- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Guanine Nucleotide Exchange Assay Using Fluorescent MANT-GDP
Published: Vol 8, Iss 7, Apr 5, 2018 DOI: 10.21769/BioProtoc.2795 Views: 14834
Reviewed by: Ralph Thomas BoettcherAkira KarasawaSarah Diermeier
Original research article
The authors used this protocol in:

Jul 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
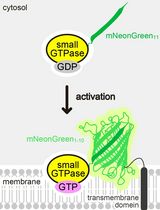
GTPases are molecular switches that cycle between the inactive GDP-bound state and the active GTP-bound state. GTPases exchange nucleotides either by its intrinsic nucleotide exchange or by interaction with guanine nucleotide exchange factors (GEFs). Monitoring the nucleotide exchange in vitro, together with reconstitution of direct interactions with regulatory proteins, provides key insights into how a GTPase is activated. In this protocol, we describe core methods to monitor nucleotide exchange using fluorescent N-Methylanthraniloyl (MANT)-guanine nucleotide.
Keywords: GTPaseBackground
GTPases are guanine nucleotide binding proteins that regulate a breadth of cellular processes, ranging from protein biosynthesis to cell-cycle progression and from cytoskeletal reorganization to membrane trafficking. GTPases can be thought of as molecular switches that cycle between a GDP-bound ‘off’ state and a GTP-bound ‘on’ state; upon GTP-binding via nucleotide exchange of GDP for GTP, GTPases become active and will bind to down-stream effector proteins to recruit and activate the biological function of these effectors. GTPases bind the γ-phosphate of GTP via interactions with a highly conserved threonine of the switch I loop (G2 domain) and a glycine within a DxxG motif of the switch II loop (G3 domain). Upon GTP hydrolysis, the loss of the interaction with the γ-phosphate causes a dynamic conformational change that turns the GTPase into an off state (Vetter and Wittinghofer, 2001). Typically, small GTPases have a very high affinity for guanine nucleotides, with dissociation constants in the nanomolar to picomolar range (Bos et al., 2007), and therefore require guanine nucleotide exchange factors (GEFs) to lower their nucleotide affinity to allow for rapid activation. Notable exceptions include several highly conserved centrosomal/ciliary small GTPases, such as Rabl2 (Kanie et al., 2017), ARL13B (Ivanova et al., 2017), Ift27/Rabl4 (Bhogaraju et al., 2011), and Arl6 (Price et al., 2012), and many large GTPases, such as the dynamin family (Gasper et al., 2009), which have lower affinities for GDP/GTP, with dissociation constants in the micromolar range. In this configuration, these GTPases can become activated without the use of GEFs via their intrinsic nucleotide exchange. Although small GTPases that show spontaneous exchange may have additional regulatory proteins, finding that a small GTPase has a micromolar affinity for GDP/GTP and is capable of spontaneous exchange may suggest the absence of a traditional GEF and suggest looking for other forms of GTPase regulatory factors (see Kanie et al. [2017] for a notable example).
Fluorescently-labeled guanine nucleotides are more suitable for monitoring nucleotide exchange than radioactive GDP/GTP, as they are safer and allow continuous spectroscopic monitoring and thus provide a more detailed analysis of kinetics. N-Methylanthraniloyl (MANT) is the most widely used fluorescent analog to label guanine nucleotides because it is smaller than most fluorophores and unlikely to cause major perturbations of protein-nucleotide interactions (Hiratsuka, 1983). Another attractive feature of this fluorescent nucleotide is that the emitted fluorescent signal increases dramatically upon binding to a GTPase (typically twice as high as the signal of unbound MANT-guanine nucleotide) (John et al., 1990), allowing one to directly monitor the association and dissociation of guanine nucleotides from GTPases. The most commonly used method to monitor nucleotide exchange is tracking the decrease in the fluorescence of protein-bound MANT-GDP upon addition of an excess amount of GppNHp, a non-hydrolyzable GTP analog. MANT-GDP-bound GTPase can be prepared by the incubation of nucleotide free GTPase with 1.5 fold excess of MANT-GDP (John et al., 1990; Eberth and Ahmadian, 2009). Although this protocol is described in great detail and allows us to save expensive MANT-GDP, it is time-consuming and limited by the accessibility to high performance liquid chromatography. Alternatively, we describe here a simpler protocol where MANT-GDP is loaded onto GTPase by incubating GTPase with 20-fold molar excess of MANT-GDP in the presence of ethylenediaminetetraacetic acid (EDTA). EDTA chelates magnesium ions, which form coordination bonds with the β and γ phosphates of GTP (or β phosphate of GDP) and with the GTPase (Pai et al., 1990; Tong et al., 1991). EDTA significantly lowers the affinity of the GTPase for guanine nucleotide. The loading reaction is stopped by the addition of excess magnesium chloride. Unbound MANT-GDP is removed by gel filtration using a NAP-5 prepacked column and the nucleotide exchange reaction is initiated by the addition of 100-fold molar excess of GppNHp. Exchange is monitored by a decrease in the fluorescent signal as recorded by a spectrometer with excitation wavelength at 360 nm and emission at 440 nm.
Variations on this method are readily accomplished by addition of biochemically pure nucleotide variants, drugs, or purified protein regulatory factors, allowing for a range of mechanistic experiments and definitive tests of biochemical mechanisms.
Materials and Reagents
- Microcentrifuge tube (Corning, Costar®, catalog number: 3621 )
- 50 ml conical tube (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 339652 )
- Oak ridge tube (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 3119-0050 )
- 15 ml conical tube (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 339650 )
- (Optional) Amicon ultra concentrator (Merck, catalog number: UFC901024 )
- Dialysis tubing (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 88242 )
- NAP-5 columns (GE Healthcare, catalog number: 17085301 )
- Aluminum foil
- 384-well microplate (Greiner Bio One International, catalog number: 784076 )
- (Optional) 96-well microplate (Corning, catalog number: 3686 )
- 0.2 µm disposable bottle top filter (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 597-4520 )
- 0.22 µm syringe filter (Merck, catalog number: SLGV033RS )
- Disposable cuvettes (Fisher Scientific, catalog number: 14-955-127 )
- Multichannel pipette tip (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 7421 )
- Rosetta2 bacterial cells (Merck, EMD Millipore, catalog number: 71403 )–Store at -80 °C
- Recombinant GTPase
Note: Ideally, the concentration should be 100 µM or greater. See the protocol below for the expression and purification of recombinant GTPases. Store at -80 °C. - Gateway-cloning compatible pGEX6p vector
- 4x LDS sample buffer (Thermo Fisher Scientific, InvitrogenTM, catalog number: NP0008 )
- 2-Mercaptoethanol (Sigma-Aldrich, catalog number: M3148 )–Store at 4 °C
- GppNHp (Abcam, catalog number: ab146659 )
Note: Dissolve in Milli-Q water to prepare a 50 mM stock solution. Store at -20 °C. - Nu-PAGE gel (Thermo Fisher Scientific, InvitrogenTM, catalog number: NP0321BOX )
- Coomassie Brilliant Blue R-250 (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 20278 )
- (Optional) Classical Laemmli sodium dodecyl sulfate (SDS) sample buffer/SDS-polyacrylamide gel electrophoresis (SDS-PAGE) gel
- Glutathione Sepharose 4B media (GE Healthcare, catalog number: 17075605 )–Store at 4 °C
- GST-PreScission (10 mg/ml)
Note: We prepare GST tagged PreScission by ourselves. 1 µl of our protease cleaves approximately 1 mg of GST-ARL3, a test protein. Alternatively, you can purchase the protease from GE Healthcare, catalog number: 27084301 . Store at -80 °C. - Bradford Reagent Concentrate (Bio-Rad Laboratories, catalog number: 5000006 )–Store at 4 °C
