- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Generating Loss-of-function iPSC Lines with Combined CRISPR Indel Formation and Reprogramming from Human Fibroblasts

Published: Vol 8, Iss 7, Apr 5, 2018 DOI: 10.21769/BioProtoc.2794 Views: 12596
Reviewed by: David CisnerosSara E HowdenGanesh Swaminathan
Original research article
The authors used this protocol in:

Aug 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
For both disease and basic science research, loss-of-function (LOF) mutations are vitally important. Herein, we provide a simple stream-lined protocol for generating LOF iPSC lines that circumvents the technical challenges of traditional gene-editing and cloning of established iPSC lines by combining the introduction of the CRISPR vector concurrently with episomal reprogramming plasmids into fibroblasts. Our experiments have produced nearly even numbers of all 3 genotypes in autosomal genes. In addition, we provide a detailed approach for maintaining and genotyping 96-well plates of iPSC clones.
Keywords: Induced pluripotent stem cellsBackground
CRISPR/Cas9 technology has allowed easy and specific targeting of a particular genomic location for gene-editing. Combining this technology with the disease modeling and regenerative medicine potential of induced pluripotent stem cells (iPSCs) will continue to have unprecedented impacts on biomedical research. However, adapting the CRISPR/Cas9 system to iPSCs has presented several challenges. The traditional approach for gene-editing in cell lines is to transfect the cells with a plasmid expressing the Cas9 protein and guide RNA (gRNA) after which single clones are generated and screened for the desired genetic alteration. Unfortunately, iPSCs are not amenable to single cell cloning. Several media supplements and cloning approaches have been developed to overcome this difficulty but are still fraught with either expensive equipment (low oxygen incubators), difficult technological steps (survival of FACS sorted single iPSCs), or labor intensive protocols (sub-cloning) (Forsyth et al., 2006; Miyaoka et al., 2014). Moreover, single cell passaging has been linked to increased genomic abnormalities in iPSCs (Bai et al., 2015). Fluorescent or antibiotic resistance genetic markers have been used to overcome issues with clonality and the overall low efficiency of gene-editing in these cells, but require homologous recombination of a large cassette via a targeting plasmid designed with long homology arms (400-800 bp) (Hendel et al., 2014). Designing these plasmids takes a great deal of time.
To overcome many of these obstacles, we utilized a combined approach of simultaneous reprogramming and CRISPR/Cas9 mutagenesis to generate both heterozygous and homozygous loss-of-function (LOF) iPSC lines. This combined approach was first presented by Howden and colleagues for homologous recombination gene-editing (Howden et al., 2015 and 2016), but we have expanded upon and further defined their original results for indel formation in a recent publication (Tidball et al., 2017). This procedure takes advantage of the cloning step inherent to iPSC reprogramming as well as the greater ease of transfection in fibroblast culture (Figure 1). The ability to efficiently generate a large number of clones with a similar proportion of all three genotypes (wild-type/wild-type, wild-type/indel, and indel/indel) will allow rapid development of LOF iPSC lines for disease modeling and basic research. 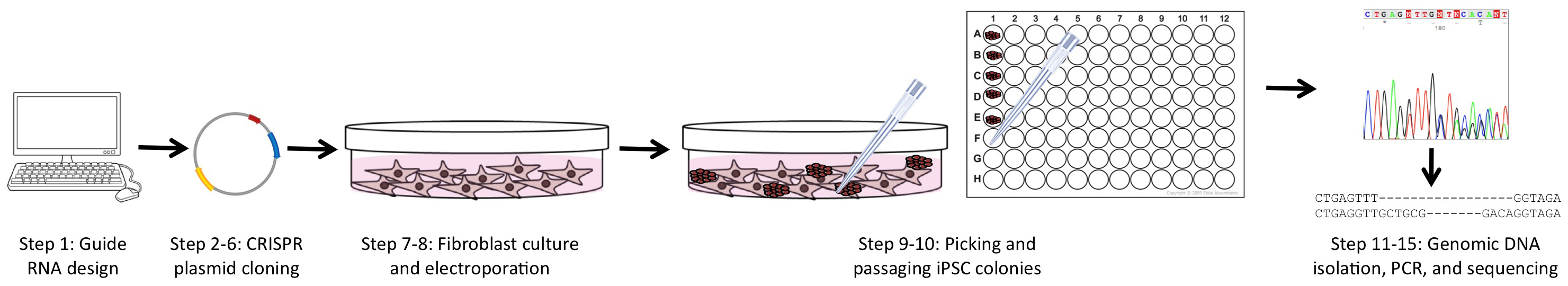
Figure 1. Overview of the experimental workflow. The protocols in this article are broken into 4 major sections, including: guide RNA and PCR primer design (Step 1), CRISPR plasmid generation and purification (Steps 2-6), fibroblast culture and electroporation with CRISPR and reprogramming plasmids (Steps 7-8), and isolating genomic DNA followed by PCR of the targeted region and sequencing (Steps 11-15).
