or test group should be compared accordingly. Results can be plotted as bars of the average values per group, with bars displaying the standard deviation or the standard error of means. As noted above, for statistical purposes, we recommend a minimum of 4 replicates per condition and the use of two-tailed unpaired t-tests for the evaluation of statistical significance. In case some values vary widely from the others within the same group of samples, outliers can be detected and removed using the Grubbs’ test (https://www.graphpad.com/quickcalcs/Grubbs1.cfm). As noted above, determining whether or not the extract has a significant effect can be subjective, and should be interpreted taking into account the experimental model used as well as the nature of the genes tested. Nevertheless, we suggest using a statistically-significant (P < 0.05), 2-fold change in gene expression as a starting point.
Notes
- The time the extract can be stored for will vary depending on the properties of each bioactive molecule. However, to avoid degradation we suggest that extracts be stored for a maximum of one month.
- The time required for complete evaporation will vary depending on the sample type, size, and water content. Solely as a reference, we expect that a sample divided into 1-ml aliquots will take about 1-2 h to completely evaporate in a Speed-Vac set at a temperature of 37 °C. A 50-ml sample in a rotatory evaporator set at the same temperature will take 2-4 h to fully evaporate.
Recipes
- 1 N HCl (100 ml)
Add 3 ml of hydrochloric acid, 36.5-38% (Sigma-Aldrich) to 97 ml of distilled water - 1 N NaOH (100 ml)
Weigh 4 g of sodium hydroxide, ≥ 98% pure (Sigma-Aldrich) and add to 100 ml of distilled water - LB broth or agar
- Weigh 25 g of LB powder and add 1 L of distilled water in a glass bottle
- For LB agar, add 15 g of agar to the solution
- Mix it well by shaking the bottle
- Measure the pH of the solution and adjust it to 7.3 using HCl or NaOH
- Sterilize the medium using an autoclave
- Weigh 25 g of LB powder and add 1 L of distilled water in a glass bottle
- LB with streptomycin
- Weigh 100 mg of streptomycin
- Add 900 µl of water
- Mix thoroughly using a vortex
- Measure the volume obtained using a micropipettor and a pipette tip
- Add more distilled water to obtain a final volume of 1 ml
- Add 1 µl of this solution per 1 ml of LB broth or agar to obtain a final concentration of 100 µg/ml
- Weigh 100 mg of streptomycin
Acknowledgments
This work was funded by the National Council for Scientific and Technological Development (CNPq-Brazil), the Rio de Janeiro State Funding Agency FAPERJ and the Ministry of Health of Brazil. The authors declare that no conflict of interest exists.
References
- Antunes, L. C. and Ferreira, R. B. (2009). Intercellular communication in bacteria. Crit Rev Microbiol 35(2): 69-80.
- Antunes, L. C., Ferreira, R. B., Buckner, M. M. and Finlay, B. B. (2010). Quorum sensing in bacterial virulence. Microbiology 156(Pt 8): 2271-2282.
- Antunes, L. C., Davies, J. E. and Finlay, B. B. (2011a). Chemical signaling in the gastrointestinal tract. F1000 Biol Rep 3: 4.
- Antunes, L. C., Han, J., Ferreira, R. B., Lolic, P., Borchers, C. H. and Finlay, B. B. (2011b). Effect of antibiotic treatment on the intestinal metabolome. Antimicrob Agents Chemother 55(4): 1494-1503.
- Antunes, L. C., McDonald, J. A., Schroeter, K., Carlucci, C., Ferreira, R. B., Wang, M., Yurist-Doutsch, S., Hira, G., Jacobson, K., Davies, J., Allen-Vercoe, E. and Finlay, B. B. (2014). Antivirulence activity of the human gut metabolome. MBio 5(4): e01183-01114.
- Kashyap, P. C., Chia, N., Nelson, H., Segal, E. and Elinav, E. (2017). Microbiome at the frontier of personalized medicine. Mayo Clin Proc 92(12):1855-1864.
- Kundu, P., Blacher, E., Elinav, E. and Pettersson, S. (2017). Our gut microbiome: the evolving inner self. Cell 171(7):1481-1493.
- Peixoto, R. J. M., Alves, E. S., Wang, M., Ferreira, R. B. R., Granato, A., Han, J., Gill, H., Jacobson, K., Lobo, L. A., Domingues, R., Borchers, C. H., Davies, J. E., Finlay, B. B. and Antunes, L. C. M. (2017). Repression of Salmonella host cell invasion by aromatic small molecules from the human fecal metabolome. Appl Environ Microbiol.
- Sekirov, I., Russell, S. L., Antunes, L. C. and Finlay, B. B. (2010). Gut microbiota in health and disease. Physiol Rev 90(3): 859-904.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Alves, E. S., Ferreira, R. B. R. and Antunes, L. C. M. (2018). Extraction of Small Molecules from Fecal Samples and Testing of Their Activity on Microbial Physiology. Bio-protocol 8(8): e2808. DOI: 10.21769/BioProtoc.2808.
Category
Microbiology > Microbial signaling > Quorum sensing
Biochemistry > Other compound > Small molecular
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.