- Liquid nitrogen
- BSA standard, 2 mg/ml (Thermo Fisher Scientific, catalog number: 23209 )–Store at 4 °C
- TritonX-100 (Acros Organics, catalog number: 215682500 )
- Milli-Q water
- Protease inhibitor tablet (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: A32965 )–Store at 4 °C
- Glycerol (Sigma-Aldrich, catalog number: G5516 )
- MANT-GDP triethylammonium salt solution (Sigma-Aldrich, catalog number: 69244 )–Store at -20 °C protected from light
- Trizma base (Sigma-Aldrich, catalog number: T6066 )
- Concentrated HCl (Aqua Solutions, catalog number: H2505-500ML )
- Glacial acetic acid (Fisher Scientific, catalog number: A490-212 )
- Methanol (Fisher Scientific, catalog number: A412-4 )
- HEPES (Sigma-Aldrich, catalog number: H3375 )
- Sodium hydroxide (Fisher Scientific, catalog number: BP359-500 )
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S3014-500G )
- Magnesium chloride hexahydrate (MgCl2·6H2O) (Sigma-Aldrich, catalog number: M2670 )
- Dithiothreitol (DTT) (Promega, catalog number: V3155 )
- Ethylenediaminetetraacetic acid (EDTA) (Fisher Scientific, catalog number: S311 )
- LB broth (Fisher Scientific, catalog number: BP9723 )
- LB agar (Thermo Fisher Scientific, InvitrogenTM, catalog number: 22700 )
- Terrific broth (Fisher Scientific, catalog number: BP24682 )
- Carbenicillin (Sigma-Aldrich, catalog number: C3416 )
- Ampicillin (Fisher Scientific, catalog number: BP1760 )
- Chloramphenicol (Sigma-Aldrich, catalog number: C0378 )
- IPTG (Thermo Fisher Scientific, InvitrogenTM, catalog number: 15529019 )–Store at -20 °C
- LB media (see Recipes)
- TB media (see Recipes)
- 100 mg/ml ampicillin (see Recipes)
- 50 mg/ml carbenicillin (see Recipes)
- 34 mg/ml chloramphenicol (see Recipes)
- LB agar plate containing 50 µg/ml carbenicillin and 34 µg/ml chloramphenicol (see Recipes)
- 1 M IPTG (see Recipes)
- 1 M Tris-HCl (pH 7.5) (see Recipes)
- 1 M HEPES-NaOH (pH 7.5) (see Recipes)
- 5 M NaCl (see Recipes)
- 1 M MgCl2 (see Recipes)
- 1 M DTT (see Recipes)
- 0.25 M EDTA (pH 8) (see Recipes)
- 10 M NaOH (see Recipes)
- Coomassie Brilliant Blue staining solution (see Recipes)
- Coomassie Brilliant Blue destaining solution (see Recipes)
- Lysis buffer (see Recipes)
- PreScission cleavage buffer (see Recipes)
- Storage buffer (see Recipes)
- Low magnesium buffer (see Recipes)
- 2x MANT-GDP loading buffer (see Recipes)
- Nucleotide exchange buffer (see Recipes)
Note: Unless otherwise noted, materials are stored at room temperature. Frozen proteins and nucleotides can be maintained for at least a year if pure.
Equipment
- Milli-Q generator (Merck, model: Milli-Q® Advantage A10, catalog number: Z00Q0V0WW )
- 250 ml culture flask (Corning, PYREX®, catalog number: 4980-250 )
- 2,800 ml culture flask (Corning, PYREX®, catalog number: 4424-2XL )
- Incubator shaker (Eppendorf, New BrunswickTM, model: Excella® E25 , catalog number: M1353-0002)
- Refrigerated centrifuge (Eppendorf, model: 5424 R )
- 500 ml centrifuge bottle (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 3141-0500 )
- JA-10 rotor (Beckman Coulter, model: JA-10 , catalog number: 369687)
- Avanti J-25I refrigerated centrifuge (Beckman Coulter, model: Avanti J-25I , catalog number: 363106)
- JA-17 rotor (Beckman Coulter, model: JA-17 , catalog number: 369691)
- Allegra X-15R refrigerated centrifuge (Beckman Coulter, model: Allegra® X-15R , catalog number: 392932)
- BioSpectrometer (Eppendorf, catalog number: 6136000010 )
- Thermomixer (Fisher Scientific, catalog number: 05-412-401 )
Note: This equipment has been discontinued. Thermomixer C (Fisher Scientific, catalog number: 05-412-503; Manufacturer: Eppendorf, catalog number: 5382000023 ) is available as an alternative. Any type of temperature-controlled mixer with cooling function should work for this experiment. - TECAN Infinite M1000 Microplate reader (TECAN, part number: 30034301 )
Note: There are a number of plates or cuvette-based fluorimeters that can be adapted to this procedure. Those include FluoroMax-4 (Horiba) (Price et al., 2012), Synergy H4 Hybrid Microplate reader (Bio Tek) (Ivanova et al., 2017), and Envision (Perkin-Elmer) (Maurer et al., 2012). - Branson Digital Sonifier (Branson)
Note: This equipment has been discontinued. Branson Ultrasonics Sonifier SFX250/SFX550 (Fisher Scientific, catalog number: 15-345-141; Manufacturer: EMERSON, Branson, catalog number: 101063969R ) is available as an alternative. - 360° vertical rotator (Grant Instruments, model: PTR-30 )
Note: This equipment has been discontinued. Grant bio 360° vertical rotator PTR-35 (Grant Instruments, model: PTR-35 ) is available as an alternative. - Lab stand (Humboldt, catalog number: H-21207 )
- Vortex mixer (Scientific Industries, model: Vortex-Genie 2 , catalog number: SI-0236)
- Multichannel electronic pipette (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 2069MTRX )
Note: This equipment has been discontinued. A newer model (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 4671020BT ) is available as an alternative. - Autoclave
Software
- Microsoft Excel (Microsoft)
- GraphPad Prism 7 software (GraphPad Software)
Procedure
- Expression and purification of recombinant GTPases from E. coli
Recombinant GTPases can be produced and purified from Escherichia coli, since the nucleotide exchange assay does not generally require post-translational modifications of the GTPase. Experimental conditions, such as temperature and culture time for inducing protein expression and Isopropyl β-D-1-thiogalactopyranoside (IPTG) concentration, need to be determined for each GTPase to obtain the maximal yield of soluble recombinant proteins. Conditions can be optimized in small scale (we typically perform a pilot experiment with 2 ml of LB media). We typically induce the protein expression with 0.1 mM IPTG at 18 °C overnight. A typical yield from 1 L culture is 10 mg of protein.- Transform Rosetta2 bacteria cells with a vector containing gene of interest. Plate the cells onto an LB agar plate containing 50 µg/ml carbenicillin (or 100 µg/ml ampicillin) and 34 µg/ml chloramphenicol. Incubate the plate at 37 °C overnight.
Note: We typically use Gateway-cloning compatible pGEX6p vector (Wright et al., 2011), which produces an N-terminally Glutathione S-transferase (GST) tagged protein with PreScission protease recognition site between GST and protein of interest. The vectors containing CDC42, a GEF-dependent GTPase, and IIGP1 and CrRabl2, GEF-independent GTPases, work as positive controls for the experiment and are provided by the authors upon request. Chloramphenicol is added into an agar plate because tRNAs for rare codons are maintained by the chloramphenicol-resistant plasmid in the Rossetta2 bacteria cells. Other antibiotics might have to be used depending on bacteria cells used. - Inoculate 50 ml of LB media containing 100 µg/ml ampicillin and 34 µg/ml chloramphenicol with a single colony of the transformed cells in a 250 ml flask. Grow the starter culture at 37 °C at 250 rpm overnight.
- Pre-warm 1 L of autoclaved TB media without antibiotics in 2,800 ml flask at 37 °C overnight in an incubator shaker, before starting a large-scale culture.
- Inoculate 1 L of pre-warmed TB media with 25 ml of the starter culture. Shake the flask at 37 °C at 250 rpm until OD600 reaches to 0.6-0.7. It takes approximately 2 h.
- As soon as OD600 reaches to 0.6-0.7, set the flask on ice and chill culture for 15 min.
- Add 100 µl of 1 M IPTG (the final concentration is 0.1 mM) to induce protein expression.
- Shake the flask at 18 °C at 250 rpm in an incubator shaker overnight.
- Place the flask on ice before harvesting the bacteria cells.