Materials and Reagents
- Materials
- 1.5 ml Eppendorf tubes (VWR, catalog number: 89000-028 )
- 10 µl, 200 µl, and 1 ml pipette tips (USA Scientific, catalog numbers: 1111-3700 , 1111-0700 , and 1111-2721 )
- 14 ml tubes for bacterial culture (Corning, Falcon®, catalog number: 352059 )
- Conical flasks (Corning, PYREX®, catalog number: 4980-500 )
- 10 cm tissue culture dish (Corning, Falcon®, catalog number: 353003 )
- 6-well plate (Corning, Costar®, catalog number: 3516 )
- 5 ml, 10 ml, and 25 ml serological pipettes (Aikali Scientific, ASI, catalog numbers: SP205 , SP210 , and SP225 )
- 96-well plate (Corning, Falcon®, catalog number: 353072 )
- Sterile 50 ml disposable pipette basins (Fisher Scientific, catalog number: 13-681-502 )
- PCR tube strips (USA Scientific, catalog number: 1402-4700 )
- 15 ml centrifuge tubes (Fisher Scientific, catalog number: 05-539-12 )
- Nalgene General Long-Term Storage Cryogenic Tubes (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 5000-1012 )
- 1.5 ml Eppendorf tubes (VWR, catalog number: 89000-028 )
- Cells
- Human foreskin fibroblasts (MTI-GlobalStem, catalog number: GSC-3002 )
- Human foreskin fibroblasts (MTI-GlobalStem, catalog number: GSC-3002 )
- Reagents
- CRISPR plasmid pX330-U6-Chmeric_BB-CBh-hSpCas9 (Addgene, catalog number: 42230 )
- Reprogramming plasmids pCXLE-hOCT3/4-p53shRNA, pCXLE-hUL, and pCXLE-hSK (Addgene, catalog numbers: 27077 , 27080 , 27078 )
- Subcloning Efficiency DH5α competent cells (Thermo Fisher Scientific, InvitrogenTM, catalog number: 18265017 )
- BbsI restriction enzyme, supplied with NEBuffer 2.1 (New England Biolabs, catalog number: R3539 )
- QIAGEN PCR purification kit (QIAGEN, catalog number: 28104 )
- Custom oligonucleotides (Thermo Fisher Scientific)
- T4 DNA ligase kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: 15224017 )
- SOC medium (Thermo Fisher Scientific, InvitrogenTM, catalog number: 15544034 )
- LB broth base (Thermo Fisher Scientific, InvitrogenTM, catalog number: 12780052 )
- LB agar (Thermo Fisher Scientific, InvitrogenTM, catalog number: 22700025 )
- Ampicillin sodium salt (Sigma-Aldrich, catalog number: A9518-25G )
- QIAGEN Miniprep Kit (QIAGEN, catalog number: 27104 )
- Glycerol (Sigma-Aldrich, catalog number: G5516-100ML )
- LKO.1 5’ sequencing primer (5’-GACTATCATATGCTTACCGT-3’)
- Endo-Free QIAGEN Maxi Prep Kit (QIAGEN, catalog number: 12362 )
- Phosphate buffered saline (PBS), pH 7.4 (Thermo Fisher Scientific, GibcoTM, catalog number: 10010023 )
- 0.25% trypsin solution (Thermo Fisher Scientific, GibcoTM, catalog number: 25200056 )
- Growth factor depleted Matrigel (Corning, catalog number: 356230 )
- Neon Transfection System 100 µl Kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: MPK10025 )
- TeSR-E7 (STEMCELL Technologies, catalog number: 05910 )
- mTeSR1 (STEMCELL Technologies, catalog number: 85850 )
- ROCK inhibitor Y-27632 (Tocris Bioscience, catalog number: 1254 )
- Accutase (Innovative Cell Technologies, catalog number: AT104 )
- ZR-96 Quick-gDNA Kit (Zymo Research, catalog number: D3010 )
- GoTaq green master mix (Promega, catalog number: M7122 )
- Molecular biology grade water (Thermo Fisher Scientific, InvitrogenTM, catalog number: 10977023 )
- ZR-96 DNA Sequencing Clean-up Kit (Zymo Research, catalog number: D4052 )
- Gel 6x Loading Dye, Purple (New England Biolabs, catalog number: B7024S )
- GeneMate LE Quick Dissolve Agarose (BioExpress, GeneMate, catalog number: E-3119-500 )
- GelRed Nucleic Acid Gel Stain 10,000x in DMSO (Biotium, catalog number: 41002 )
- Accugene 10 TBE Buffer (Lonza, catalog number: 50843 )
- Quant-IT PicoGreen dsDNA Assay Kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: P7589 )
- mFreSR (STEMCELL Technologies, catalog number: 05855 )
- Liquid nitrogen
- Dulbecco modified Eagle’s medium (high glucose) (Thermo Fisher Scientific, GibcoTM, catalog number: 11965118 )
- Fetal bovine serum (FBS) (Thermo Fisher Scientific, GibcoTM, catalog number: 10437010 )
- Penicillin-streptomycin solution (10,000 U/ml) (Thermo Fisher Scientific, GibcoTM, catalog number: 15140122 )
- MEM Non-essential amino acids solution 100x (Thermo Fisher Scientific, GibcoTM, catalog number: 11140050 )
- Fibroblast growth medium (see Recipes)
- CRISPR plasmid pX330-U6-Chmeric_BB-CBh-hSpCas9 (Addgene, catalog number: 42230 )
Equipment
- Portable Pipet-aid (Drummond, model: DP-101, catalog number: 4-000-101 )
- Laminar flow hood (Thermo Fisher Scientific, Thermo ScientificTM, model: 1300 Series Class II Type A2 , catalog number: 1335)
- Neon electroporator (Thermo Fisher Scientific, InvitrogenTM, catalog number: MPK5000 )
- Heraguard ECO Clean Bench (Thermo Fisher Scientific, Thermo ScientificTM, model: HeraguardTM ECO, catalog number: 51029701 )
- NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Thermo ScientificTM, model: NanoDropTM 2000 )
- iCycler Thermal cycler (Bio-Rad Laboratories, model: iCycler Thermal cycler )
- Water bath (Thermo Electron Corporation, catalog number: 51221073 )
- Refrigerator (Frigidaire, model: FFTR2021QW1 )
- Tissue culture incubator set at 37 °C, 5% CO2 (Thermo Fisher Scientific, Thermo ScientificTM, model: HeracellTM 150i , catalog number: 51026283)
- Thermo Scientific Sorvall St8 120v cell culture centrifuge with plate adapters (Thermo Fisher Scientific, Thermo ScientificTM, model: SorvallTM St 8 , 120v)
- Bright-Line Hemocytometer (Hausser Scientific, catalog number: 3110 )
- HandEvac Handheld Aspirator with 8-channel adapter (Argos Technologies, catalog number EV500 )
- Olympus CKX41 inverted cell culture microscope (Olympus, model: CKX41 )
- Multichannel pipettes (Thermo Fisher Scientific, Thermo ScientificTM, catalog numbers: 4662000 and 4662010 )
- Sub-cell GT Electrophoresis cell (Bio-Rad Laboratories, model: Sub-Cell® GT Cell )
- Gel Imager
- Beckman Coulter DTX 880 Multimode Detector Microplate Reader (Beckman Coulter, model: DTX 880 )
- CoolCell LX Freezing Container (Biocision, catalog number: BCS-405 )
Procedure
- Designing guide RNA (gRNA) and sequencing primers
- Using a genome browser (genome.ucsc.edu), the first conserved exon in all transcript isoforms of the gene (unless an isoform specific LOF mutation is desired) should be identified and the sequence copied. This sequence can then be inserted into a guide RNA design tool such as crispor.tefor.net or crispr.mit.edu.