- To confirm the expression level and the solubility of the protein, transfer 1 ml of the bacterial suspension into a microcentrifuge tube. Collect the cell pellet by centrifugation at 8,000 rpm (6,010 x g) at 4 °C for 1 min using a refrigerated centrifuge. Re-suspend the cell pellet in 300 µl of pre-chilled lysis buffer and lyse the cells by sonication for 1 min on ice (0.6 sec/0.4 sec on/off cycle at 30% amplitude), followed by centrifugation at 15,000 rpm (21,130 x g) at 4 °C for 10 min. Transfer the supernatant into a microcentrifuge tube and add 100 µl of 4x LDS sample buffer and 10 µl of 2-mercaptoethanol to prepare the soluble fraction. To prepare the insoluble fraction, add 290 µl of the lysis buffer, 100 µl of 4x LDS sample buffer and 10 µl of 2-mercaptoethanol to the pellet. Incubate the samples at 70 °C for 10 min and run them on Nu-PAGE gel at 200 V until the dye front reaches to the bottom of the gel. Stain the gel in Coomassie Brilliant Blue staining solution for 1 h at room temperature with gentle agitation, followed by destaining in Coomassie Brilliant Blue destaining solution overnight at room temperature with gentle agitation. Your protein of interest should appear in the soluble fraction.
Note: Classical Laemmli sodium dodecyl sulfate (SDS) sample buffer/SDS-polyacrylamide gel electrophoresis (SDS-PAGE) can be used instead of 4x LDS sample buffer/Nu-PAGE system. - Harvest bacteria by centrifugation in four 500 ml centrifuge bottles at 6,000 rpm (6,371 x g) in JA-10 rotor using Avanti J-25I refrigerated centrifuge at 4 °C for 10 min.
Note: Put a maximum of 300 ml in each tube, to prevent spilling during centrifugation. - Discard supernatant and freeze the cell pellet at -20 °C until you confirm the expression and the solubility of the protein in Step A9.
Note: Bacterial pellet can be stored at -20 °C for at least one week. Alternatively, you may keep the bacteria pellet on ice for same day confirmation of the expression/solubility then proceed immediately to the next step. - Re-suspend the 1 L cell pellet in 30 ml of pre-chilled lysis buffer and transfer suspension to a 50 ml conical tube.
- Sonicate the cells on ice until the suspension becomes watery (approximately 2 min; 0.6 sec/0.4 sec on/off cycle at 80% amplitude).
- Transfer the lysate into an oak ridge tube and clarify the lysate by centrifugation at 15,000 rpm (30,996 x g) at 4 °C in the JA-17 rotor using the Avanti J-25I refrigerated centrifuge for 30 min.
- During centrifugation, wash 2 ml bed volume of glutathione Sepharose 4B media twice in 20 ml of ice-cold lysis buffer in a 50 ml conical tube. To wash the beads, pellet the beads by centrifugation at 1,466 rpm (500 x g) at 4 °C for 5 min using the Allegra X-15R refrigerated centrifuge and aspirate the supernatant. Add 20 ml of the lysis buffer and mix by inverting the tube. All washes in the following steps will be done in the same manner.
Note: 1 ml bed volume of beads bind approximately 5 mg of GST. - Add clarified lysate into the washed glutathione Sepharose beads and rotate the 50 ml conical tube on a 360° vertical rotator at 4 °C for 2 h.
- Wash the beads three times with 40 ml (20 resin volumes) of lysis buffer without protease inhibitor.
- Wash the beads twice with 40 ml (20 resin volumes) of PreScission cleavage buffer.
- Add 4 ml (2 resin volumes) of PreScission cleavage buffer and transfer them into a 15 ml conical tube.
- Add 10 µl of 10 mg/ml GST-PreScission to elute the protein. Rotate the 15 ml conical tube on a 360° vertical rotator at 4 °C for 16 h.
Note: 10 µl of 10 mg/ml GST-PreScission is added, because we expect the final yield 10 mg and 1 µl of our GST-PreScission cleaves 1 mg of GST tagged protein. The amount of GST-PreScission that should be added depends on the expression level of the protein and/or how much protein is desired. The amount of proteins contained in the lysate can be estimated based on the amount of protein in the test sample taken at Step A9. - Pellet the beads by centrifugation at 1,466 rpm (500 x g) at 4 °C for 5 min using the Allegra X-15R refrigerated centrifuge and collect supernatant into a 15 ml conical tube.
- Wash the beads with 2 ml (one resin volume) of PreScission cleavage buffer and collect supernatant following centrifugation at 1,466 rpm (500 x g) at 4 °C for 5 min. Repeat wash one more time and combine with the previous supernatant.
- Measure the concentration of the protein using the Bradford assay (see Step B8 for detail).
Note: If the concentration is less than 100 µM, concentrate the eluted protein using the Amicon ultra concentrator (Merck, catalog number: UFC901024). - Check the quality of the protein by Nu-PAGE and Coomassie Brilliant Blue staining.
Note: Classical Laemmli SDS sample buffer/SDS-PAGE can be used instead of 4x LDS sample buffer/Nu-PAGE system. - Dialyze the protein using dialysis tubing in at least 100-fold protein volumes of storage buffer at 4 °C overnight.
- Prepare 200 µl aliquots of the purified protein (the concentration should be 100 µM or greater) and snap freeze the aliquots in liquid nitrogen. Store at -80 °C.
- Transform Rosetta2 bacteria cells with a vector containing gene of interest. Plate the cells onto an LB agar plate containing 50 µg/ml carbenicillin (or 100 µg/ml ampicillin) and 34 µg/ml chloramphenicol. Incubate the plate at 37 °C overnight.
- Buffer exchange to low magnesium buffer
Loading MANT-GDP on GTPase is performed in the buffer containing 0.5 mM MgCl2 in the presence of 5 mM EDTA. Since our storage buffer contains 5 mM MgCl2 (see Recipes) for the stability of GTPase, we first exchange the storage buffer for the low magnesium buffer (see Recipes) using a gel filtration column.- Thaw 200 µl of purified GTPase (100 µM or greater) on ice.
Note: 100 µM of GTPase is generally a good starting point. - Clamp a NAP-5 column onto a lab stand.
- Remove the top and bottom cap from the NAP-5 column to allow the excess manufacturer storage buffer to flow through.
- Equilibrate the NAP-5 gel filtration column with low magnesium buffer by three complete refills of the column. The volume of buffer used for equilibration is approximately 10 ml in total.
- Apply 200 µl of purified GTPase onto the center of the equilibrated NAP-5 column and allow the sample to enter the gel bed completely by gravity flow.
- Apply 500 µl of low magnesium buffer to the column and collect the flow through in a microcentrifuge tube.
Note: This fraction should not contain any proteins. - Apply 1 ml of low magnesium buffer to elute the protein. Collect eluate 100 µl at a time in microcentrifuge tubes.
Note: Most of the protein should be eluted in the first 4 fractions. - Measure the concentration of each fraction including the flow through by the Bradford assay.
- Briefly, dilute one part of Bradford Reagent Concentrate with 4 parts of Milli-Q water, and add 1 ml of diluted Bradford reagent to disposable cuvettes. Add 10 µl of Milli-Q water or BSA standards (at 0.1, 0.2, 0.4, and 0.8 mg/ml), or 1-2 µl of each fraction of buffer exchanged GTPase into each cuvette. Mix the cuvettes by vortexing and incubate at room temperature for 5 min. Measure the concentration of each fraction on the BioSpectrometer.
- Calculate the molar concentration from the measured concentration. Use the fraction containing the highest concentration of protein for further experiment. The protein concentration following buffer exchange is generally half as much as the starting concentration.
- Briefly, dilute one part of Bradford Reagent Concentrate with 4 parts of Milli-Q water, and add 1 ml of diluted Bradford reagent to disposable cuvettes. Add 10 µl of Milli-Q water or BSA standards (at 0.1, 0.2, 0.4, and 0.8 mg/ml), or 1-2 µl of each fraction of buffer exchanged GTPase into each cuvette. Mix the cuvettes by vortexing and incubate at room temperature for 5 min. Measure the concentration of each fraction on the BioSpectrometer.
- Thaw 200 µl of purified GTPase (100 µM or greater) on ice.
- Loading MANT-GDP on GTPase
- Mix 100 µl of buffer exchanged GTPase with 100 µl of 2x MANT-GDP loading buffer (see Recipes) in microcentrifuge tubes. The final reaction contains 20 mM HEPES-NaOH (pH 7.5), 50 mM NaCl, 0.5 mM MgCl2, 5 mM EDTA, 1 mM dithiothreitol (DTT), GTPase, and 20-fold molar excess of MANT-GDP.
Note: The concentration of MANT-GDP depends on the protein concentration and must be calculated in every experiment. - Incubate the reaction at 20 °C in a thermomixer without shaking for 90 min. Protect the reaction from the light with aluminum foil.
- Stop the reaction by adding 2 µl of 1 M MgCl2 (the final concentration is 10 mM). Mix the reaction by inverting the tubes, followed by centrifugation then incubate the tubes at 20 °C for 30 min.
- During the incubation, equilibrate a NAP-5 gel filtration column with three complete refills of nucleotide exchange buffer (see Recipes). The volume of buffer used for equilibration is approximately 10 ml in total.
- To remove unbound MANT-GDP, apply the entire volume (200 µl) of MANT-GDP loaded GTPase onto the NAP-5 column. Allow the sample to enter the gel bed completely by gravity flow.
- Apply 500 µl of nucleotide exchange buffer and collect flow through.
Note: This fraction should not contain any proteins. - Apply 1 ml of nucleotide exchange buffer to elute the protein. Collect 100 µl at a time in microcentrifuge tubes. Keep MANT-GDP loaded GTPases on ice, protected from light (by aluminum foil).
Note: Most of the protein should be eluted in the first 4 fractions. - Measure the concentration of each fraction by the Bradford assay as described above.
- Mix 100 µl of buffer exchanged GTPase with 100 µl of 2x MANT-GDP loading buffer (see Recipes) in microcentrifuge tubes. The final reaction contains 20 mM HEPES-NaOH (pH 7.5), 50 mM NaCl, 0.5 mM MgCl2, 5 mM EDTA, 1 mM dithiothreitol (DTT), GTPase, and 20-fold molar excess of MANT-GDP.
- Measure the loading efficiency on the fluorimeter
- Prepare MANT-GDP standards by making two-fold serial dilutions of free MANT-GDP from 40 µM to 0.15625 µM in nucleotide exchange buffer.
Note: The minimal concentration of free MANT-GDP that can be detected on TECAN Infinite M1000 Microplate reader is around 0.1 µM. - Pipette 15 µl of nucleotide exchange buffer, free MANT-GDP standards, or MANT-GDP loaded GTPase into 384-well microplate.
Note: This measurement also works with 96-well format. If you use 96-well format, pipette 50 µl of each sample into a 96-well microplate (Corning). - Read the fluorescent signal every 15 sec, 10 times in total, at room temperature (25 °C). The parameters used for the detection are listed (Table 1).
Table 1. The parameters used for the detection of MANT fluorescence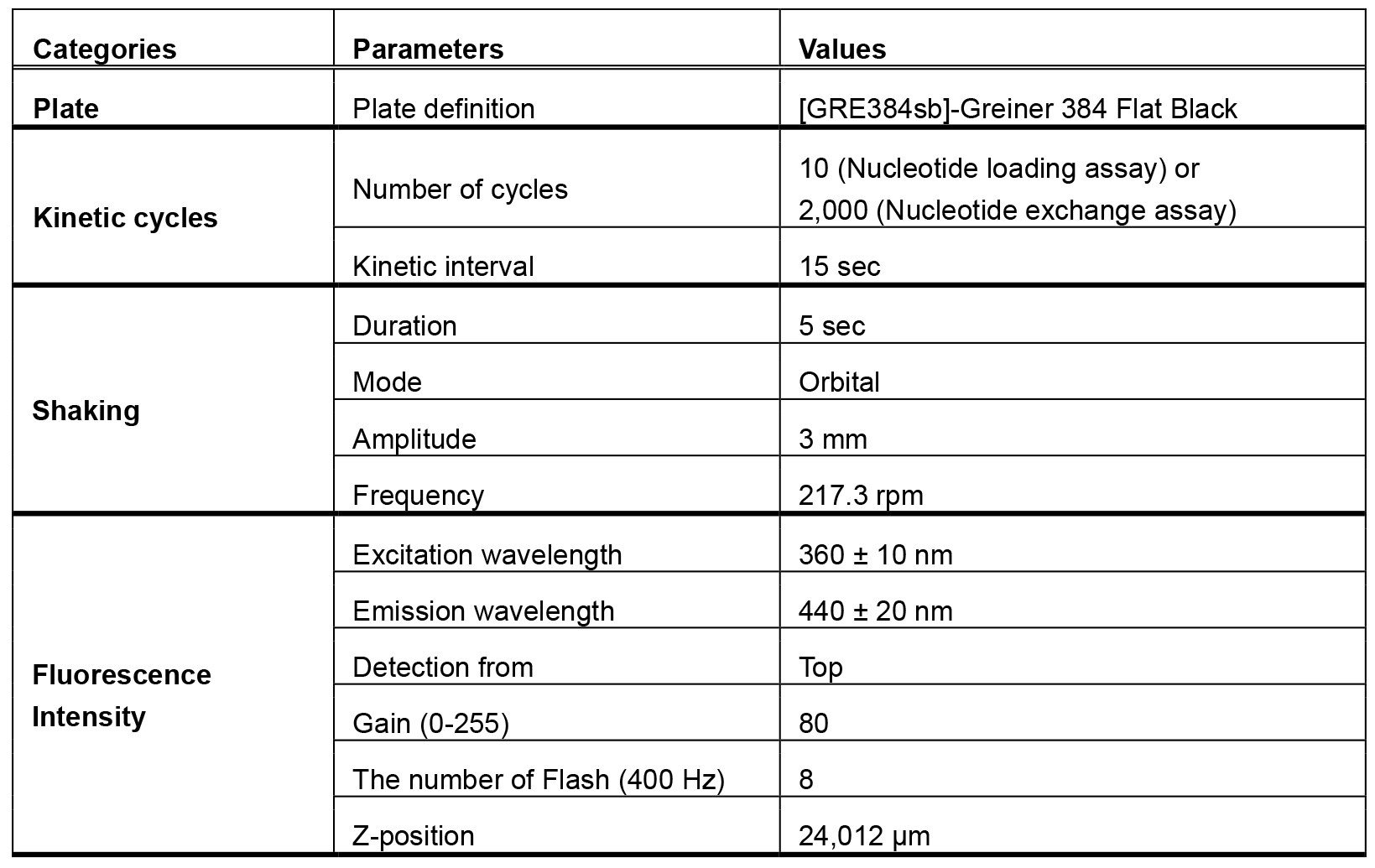
- To calculate the loading efficiency, average the fluorescence intensity for each sample from all 10 time points, then calculate the normalized average by subtracting the background fluorescence value measured for the buffer-only control.
- Plot the free MANT-GDP concentration vs. measured fluorescence and fit the standard curve to a linear function using Microsoft Excel. Use the standard curve to determine the concentration of the MANT-GDP-loaded GTPase from its measured fluorescence; since MANT-GDP signal typically increases approximately two-fold upon binding to protein, first divide the fluorescent signal by two then estimate the concentration of loaded GTPase using the standard curve function. A representative example of this data (Table 2) and the standard curve of free MANT-GDP created from the data (Figure 1) are shown.