Note: The most important determinate is the specificity score. Use the highest score possible. Our previously reported data had an average score of 88 (Tidball et al., 2017). Record the most likely off-target sites as well to potentially screen for off-target indels in iPSC lines. The crispr.mit.edu does not identify off-targets with highly repetitive sequences (triplet repeats). The second score to be aware of is the out-of-frame score. This is a calculated probability that any given indel formed at the cut site will be out-of-frame, thus resulting in a frame-shift mutation and loss-of-function (presumably). Our previously reported data had an average score of 63. Lastly, the efficiency score should be > 50 to ensure a high rate of indel formation. Our previously reported data had an average score of 59. - The gRNA can be ordered as standard desalted oligo-nucleotides with the sequence CACCG- on the 5’ end of the forward sequence and AAACC- on the 5’ end of the reverse sequence. These provide the appropriate overhangs for insertion into the px330 (or related) plasmid as well as a G at the beginning of the gRNA to initiate transcription from the U6 promoter.
Note: DO NOT include the PAM sequence (NGG) in the gRNA oligo design. - Sequencing primers should be designed with a ~200 bp amplicon size that nearly evenly flanks the CRISPR cut site, if possible. The crispor.tefor.net site gives a good set of sequencing primers. Confirm the melting temperature and specificity of these primers by using the NCBI primer blast site (www.ncbi.nlm.nih.gov/tools/primer-blast/) (be sure to use human genomic reference sequence).
- Using a genome browser (genome.ucsc.edu), the first conserved exon in all transcript isoforms of the gene (unless an isoform specific LOF mutation is desired) should be identified and the sequence copied. This sequence can then be inserted into a guide RNA design tool such as crispor.tefor.net or crispr.mit.edu.
- Restriction digest of the vector plasmid
- Add the following to a 1.5 ml tube:
2 µg px330
5 µl Buffer 2.1
2 µl BbsI enzyme
Water to 50 µl (add water to tube first) - Perform the digestion overnight at 37 °C.
- On the following day, heat inactivate the mix at 65 °C for 20 min to stop the digestion reaction.
- Purify the cut plasmid using QIAGEN PCR purification kit. Elute the purified plasmid in water.
Note: The PCR purification kit is more efficient than gel extraction in isolating the cut plasmid and removes the majority of the small cut piece. When performing ligation for the first time, use BbsI cut plasmid without insert as a control to confirm lack or near lack of colonies. Cut plasmid can be stored for several months at -20 °C. - Use a NanoDrop Spectrophotometer to determine the concentration of the cut plasmid.
- Add the following to a 1.5 ml tube:
- Annealing the gRNA oligos to make a double-stranded insert
- Add the following to a 1.5 ml tube:
1 µl of Oligo1 (100 µM)
1 µl of Oligo2 (100 µM)
7 µl water
1 µl of Buffer 2 - Anneal primers in a PCR thermocycler using the following program:
Step 1: 95 °C for 5 min
Step 2: Ramp at -0.1 °C/sec from 95 °C to 25 °C
Step 3: 25 °C for 30 sec
Step 4: Hold at 4 °C. - Dilute 1:250 in water before proceeding.
- Add the following to a 1.5 ml tube:
- Ligation reaction of cut vector plasmid and annealed gRNA insert.
- Add the following components to a 1.5 ml tube:
100 ng of cut px330 plasmid from Step 2
2 µl of diluted oligo duplex from Step 3c
4 µl of 5x T4 buffer
1 µl of T4 ligase
Water to 20 µl. - Incubate the ligation mix at room temperature for 2 h.
- Add the following components to a 1.5 ml tube:
- Bacterial transformation of CRISPR-gRNA ligated construct
- Gently add 2 µl of ligation reaction into 50 µl of DH5α heat shock competent cells (on the ice). Mix by flicking the tube a few times.
- Place the tube on ice for 30 min.
- Heat shock the transformation mixture by placing the tube in a 42 °C water bath for 20 sec.
- Incubate the tube on ice for 2 min.
- Add 950 µl of SOC medium and place the tube in a shaker at 37 °C for 1 h at 200 rpm.
- Plate 40 µl and 400 µl of cells on to two LB plates containing 50 µg/ml ampicillin.
- Incubate the plates at 37 °C overnight.
- Gently add 2 µl of ligation reaction into 50 µl of DH5α heat shock competent cells (on the ice). Mix by flicking the tube a few times.
- Sequencing clones
- Pick 3 clones (using P200 pipette tips) per transformation reaction and inoculate each into a 14 ml tube containing 2 ml of LB broth with 50 µg/ml ampicillin.
- Place the 14 ml tubes with inoculant in a shaker at 37 °C and grow the bacterial culture for 7 h (or until cloudy).
- Use QIAGEN Miniprep kit according to the manufacturer’s instructions to isolate plasmid from 1.5 ml of bacteria.
- Mix remaining 0.5 ml of bacterial cultures with 0.5 ml 50% glycerol and store in -80 °C freezer until obtaining sequencing results.
- Determine the concentration of the isolated plasmids using a NanoDrop Spectrophotometer. Concentrations should be > 15 ng/µl to ensure starting material is adequate for sequencing.
- Send samples for Sanger sequencing. Typically, the sample will be diluted to 1 ng/µl in ddH2O. Use 1 µM of LKO.1 5’ sequencing primer (5’-GACTATCATATGCTTACCGT-3’).
- Based on results from sequencing, pick a clone containing the gRNA sequence to set up for a maxi culture. Inoculate 0.1 ml from the miniprep culture (Step 6d) into 100 ml of LB-Amp in a conical flask for overnight culture in a shaker at 37 °C and 200 rpm.
- Perform DNA isolation from overnight bacterial cultures using the Endotoxin-free QIAGEN Maxi prep kit according to the manufacturer’s instructions.
- Determine the concentration of the plasmid using NanoDrop.
- Pick 3 clones (using P200 pipette tips) per transformation reaction and inoculate each into a 14 ml tube containing 2 ml of LB broth with 50 µg/ml ampicillin.
- Fibroblast culture
- Change fibroblast medium (see Recipes) every other day.
- Passage cells before they become fully confluent approximately every 4 days.
- Wash once with 10 ml of 1x PBS.
- Add 2 ml of 0.25% trypsin per 10 cm (for a 10 cm dish).