Table 2. An example of the data obtained from loading of MANT-GDP on GTPase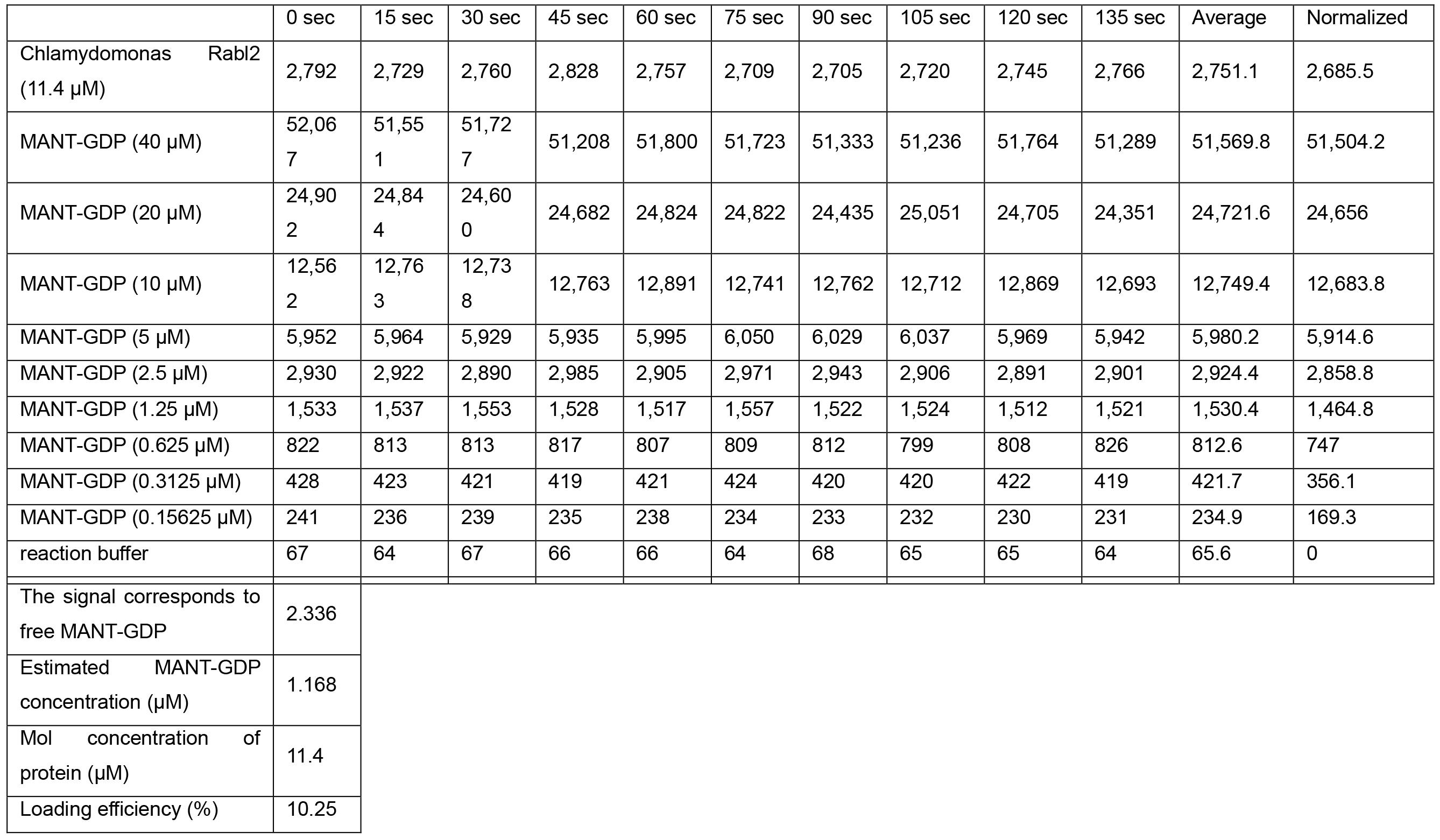
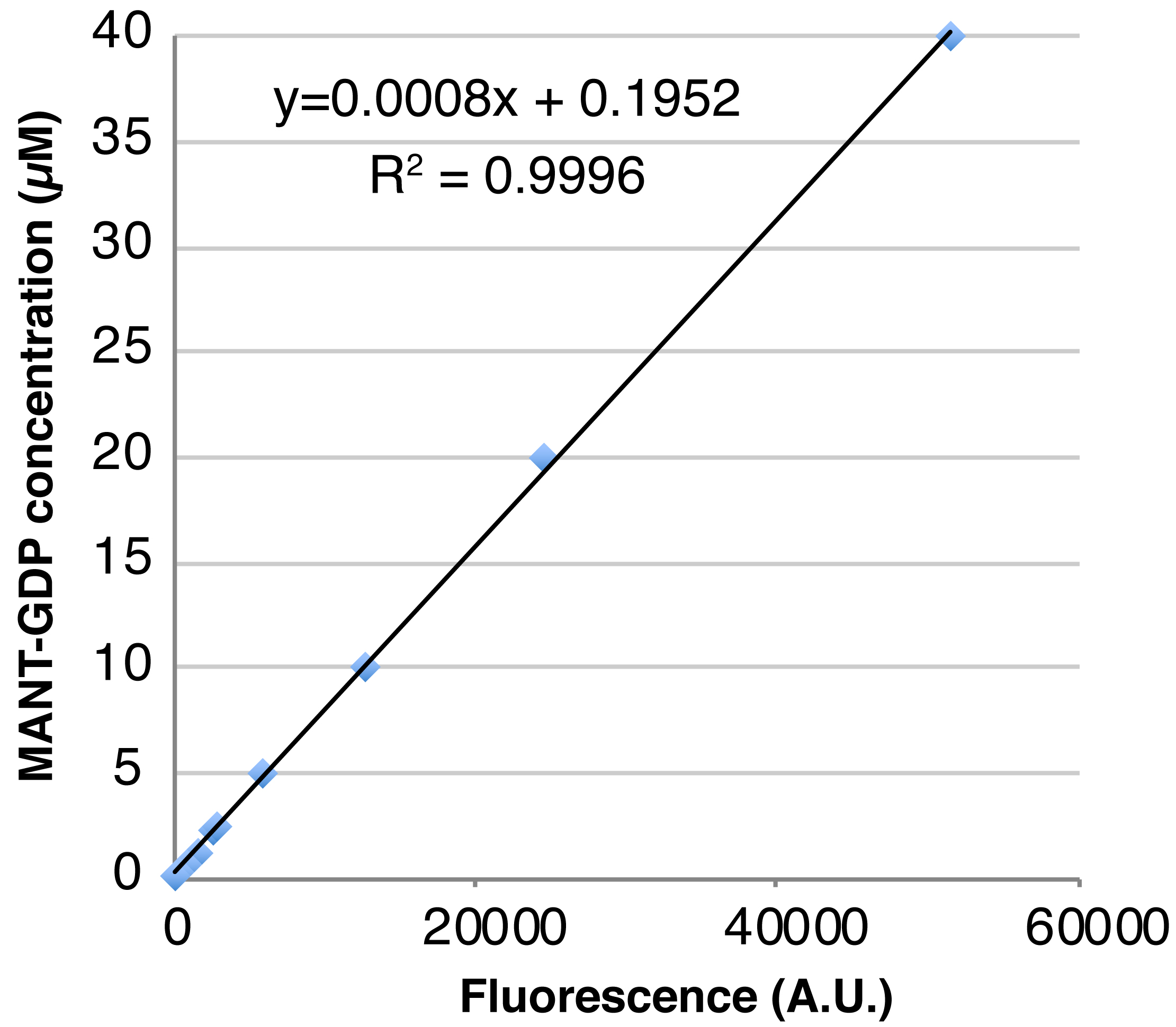
Figure 1. A standard curve of free MANT-GDP created using the data shown in Table 2 - Prepare MANT-GDP standards by making two-fold serial dilutions of free MANT-GDP from 40 µM to 0.15625 µM in nucleotide exchange buffer.
- Nucleotide exchange assay
- Add 14 µl of nucleotide exchange buffer or MANT-GDP loaded GTPase into an appropriate number (the number of conditions x 4 technical replicates) of wells of a 384-well microplate.
Note: The concentration of protein that is added to the well depends on the loading efficiency calculated above, since the starting fluorescence intensity of the sample should be high enough to detect dynamic changes in the fluorescence. If the loading efficiency is high (more than 50%), 1 µM or less GTPase can be used. If the loading efficiency is low (less than 10%), the highest concentration of GTPase should be added. - Before measuring the fluorescence, prepare a working solution of GppNHp, which has 15 x 100 times higher concentration than that of the GTPase, with nucleotide exchange buffer. If you use 1 µM of GTPase, prepare 1.5 mM GppNHp.
- Read the fluorescence for 150 sec at room temperature (25 °C) and confirm the fluorescent signal is stable.
Note: The same detection parameters used in the Step D3 (see Table 1) can be used in the nucleotide exchange assay. - Press the ‘Pause’ button on the display to eject the plate, and then add 1 µl of nucleotide exchange buffer or GppNHp working solution (the final concentration is 100 times higher than that of GTPase) to respective wells. If GEF mediated nucleotide exchange is to be measured, add purified GEF in addition to GppNHp.
Note: A multichannel pipet should be used to simultaneously add buffer or GppNHp to start all reactions simultaneously. To ensure rapid addition of the buffer or GppNHp, we pre-fill a multichannel pipette with 1 µl of the buffer/GppNHp solution from another 384-well plate before ejecting the assay plate, and then pipette reagent into the assay plate immediately after ejection of the plate. To avoid making any bubble, we set the fill volume to 1.5 µl and the dispense volume to 1 µl. - Press the ‘Continue’ button to insert the plate and continue reading fluorescence every 15 sec at room temperature (25 °C) for at least 30 min.
- Add 14 µl of nucleotide exchange buffer or MANT-GDP loaded GTPase into an appropriate number (the number of conditions x 4 technical replicates) of wells of a 384-well microplate.
Data analysis
- Export the fluorimeter data to a Microsoft Excel spreadsheet.
- Subtract the background intensity of the nucleotide exchange buffer from each fluorescence intensity.
- Set the fluorescence intensity at the first time point after the addition of either buffer or GppNHp to 1 and calculate the relative fluorescence of each of the later time points.
- A pseudo first order rate constant (observed rate constant or Kobs) can be calculated from the data, since the reaction contains a large excess of GppNHp over GTPase (100 times or more).
Because the fluorescence of MANT-bound GTPase decreases exponentially with time, software programs that are capable of fitting data with nonlinear regression can be used for calculating Kobs. Those programs include, but are not limited to, GraphPad Prism, KaleidaGraph and Grafit. We describe here how to do the analysis by using GraphPad Prism 7 as an example.- Open GraphPad Prism 7 and create a new XY table.
- Enter the values into each column, click the ‘Analyze’ button and choose Nonlinear regression from the list of XY analyses.
- Select ‘Dissociation-One phase exponential decay’ to obtain the Kobs value.
- Open GraphPad Prism 7 and create a new XY table.
Notes
In our hand, the loading efficiency of GTPases with low guanine nucleotide affinity was much lower than that of typical small GTPases, which have a high affinity for guanine nucleotides. Presumably that reflects the higher dynamics of the low affinity GTPase. In the former case, the fluorescence signal is largely affected by dilution of the protein, probably because dilution itself changes the equilibrium.
Recipes
- LB media (1 L)
Dissolve 25 g of LB broth in 900 ml of Milli-Q water
Adjust pH to 7.2 with 10 M NaOH and bring volume with Milli-Q water to 1 L
Sterilize by autoclave and store at room temperature - TB media (1 L)
Dissolve 50.8 g of terrific broth in 900 ml of Milli-Q water and bring volume with Milli-Q water to 1 L
Transfer into a 2.8 L flask and rotate the flask at 250 rpm at 28 °C in an incubator shaker until the powder is completely dissolved
Note: It may take less than 30 min.
Sterilize by autoclave - 100 mg/ml ampicillin (10 ml)
Dissolve 1 g of ampicillin in 8 ml of Milli-Q water and bring volume with Milli-Q water to 10 ml
Sterilize with a 0.22 µm syringe filter
Dispense into 1 ml aliquots, and store at -20 °C - 50 mg/ml carbenicillin (10 ml)
Dissolve 500 mg of carbenicillin in 8 ml of Milli-Q water and bring volume with Milli-Q water to 10 ml
Sterilize with a 0.22 µm syringe filter
Dispense into 1 ml aliquots
Store at -20 °C - 34 mg/ml chloramphenicol (10 ml)
Dissolve 340 mg of chloramphenicol in 8 ml of 100% ethanol and bring volume with 100% ethanol to 10 ml
Sterilize with a 0.22 µm syringe filter
Dispense into 1 ml aliquots
Store at -20 °C - LB agar plate containing 50 µg/ml carbenicillin and 34 µg/ml chloramphenicol
Dissolve 32 g of LB agar in 900 ml of Milli-Q water and bring volume with Milli-Q water to 1 L
Sterilize by autoclave and cool down to 50 °C
Add 1 ml of 50 mg/ml carbenicillin and 1 ml of 34 mg/ml chloramphenicol
Pour into plates and store at 4 °C - 1 M IPTG (10 ml)
Dissolve 2.383 g of IPTG in 8 ml of Milli-Q water and bring volume with Milli-Q water to 10 ml
Sterilize with a 0.22 µm syringe filter
Dispense into 1 ml aliquots
Store at -20 °C - 1 M Tris-HCl (pH 7.5, 1 L)
Dissolve Trizma base in 800 ml Milli-Q water
Adjust pH with concentrated HCl to 7.5 and bring volume with Milli-Q water to 1 L
Autoclave or filter to sterilize - 1 M HEPES-NaOH (pH 7.5, 1 L)
Dissolve 238.30 g of HEPES base in 800 ml of Milli-Q water
Adjust pH to 7.5 with 10 M NaOH and bring volume with Milli-Q water to 1 L
Filter sterilize through a 0.2 µm disposable bottle top filter
Store at 4 °C - 5 M NaCl (1 L)
Dissolve 292.2 g of NaCl in 700 ml of Milli-Q water and bring volume with Milli-Q water to 1 L
Sterilize by autoclave and store at room temperature - 1 M MgCl2 (100 ml)
Dissolve 20.33 g of MgCl2·6H2O in 70 ml of Milli-Q water and bring volume with Milli-Q water to 100 ml
Sterilize by autoclave and store at room temperature - 1 M DTT (30 ml)
Dissolve 4.63 g of DTT in 20 ml of Milli-Q and bring volume with Milli-Q water to 30 ml
Dispense into 1 ml aliquots
Store at -20 °C - 0.25 M EDTA (pH 8.0, 1 L)
Dissolve 93.06 g of EDTA·2H2O in 800 ml of Milli-Q water
Adjust pH to 8.0 with NaOH
Note: EDTA will not dissolve unless pH is ~8.