- Place in a 37 °C incubator for 5 min.
- Once the cells detach, add 8 ml of fibroblast media and transfer the cells to a 15 ml conical tube.
- Centrifuge at 100 x g for 5 min at room temperature.
- Add 10 ml of fresh fibroblast medium and split cells at a 1:3-1:6 dilution factor on to a new 10 cm dish and add enough additional medium to each plate for 10 ml total.
- Wash once with 10 ml of 1x PBS.
- Change fibroblast medium (see Recipes) every other day.
- Transfecting fibroblasts with CRISPR and reprogramming plasmids
- 1-2 h prior to transfection, coat a 6-well plate with Matrigel diluted 100 µg/ml in ice-cold DMEM/F12. Add 1 ml per well and incubate at 37 °C.
- Aspirate medium from sub-confluent fibroblasts (confluency 80% or less).
- Wash once with 10 ml of 1x PBS.
- Add 2 ml of 0.25% trypsin and incubate at 37 °C for 5 min.
- Add 8 ml of fibroblast medium to the detached fibroblasts.
- Pipette up and down several times with 5 ml Serological pipette to ensure even single cell suspension.
- Perform cell count using a hemocytometer.
- Transfer 2.5 x 105 cells into a 15 ml conical tube and centrifuge at 100 x g for 5 min at room temperature.
- Pipette 250 µl of Buffer R into a sterile 1.5 ml tube. Add 2.5 µg each of the three reprogramming plasmid and 2.5 µg of CRISPR plasmid. (100 µl of the suspension is for electroporation, 50 µl is to limit bubble formation in Neon tip, and 100 µl is for an additional electroporation if arcing occurs).
- Resuspend the fibroblast pellet in 250 µl of Buffer R containing reprogramming and CRISPR plasmids.
- Using a 100 µl Neon electroporation tip electroporate in the Neon at 1,650 V, 10 msec, 3 pulses.
- If the electroporation arcs (indicated by a flash of light and popping sound), discard the cell suspension in the tip and repeat with the extra 150 µl of cell suspension.
- Pipette successfully electroporated cell suspension from the Neon tip into 12 ml of warm fibroblast medium.
- Remove Matrigel solution from a Matrigel-coated 6-well plate, and add 2 ml of cell suspension to each desired well.
- Incubate the plate at 37 °C, and change fibroblast medium 18-24 h later.
- Three days after plating, change the medium to TeSR-E7 reprogramming medium.
- Replace TeSR-E7 medium daily.
- ‘Pick-able’ colonies appear between 15-20 days after electroporation (see Figure 2).
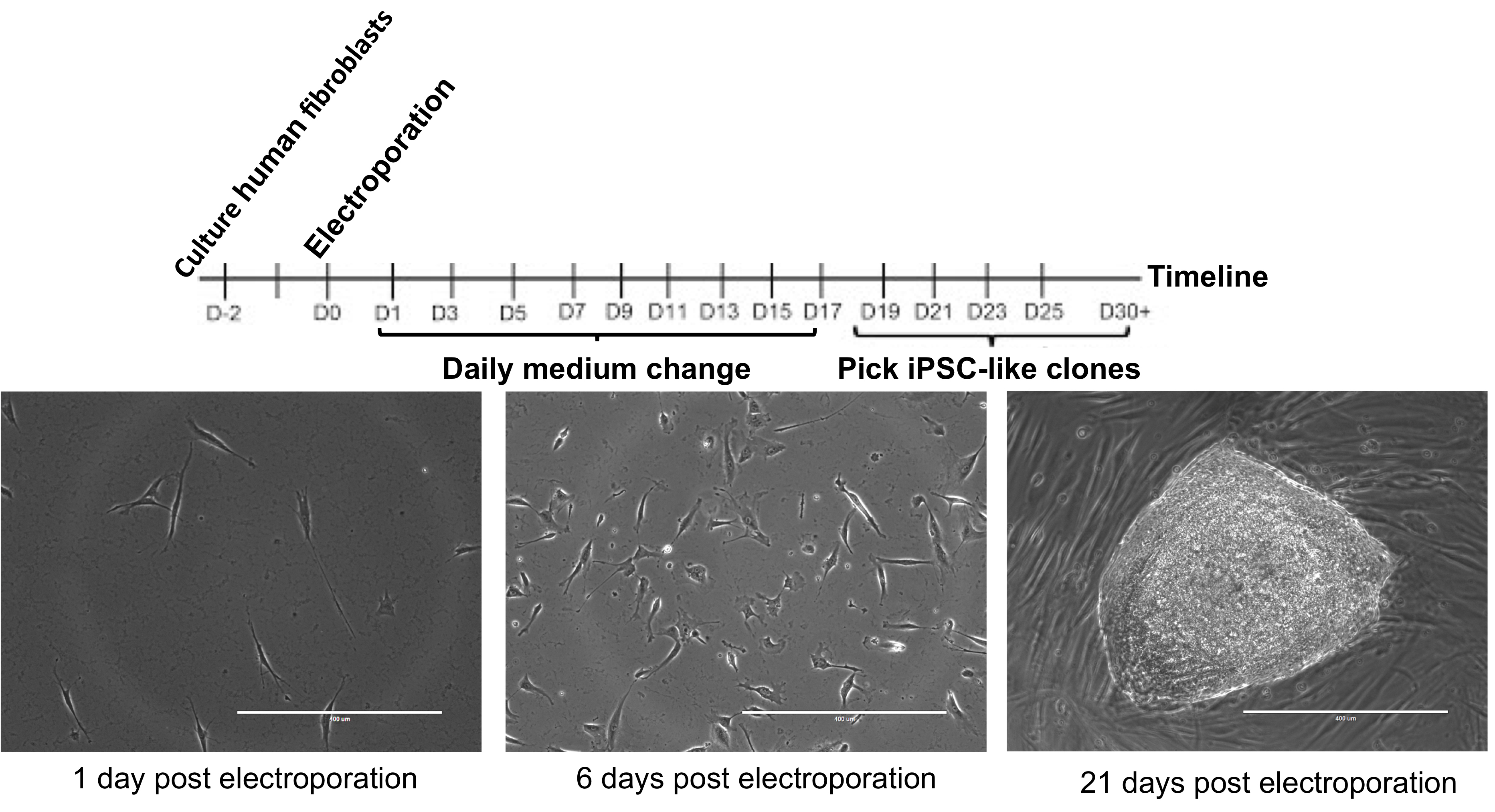
Figure 2. iPSC reprogramming timeline. The day 21 image is good for picking because of the well-defined margins, relative circularity, and lack of nearby colonies. Scale bars = 400 µm.