Bring volume with Milli-Q water to 1 L
Sterilize by autoclave and store at room temperature - 10 M NaOH (500 ml)
Slowly add 200 g of sodium hydroxide into 400 ml of Milli-Q water while stirring continuously
Bring volume with Milli-Q water to 500 ml - Coomassie Brilliant Blue staining solution (1 L)
Dissolve 2.5 g Coomassie Brilliant Blue R-250 in 500 ml methanol
Add 400 ml of Milli-Q water and 100 ml of glacial acetic acid
Filter through a 0.2 µm disposable bottle top filter - Coomassie Brilliant Blue destaining solution (1 L)
Add 500 ml Milli-Q H2O and 400 ml methanol to a 1 L bottle
Add 100 ml glacial acetic acid slowly to solution - Lysis buffer (500 ml)
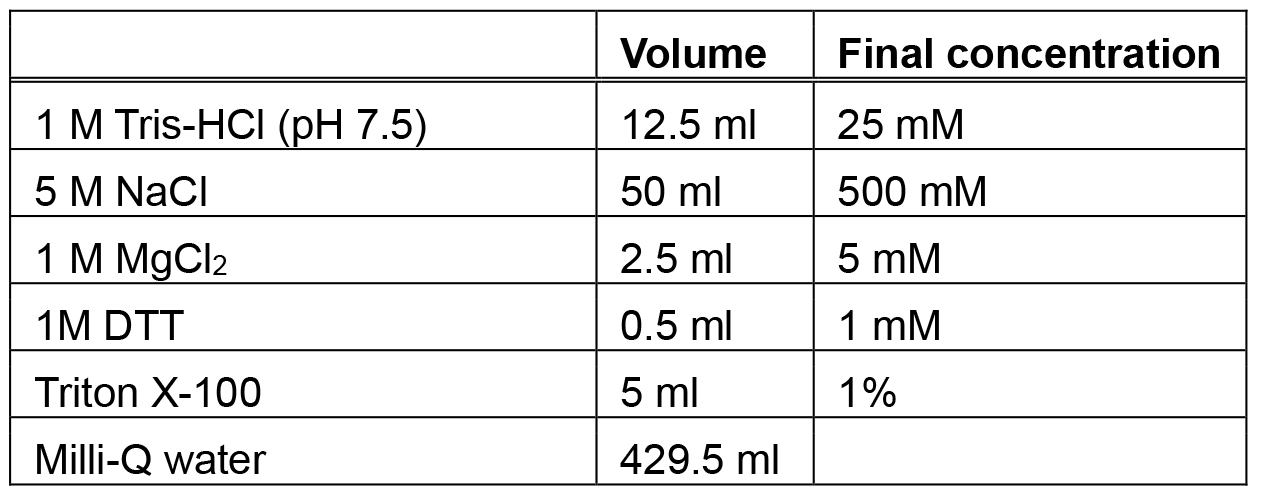
One tablet of a protease inhibitor is added in 50 ml of lysis buffer immediately before use
Make fresh each time - PreScission cleavage buffer
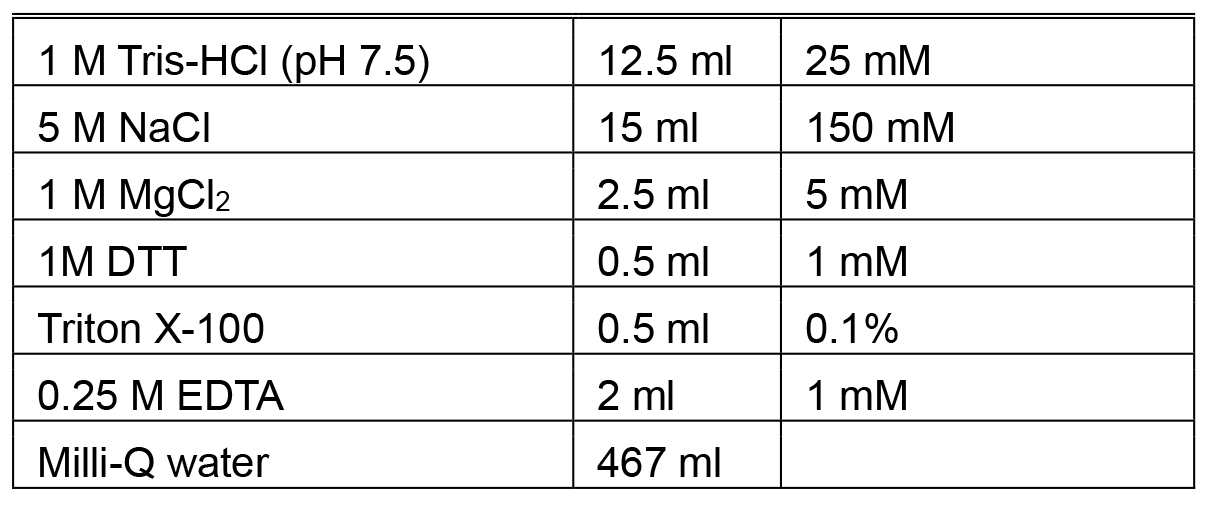
Make fresh each time - Storage buffer (500 ml)
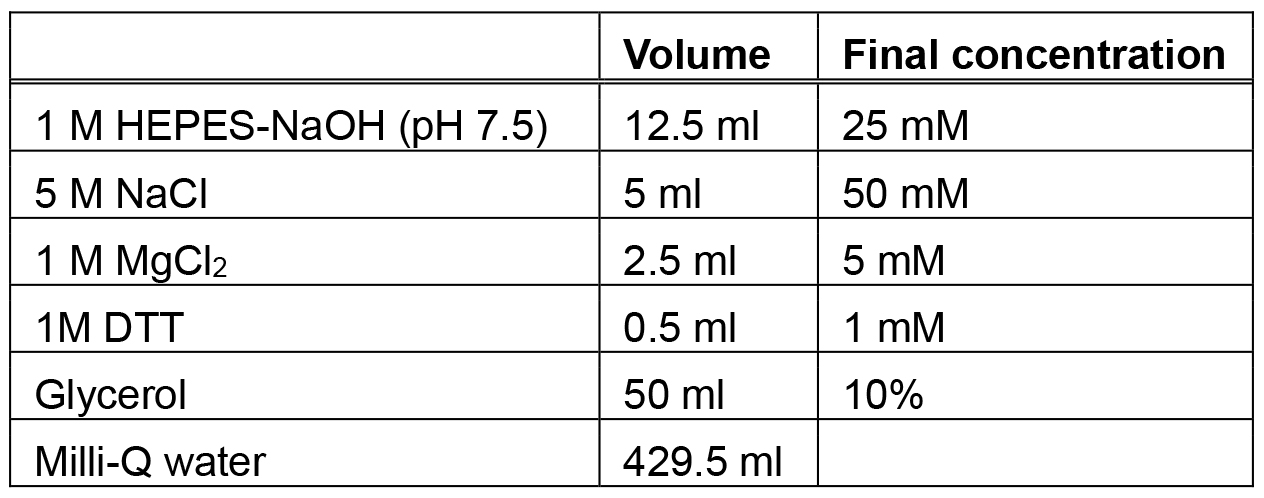
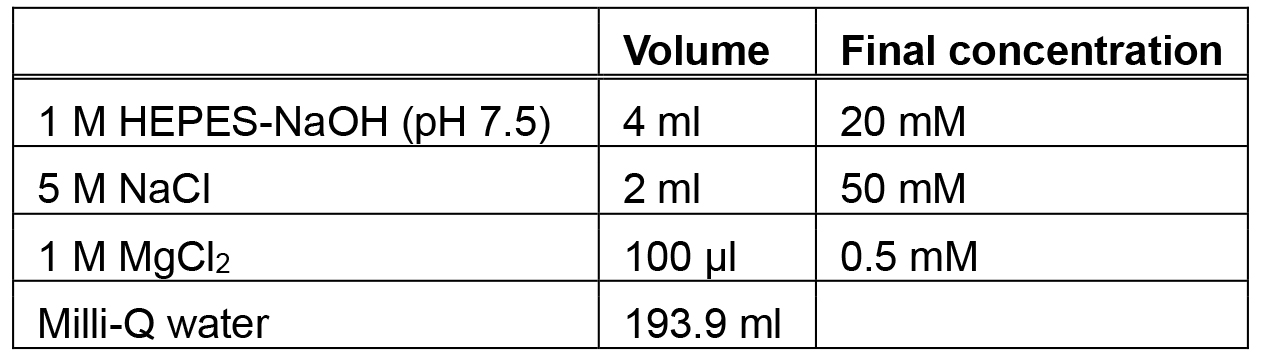
Make fresh each time - Low magnesium buffer (200 ml)
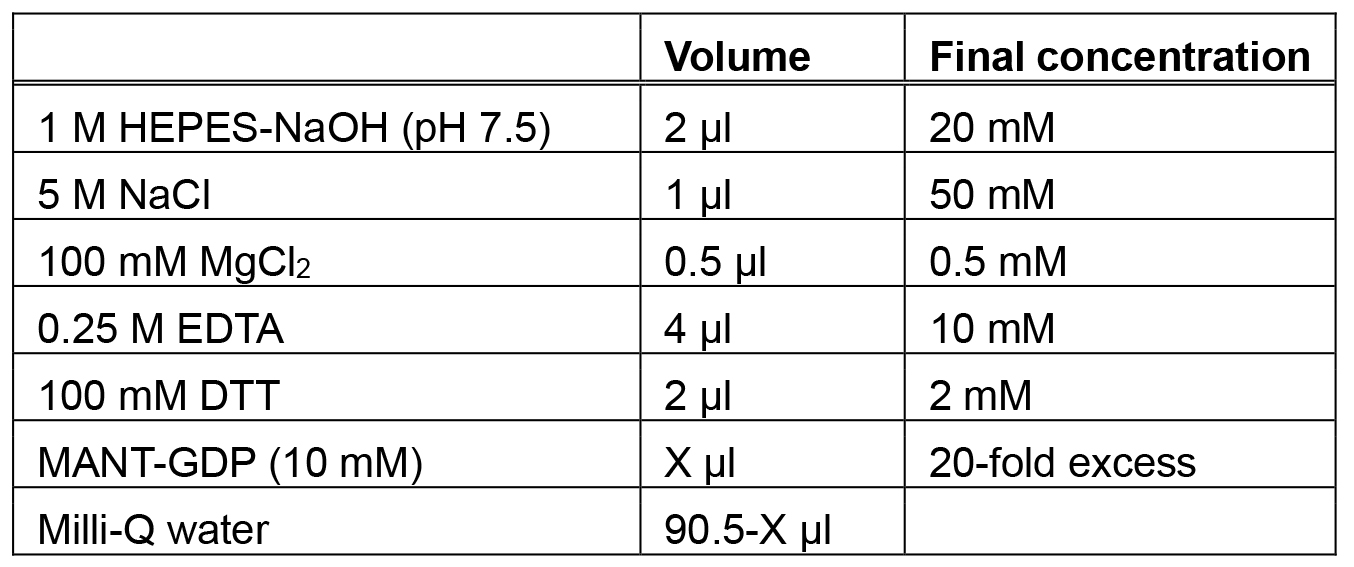
Store at room temperature - 2x MANT-GDP loading buffer (100 µl)
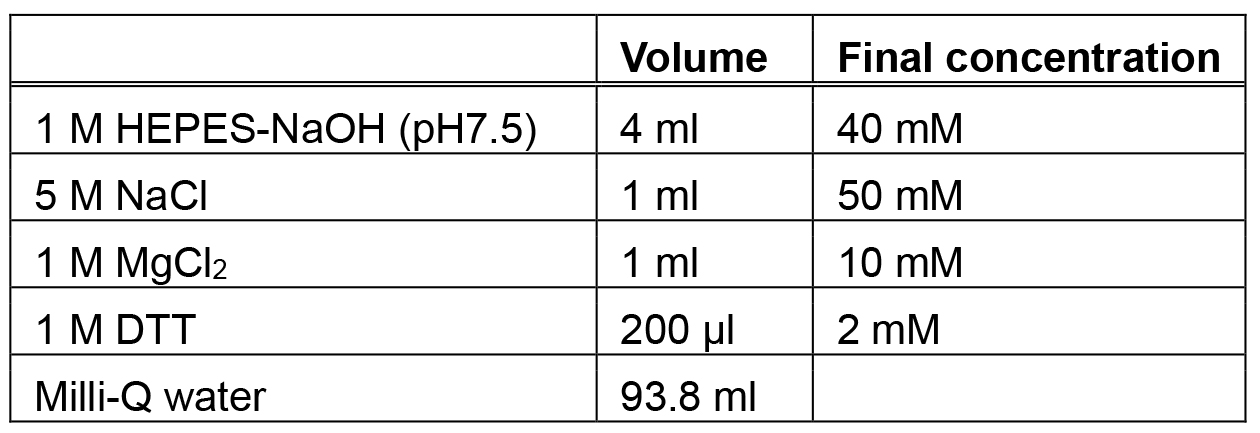
Make fresh each time. The concentration of MANT-GDP should be calculated from the GTPase concentration - Nucleotide exchange buffer (100 ml)
Make fresh each time
Acknowledgments
This protocol was adapted from the previous work (Kanie et al., 2017). The authors would like to thank Keene Abbott and Henrietta Bennett for careful reading of the manuscript. This work was support by funds from the Baxter Laboratory for Stem Cell Research, the Stanford Department of Research, the Stanford Cancer Center, NIH grants R01GM114276 and R01GM121565 to PKJ, and postdoctoral support from the Uehara memorial foundation and Human Frontier Science Program to TK. The authors declare no conflict of interest.
References
- Bhogaraju, S., Taschner, M., Morawetz, M., Basquin, C. and Lorentzen, E. (2011). Crystal structure of the intraflagellar transport complex 25/27. EMBO J 30(10): 1907-1918.
- Bos, J. L., Rehmann, H. and Wittinghofer, A. (2007). GEFs and GAPs: critical elements in the control of small G proteins. Cell 129(5): 865-877.
- Eberth, A. and Ahmadian, M. R. (2009). In vitro GEF and GAP assays. Curr Protoc Cell Biol Chapter 14: Unit 14.9.
- Gasper, R., Meyer, S., Gotthardt, K., Sirajuddin, M. and Wittinghofer, A. (2009). It takes two to tango: regulation of G proteins by dimerization. Nat Rev Mol Cell Biol 10(6): 423-429.
- Hiratsuka, T. (1983). New ribose-modified fluorescent analogs of adenine and guanine nucleotides available as substrates for various enzymes. Biochim Biophys Acta 742(3): 496-508.
- Ivanova, A. A., Caspary, T., Seyfried, N. T., Duong, D. M., West, A. B., Liu, Z. and Kahn, R. A. (2017). Biochemical characterization of purified mammalian ARL13B protein indicates that it is an atypical GTPase and ARL3 guanine nucleotide exchange factor (GEF). J Biol Chem 292(26): 11091-11108.
- John, J., Sohmen, R., Feuerstein, J., Linke, R., Wittinghofer, A. and Goody, R. S. (1990). Kinetics of interaction of nucleotides with nucleotide-free H-ras p21. Biochemistry 29(25): 6058-6065.
- Kanie, T., Abbott, K. L., Mooney, N. A., Plowey, E. D., Demeter, J. and Jackson, P. K. (2017). The CEP19-RABL2 GTPase complex binds IFT-B to initiate intraflagellar transport at the ciliary base. Dev Cell 42(1): 22-36 e12.
- Maurer, T., Garrenton, L. S., Oh, A., Pitts, K., Anderson, D. J., Skelton, N. J., Fauber, B. P., Pan, B., Malek, S., Stokoe, D., Ludlam, M. J., Bowman, K. K., Wu, J., Giannetti, A. M., Starovasnik, M. A., Mellman, I., Jackson, P. K., Rudolph, J., Wang, W. and Fang, G. (2012). Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc Natl Acad Sci U S A 109(14): 5299-5304.
- Pai, E. F., Krengel, U., Petsko, G. A., Goody, R. S., Kabsch, W. and Wittinghofer, A. (1990). Refined crystal structure of the triphosphate conformation of H-ras p21 at 1.35 A resolution: implications for the mechanism of GTP hydrolysis. EMBO J 9(8): 2351-2359.
- Price, H. P., Hodgkinson, M. R., Wright, M. H., Tate, E. W., Smith, B. A., Carrington, M., Stark, M. and Smith, D. F. (2012). A role for the vesicle-associated tubulin binding protein ARL6 (BBS3) in flagellum extension in Trypanosoma brucei. Biochim Biophys Acta 1823(7): 1178-1191.
- Tong, L. A., de Vos, A. M., Milburn, M. V. and Kim, S. H. (1991). Crystal structures at 2.2 A resolution of the catalytic domains of normal ras protein and an oncogenic mutant complexed with GDP. J Mol Biol 217(3): 503-516.
- Vetter, I. R. and Wittinghofer, A. (2001). The guanine nucleotide-binding switch in three dimensions. Science 294(5545): 1299-1304.
- Wright, K.J., Baye, L.M., Olivier-Mason, A., Mukhopadhyay, S., Sang, L., Kwong, M., Wang, W., Pretorius, P.R., Sheffield, V.C., Sengupta, P., et al. (2011). An ARL3-UNC119-RP2 GTPase cycle targets myristoylated NPHP3 to the primary cilium. Genes Dev 25: 2347-2360.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Kanie, T. and Jackson, P. K. (2018). Guanine Nucleotide Exchange Assay Using Fluorescent MANT-GDP. Bio-protocol 8(7): e2795. DOI: 10.21769/BioProtoc.2795.
Category
Biochemistry > Protein > Fluorescence
Biochemistry > Protein > Activity
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.