- 1-2 h prior to transfection, coat a 6-well plate with Matrigel diluted 100 µg/ml in ice-cold DMEM/F12. Add 1 ml per well and incubate at 37 °C.
- Picking iPSC colonies
Note: Typical yield will be ~20-40 colonies per well. If the number is substantially lower, this may indicate the starting fibroblast cultures were suboptimal (late passage, chromosomal abnormality, etc.). If the number is substantially higher, a dilution error may have occurred during the electroporation and single clones will be difficult to obtain.- 1-2 h prior to picking, coat a 96-well plate with Matrigel diluted 100 µg/ml in ice-cold DMEM/F12. Add 60 µl to each well and incubate at 37 °C.
- Immediately before picking colonies, remove Matrigel solution and add 100 µl of mTeSR1 with 10 µM ROCK inhibitor (Y-27632).
- In a HEPA workstation containing an inverted cell culture microscope (4x objective), pick individual colonies by dissecting with a 200 µl pipette tip on a micropipettor. After lifting the colony from the dish, remove the colony piece (or pieces) with the pipette in 50 µl volume, and place in one well of the 96-well plate.
Note: Use standard 200 µl pipette tips, gel-loading tips or similar tips with small bore size will likely shear the colony too much. An excellent video of this technique can be found at: https://www.youtube.com/watch?v=B0lXzsHIcK8. - Bias picking toward round isolated colonies to decrease the risk of picking merged colonies leading to heterogeneous genotypes. (see Figure 2)
Note: With typical reprogramming efficiency, 10-20 colonies can easily be picked from each well. Picking can be performed on consecutive days; however, do not pick from the same well of the 6-well plate on multiple days since secondary colonies could form from leftover pieces of picked colonies. - Change media to mTeSR1 without Y-27632 daily.
- 1-2 h prior to picking, coat a 96-well plate with Matrigel diluted 100 µg/ml in ice-cold DMEM/F12. Add 60 µl to each well and incubate at 37 °C.
- Passaging 96-well plates of iPSC clones
- Passage 96-well plates when the majority of wells are at or near 100% confluency.
- Coat each new plate with 60 µl of 100 µg/ml Matrigel diluted in ice-cold DMEM/F12 media per well. Incubate plates for 1-2 h in a cell culture incubator.
- Aspirate all media from 96-well plate of cells using the 8-channel aspiration attachment.
- Wash the plate once with 150 µl of 1x PBS.
- Aspirate and add 30 µl of room temperature Accutase to each well of the plate. Incubate for 5 min in the cell culture incubator.
- Add 210 µl of mTeSR1 containing 10 µM ROCK inhibitor, Y-27632 to each of the wells.
- Aspirate the Matrigel from previously coated plates.
- Transfer 50 µl of cells to the corresponding well of each new plate to be generated.
- After transferring the cells, add 100 µl of additional mTeSR1 with 10 µM ROCK inhibitor to each well to further dilute the Accutase. The cell confluency 24 h later should be between 10-30%.
- Passage 96-well plates when the majority of wells are at or near 100% confluency.
- Isolating genomic DNA from 96-well plate using ZR-96 Quick gDNA kit from Zymo
- Remove medium and wash once with 100 µl of PBS.
- Add 300 µl of Genomic Lysis Buffer and incubate at room temperature for 10 min.
- Pipette lysate into Silicon-A Plate placed on a Collection plate.
- Using the plate adapters, centrifuge the plate at 2,500 x g for 5 min.
Note: Be sure to properly balance centrifuge if only using 1 plate. - Add 200 µl of DNA Pre-Wash Buffer to each well and centrifuge at 2,500 x g for 5 min. Discard flow through.
- Add 300 µl of g-DNA Wash Buffer to each well and centrifuge at 2,500 x g for 5 min. Discard flow through.
- Centrifuge dry at 2,500 x g for 5 min to remove any residual buffer.
- Place the Silicon-A Plate over the Elution plate.
- Add 40 µl of DNA Elution Buffer or water and incubate for 5 min at room temperature.
- Centrifuge at 2,500 x g for 5 min.
- Check that DNA was purified by a NanoDrop Spectrophotometer.
- Remove medium and wash once with 100 µl of PBS.
- PCR to amplify the target region
- For each 25 µl reaction mix, combine the following in a 1.5 ml Eppendorf tube:
12.5 µl of GoTaq 2x Master Mix
0.1 µl of 100 µM of each primer
10.3 µl of Molecular Biology grade water - Add 23 µl of the mixture to each well of a 96-well PCR plate or 8-well PCR tube strip for PCR.
- Add 2 µl of the template to each PCR well.
- Use optimized PCR annealing temperature and standard GoTaq protocol to perform the PCR. Use a 2 min denaturing step at 95 °C followed by 40 cycles of the following: 95 °C for 30 sec, optimized annealing temperature for 30 sec, and 72 °C for 30 sec. Finally, a step of 72 °C for 5 min will finish incomplete extension of the PCR products.
- For each 25 µl reaction mix, combine the following in a 1.5 ml Eppendorf tube:
- Purifying PCR products with ZR-96 DNA Sequencing Clean-Up Kit (Zymo)
- Add 240 µl of Sequencing Binding Buffer to PCR reactions.
- Transfer the mixture to Zymo-Spin IB-96 plate on a collection plate.
- Centrifuge at 2,500 x g for 5 min.
- Add 300 µl of Sequencing Wash Buffer to each well. Centrifuge at 2,500 x g for 5 min.
- Add 20 µl of water to each well and mount plate onto 96-well PCR plate.
- Centrifuge at 2,500 x g for 5 min.
- Purified PCR products on the 96-well plate can be sealed and stored at -20 °C.
- Run 5 µl of the purified PCR products mixed with 1 µl of 6x loading dye on a 2% agarose gel containing 1x GelRed Nucleic Acid Stain. Separate bands by electrophoresis in 1x TBE buffer at 100 V for 1-2 h and check for correct amplicon size using a gel imager.
- Add 240 µl of Sequencing Binding Buffer to PCR reactions.
- Quantifying DNA concentrations using PicoGreen
Note: Alternatively, several wells could be quantified using the NanoDrop Spectrophotometer; however, the Zymo kit has a tendency for high 230 nm peaks making DNA quantification difficult.- Thaw Quant-IT PicoGreen dsDNA Assay Kit components.
- Prepare 1x TE buffer by adding 1 ml of 20x TE buffer with 19 ml of water.
- Dilute PicoGreen 1:200 in 1x TE buffer (50 µl of diluted PicoGreen per well).
- Make a set of DNA concentration standards by serial diluting the DNA standard to 5, 1, 0.2, 0.04, and 0 ng/µl in 1x TE buffer.
- In a clear 96-well plate, add 50 µl of diluted PicoGreen to 15 wells.
- Add 50 µl of each standard in triplicate.
- After 5 min, use a fluorescence plate reader to measure the fluorescence with an excitation of ~480 nm and emission of ~520 nm.
- Make a standard curve by inputting the standard DNA concentrations and fluorescence intensities into side-by-side columns in Excel. Generate an XY scatter-plot graph, right click on a data-point and select ‘Add Trendline’. In the ‘Options’ tab, select ‘Display equation on chart’. Record the equation.
Note: If the graph is not linear, delete data-points in the non-linear regions and only use the standard curve equation for samples having fluorescence readings in the linear region. - Add 50 µl of diluted PicoGreen to each well of a 96-well plate.
- Add 45 µl of 1x TE to each well.
- Add 5 µl of DNA from Step 13.
- Read on the plate reader and quantify the concentration using the equation generated in Step 14h and multiplying by 10.
- Thaw Quant-IT PicoGreen dsDNA Assay Kit components.
- Sanger or Next-Gen sequencing
- The PCR products can now be sequenced. Sanger sequencing is economical; however, the indels will cause overlapping peaks. These can be used to identify mutations using a program like Mutation Surveyor (Softgenetics.com) or manually to identify the indels formed.
- A less labor-intensive analysis solution is the use of Next-Gen sequencing. Samples can be submitted to the MGH (Massachusetts General Hospital) DNA Core for CRISPR Sequencing analysis. Briefly, barcoded Next-Gen sequencing adaptors are added to the ends of each sample and multiplex. 100 bp sequencing reads in the forward and reverse direction are performed. Each DNA species is then quantified as a percentage of total reads for each barcode, and aligned sequences with the percentage are listed for each sample.
- The PCR products can now be sequenced. Sanger sequencing is economical; however, the indels will cause overlapping peaks. These can be used to identify mutations using a program like Mutation Surveyor (Softgenetics.com) or manually to identify the indels formed.
- Suggested characterization steps
- Test for episomal vector integration using qPCR for plasmid DNA.
- G-band karyotyping or SNP microarray to test for genomic abnormalities.
- CRISPR off-target PCR and sequencing for highest off-target site scores from the gRNA design tool. This can be done for exonic and intronic or exonic-only off-targets.
- Test for mycoplasma contamination using myco-specific primers on cell lysates.
- Test for episomal vector integration using qPCR for plasmid DNA.
- Making frozen stock iPSC vials
Note: The most important aspect for the high viability of frozen iPSC is maintaining large colonies. Minimize pipetting whenever possible and use a larger scrapper to minimize colony breaking.- Aspirate medium from iPSC culture in 1 well of 6-well plate that is ~50% confluent (this is higher than optimal for normal passaging but increases viability when freezing).
- Wash 1x with 2 ml of 37 °C DMEM/F12.
- Aspirate, add 1 ml of 37 °C DMEM/F12, and add 1 ml of cold 2 U/ml Dispase solution.
- Incubate at 37 °C for 8 min.
- Aspirate and wash 1x with 2 ml of 37 °C DMEM/F12.
- Add 2 ml of mTesr1 medium and scrap into the medium.
- Pipette gently into a 15 ml centrifuge tube and centrifuge at 50 x g for 3 min in a 15 ml conical tube.
- Aspirate medium and add 1 ml of mFreSR.
- Pipette gently into a 1.5 ml cryovial, place into a CoolCell cell freezing container, which is then placed in a -80 °C freezer overnight.
- Transfer cryovials into a liquid nitrogen storage tank the next day.
- Aspirate medium from iPSC culture in 1 well of 6-well plate that is ~50% confluent (this is higher than optimal for normal passaging but increases viability when freezing).
Data analysis
- Sanger sequencing analysis
The main data analysis for this protocol is to analyze the sequencing data and perform quality controls. First, samples with insufficient DNA concentrations on the PicoGreen DNA quantification (below limit of detection) should be excluded. Often sequencing these samples will result in odd indel identification (large deletions with sizes not found in other clones) in these samples (data not shown). When using Sanger sequencing, samples with 3 sets of overlapping peaks should also be excluded since these samples are either not clonal or may be a result of DNA contamination prior to PCR. Indels from Sanger sequencing are identified manually by aligning the overlapping peak calls with the sequence downstream from the start of the indel (where peaks begin to overlap) (Figure 3A). Wild-type or homozygous indels are easily identified by using online BLAST analysis (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Be sure to use a human genome reference sequence and the ‘discontiguous megablast’ option to allow for indel alignment. Heterozygous indel identification can be aided by programs like Mutation Surveyor (Softgenetics.com), which separates the two sets of peaks based on a wild-type reference chromatograph and aligns to find the indel position. Unfortunately, this program is not very effective for compound heterozygous indels. Typically, indels form adjacent to the Cas9 cut site with small deletions being the most common (Table 1). When generating tables of indels, use Courier New. This font uses identical sizing for each character, allowing sequence alignment.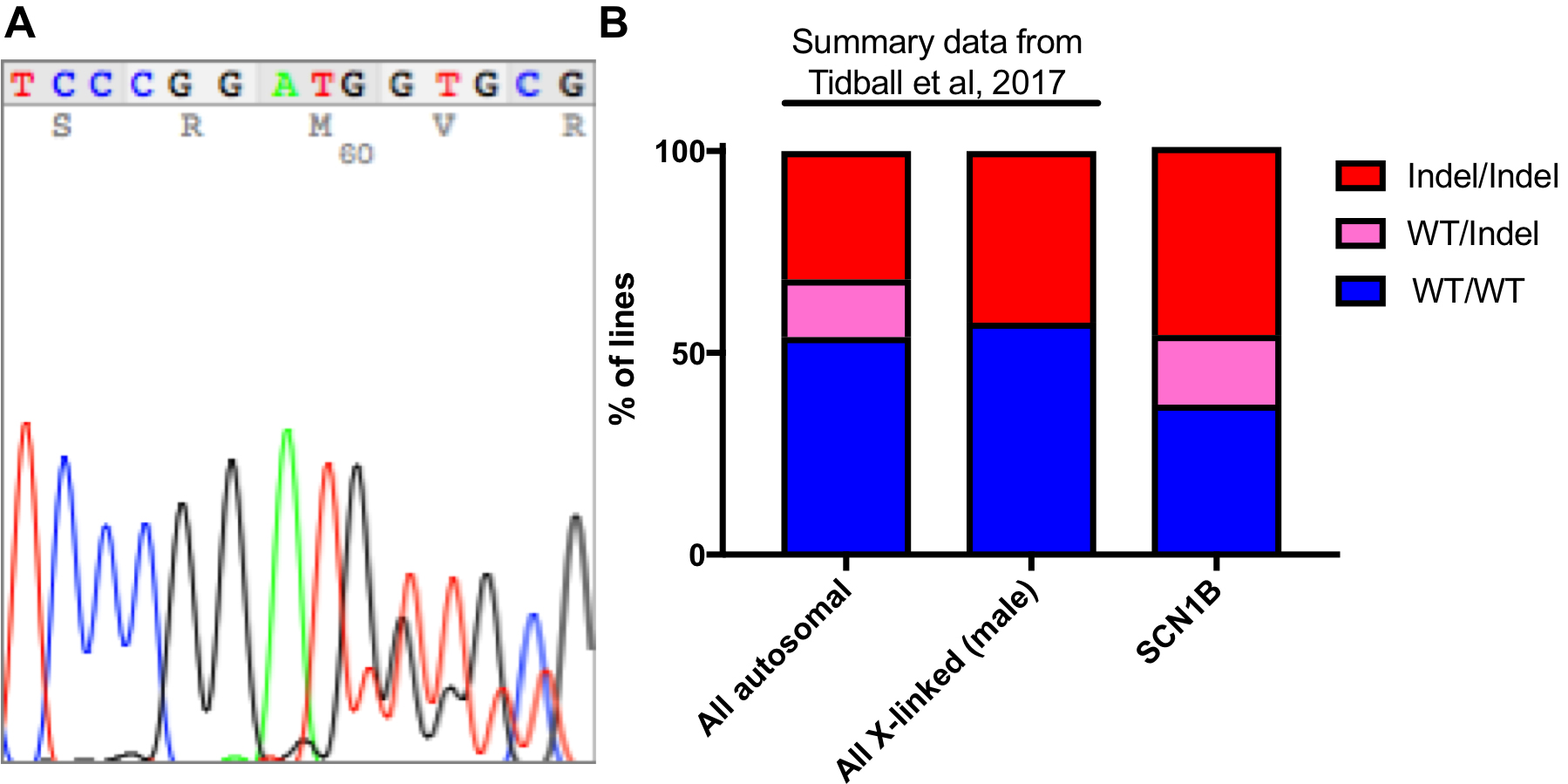
Figure 3. Typical indel formation data. A. Typical Sanger sequencing chromatogram for a heterozygous 2 bp deletion. B. Summary data from Tidball et al. (2017) is compared to a previously unpublished data set for SCN1B CRISPR/reprogramming. The data includes 22 WT/WT, 10 WT/Indel, and 27 Indel/Indel lines.
Table 1. Typical indels formed by a single experiment with > 50 clones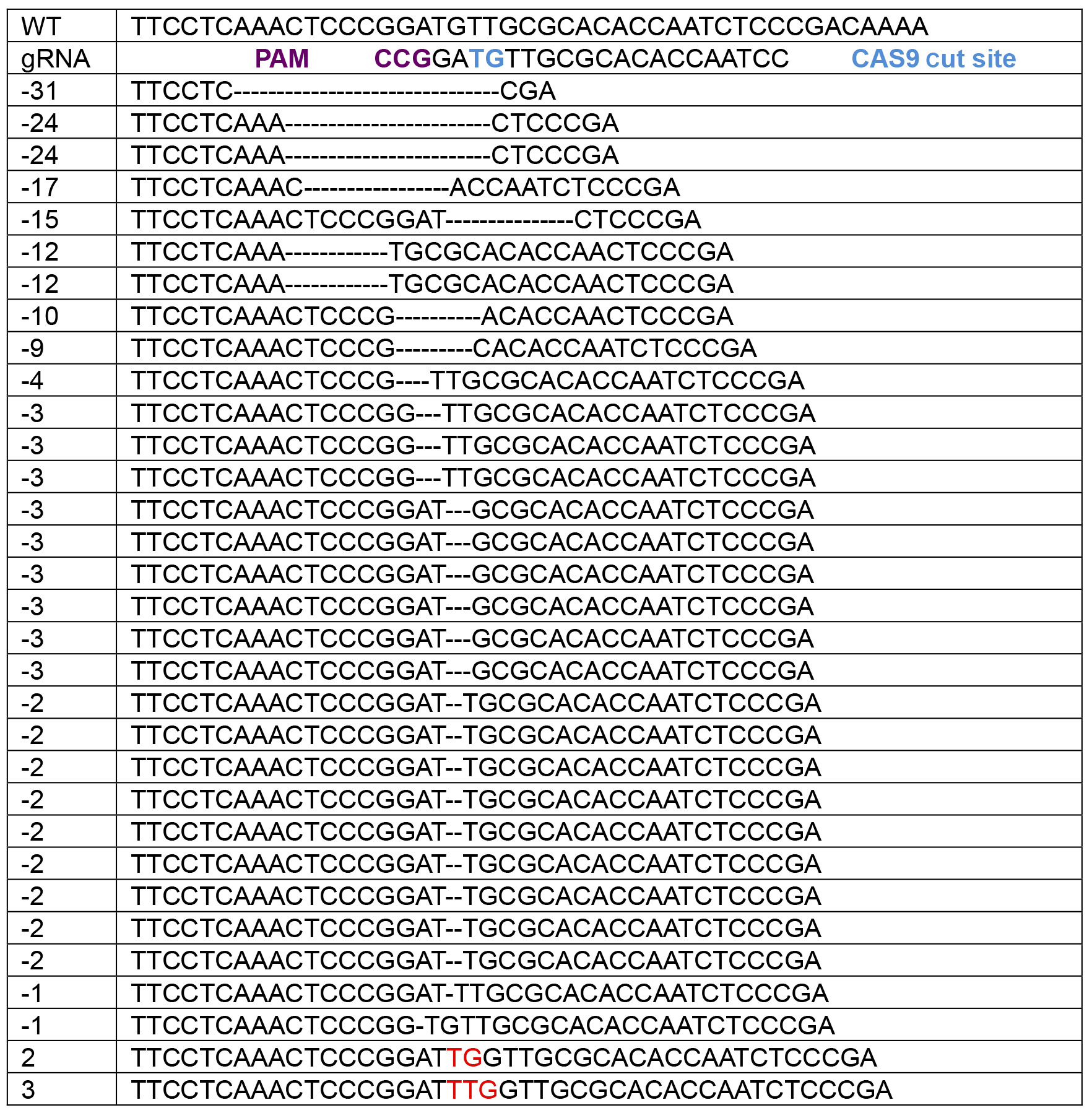
- Next-Gen sequence analysis
Next-Gen sequencing will provide the sequence for each DNA species found in the samples and the percentage of reads. If the PCR is not specific, a small percentage of off-target DNA may also be present. Besides indel identification, the quantitative nature of next-Gen sequencing also allows for identifying heterogeneous lines (2 or more clones grown or picked together). For example, if a heterozygous line diverges from a 50:50 ratio, this can indicate heterogeneity. Note that large indels can slightly skew this ratio (for example a 59 bp deletion heterozygous line resulted in nearly > 70% WT quantification). - Comparing the ratio of genotypes
While our previously published results for 5 epilepsy genes showed remarkable reproducibility of the ratio of all three genotypes (Tidball et al., 2017 and Figure 3B), this may vary. If many WT/indel lines are identified but no indel/indel lines, it may indicate that complete LOF results in inability to replicate or become pluripotent. To determine whether this is true, the experiment should be compared to a simultaneously performed experiment on another gene. For such experiments, a guide RNA for the genes mutated in Tidball et al. (2017) could be used. Comparisons in the ratio of the 3 genotypes can be performed using Chi-square analysis in Prism 7 (GraphPad Software).
Notes
- Depending on the gene being targeted, the efficiency can deviate from what we have published. For instance, when a gene function is necessary for proliferation, survival, or maintenance of the pluripotent state, you can expect a low number of full indel/indel LOF lines because the LOF makes these iPSC colonies less likely to form. Interestingly, the large number of clones generated and the reproducibility of our data does allow for some comparison between genes. For instance, one gene had at least one in-frame mutation in each indel/indel iPSC line. Since this deviated drastically from what we have previously seen and from the expected Out-Frame mutation percentage (CRISPOR.org), we hypothesize that expression of this gene is necessary for iPSC formation.
- We have been careful to use the same plasmid preparations and electroporation kits throughout our experiments. If changes are made to the protocol, the amount of plasmid introduced could vary potentially resulting in changes in the percentages of each genotype group.
Recipes
- Fibroblast growth medium
450 ml high glucose DMEM
50 ml fetal bovine serum (FBS)
5 ml non-essential amino acids
2.5 ml Penn/Strep
Acknowledgments
This work was supported by grants from the NIHNS090364 and Citizens United for Research in Epilepsy (CURE) to J.M.P. We thank the Center for Computational and Integrative Biology (CCIB) DNA Core Facility at Massachusetts General Hospital (Cambridge, MA) for providing sequencing services. The authors do not have any financial or non-financial interest in the subject matter or materials discussed in this manuscript.
References
- Bai, Q., Ramirez, J. M., Becker, F., Pantesco, V., Lavabre-Bertrand, T., Hovatta, O., Lemaitre, J. M., Pellestor, F. and De Vos, J. (2015). Temporal analysis of genome alterations induced by single-cell passaging in human embryonic stem cells. Stem Cells Dev 24(5): 653-662.
- Forsyth, N. R., Musio, A., Vezzoni, P., Simpson, A. H., Noble, B. S. and McWhir, J. (2006). Physiologic oxygen enhances human embryonic stem cell clonal recovery and reduces chromosomal abnormalities. Cloning Stem Cells 8(1): 16-23.
- Hendel, A., Kildebeck, E. J., Fine, E. J., Clark, J., Punjya, N., Sebastiano, V., Bao, G. and Porteus, M. H. (2014). Quantifying genome-editing outcomes at endogenous loci with SMRT sequencing. Cell Rep 7(1): 293-305.
- Howden, S. E., Maufort, J. P., Duffin, B. M., Elefanty, A. G., Stanley, E. G. and Thomson, J. A. (2015). Simultaneous reprogramming and gene correction of patient fibroblasts. Stem Cell Reports 5(6): 1109-1118.
- Howden, S. E., McColl, B., Glaser, A., Vadolas, J., Petrou, S., Little, M. H., Elefanty, A. G. and Stanley, E. G. (2016). A Cas9 variant for efficient generation of indel-free knockin or gene-corrected human pluripotent stem cells. Stem Cell Reports 7(3): 508-517.
- Miyaoka, Y., Chan, A. H., Judge, L. M., Yoo, J., Huang, M., Nguyen, T. D., Lizarraga, P. P., So, P. L. and Conklin, B. R. (2014). Isolation of single-base genome-edited human iPS cells without antibiotic selection. Nat Methods 11(3): 291-293.
- Tidball, A. M., Dang, L. T., Glenn, T. W., Kilbane, E. G., Klarr, D. J., Margolis, J. L., Uhler, M. D. and Parent, J. M. (2017). Rapid generation of human genetic loss-of-function iPSC lines by simultaneous reprogramming and gene editing. Stem Cell Reports 9(3): 725-731.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Tidball, A. M., Swaminathan, P., Dang, L. T. and Parent, J. M. (2018). Generating Loss-of-function iPSC Lines with Combined CRISPR Indel Formation and Reprogramming from Human Fibroblasts. Bio-protocol 8(7): e2794. DOI: 10.21769/BioProtoc.2794.
Category
Stem Cell > Pluripotent stem cell > Cell induction
Cell Biology > Cell engineering > CRISPR-cas9
Molecular Biology > DNA > Mutagenesis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.