- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Fluorescein Transport Assay to Assess Bulk Flow of Molecules Through the Hypocotyl in Arabidopsis thaliana
Published: Vol 8, Iss 7, Apr 5, 2018 DOI: 10.21769/BioProtoc.2791 Views: 7541
Reviewed by: Jyotiska ChaudhuriSmita NairAdam Idoine
Original research article
The authors used this protocol in:

Jul 2017
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
The bulk transport of molecules through plant tissues underpins growth and development. The stem acts as a conduit between the upper and low domains of the plant, facilitating transport of solutes and water from the roots to the shoot system, and sugar plus other elaborated metabolites towards the non-photosynthetic organs. In order to perform this function efficiently, the stem needs to be optimized for transport. This is achieved through the formation of vasculature that connects the whole plant but also through connectivity signatures that reduce path length distributions outside the vascular system. This protocol was devised to characterize how cell connectivity affects the bulk flow of molecules traversing the stem. This is achieved by exposing young seedlings to fluorescein, for which no specific transporter is assumed to be present in A. thaliana, and assessing the relative concentration of this fluorescent compound in individual cells of the embryonic stem (hypocotyl) using confocal microscopy and quantitative 3D image analysis after a given exposure time.
Keywords: ConnectomicsBackground
The link between structure and function has always fascinated biologists, from the design spaces of organs (Eldredge, 1989) to the convergence or divergence of evolutionary paths (Morris, 2003). At a smaller scale, cells are also organized in a robust and tightly controlled manner, intimately related to the functions the tissue performs (Jackson et al., 2017a). The collection of cellular physical interactions that makes up a specific tissue can also be regarded as a network, a cellular connectome. This connectome is especially interesting in plants as shared cell walls impede cellular movement, thus the network dynamics depend only on cell death and replication.
We hypothesize that tissue architecture and thus cell connectomes are relevant to physiological features and organ function. This way, network metrics and quantitative networks analysis can be used to make predictions and gain understanding of biological systems (Duran-Nebreda and Bassel, 2017; Jackson et al., 2017b).
In the current example (Jackson et al., 2017a), we investigated the topological properties of different cell types in the embryonic stem by digitally capturing global cellular interactions using confocal microscopy, revealing systematic arrangements of reduced path length in the atrichoblast (non-hair forming) cell files. To address a possible functional link between the preferential movement of small molecules and this path length distribution, we developed a fluorescein transport assay. This involves exposing the embryos to fluorescein in a non-saturating manner and quantifying cell-type specific fluorescence following cell segmentation. Similar assays using fluorescein either with specific (Konishi et al., 2002; De Bruyn et al., 2011) or non-specific interactions (Wang and Fisher, 1994; Tichauer et al., 2015) exist, some using caged variants that allow for more control in activation and release (Kobayashi et al., 2007; Christensen et al., 2009). However, these did not provide a connection to global cellular connectivity and thus producing a general quantitative framework for structure-function relationships in tissue architecture and bulk transport processes.
Materials and Reagents
- 94 x 16 mm sterile Petri dishes (Greiner Bio One International, catalog number: 633181 )
- Barky Ultipette capillary tips (Barky Instruments International, catalog number: CP-100 )
- Aluminum foil or opaque container
- Cellview cell culture dish, 35 x 10 mm glass bottom (Greiner Bio One International, catalog number: 627861 )
- 1.5 ml Eppendorf tube
- Arabidopsis thaliana seeds
- Sterile distilled water (type I water pH 7.0)
- Bleach (Domestos)
- Ethanol
- Propidium iodide solution (Sigma-Aldrich, catalog number: P4864-10ML )
- Murashige and Skoog basal salt mixture with vitamins (Duchefa Biochemie, catalog number: M0222 )
- Agar-agar granular powder (Fisher Scientific, catalog number: A/1080/53 )
- Fluorescein (Alfa Aesar, catalog number: L13251 )
- Potassium hydroxide (Fisher Scientific, catalog number: P/5640/53 )
- ½ Murashige and Skoog medium (see Recipes)
- Fluorescein plates (see Recipes)
Equipment
- Tissue culture hood (Azbil Telstar, model: AH-100 )
- 20 μl pipette (Gilson, model: PIPETMAN P20L )
- Small forceps (IDEAL-TEK, model: 4.SA.0 )
- Inverted confocal microscope (ZEISS, model: LSM 710 )
- Dissection microscope (Leica Microsystems stereo microscope, Leica Microsystems, model: Leica S6 E )
- 1,000 μl pipette (Gilson, model: PIPETMAN P1000L )
- Heated bath or microwave
- Water purification system (ELGA LabWater, model: Option-R 7 )
- 500 ml Pyrex bottles (DWK Life Sciences, DURAN, catalog number: 21 801 44 )
- Autoclave (Dixons, catalog number: ST 2228 )
- pH-meter (Hanna Instruments, model: pH 210 )
- Growth cabinet or room (16 h light photoperiod with light intensity at 150-175 mmol m2 sec-1 at 23 °C and 8 h dark at 18 °C)
Software
- ImageJ (Schindelin et al., 2012) with the Bio-formats plug-in
- MorphoGraphX (Barbier de Reuille et al., 2015)
Note: Uses the CUDA toolkit, software developers recommend NVidia graphics card and enough memory to handle the stacks being processed.
Procedure
- Germinating the seeds
- Prepare in advance ½ MS Petri dishes as described in the ‘Recipes’ section.
- Prepare a fresh 1/10 dilution of the commercial bleach with distilled water to sterilize the seeds.
- Place 60-100 seeds of each ecotype or species in a separate 1.5 ml Eppendorf tube and add 500 μl of the bleach solution to each tube.
- Incubate at RT for 5 min, mix by inverting the tube every minute.
- Move to the tissue culture hood.
- Sterilize the flexible pipette tips with ethanol.
- Pipette out the bleach and wash the seeds three times with 500 μl of sterile distilled water.
- Pick a string (10-30) of sterile seeds with a P20 pipette and the flexible pipette tips and put the seeds one by one and approximately 5 mm apart in a ½ MS plate.
- Cover the Petri dishes with aluminum foil or an opaque container and take them to the growth room or cabinet at 23 °C.
- Incubate in complete darkness for 4 days.
Note: This causes the seedlings to be elongated and to contain very little chlorophyll, which reduces autofluorescence of the sample during imaging, yielding better signal.
- Prepare in advance ½ MS Petri dishes as described in the ‘Recipes’ section.
- Fluorescein incubation treatment
- Prepare on the same day a batch of fluorescein-agar plates as described in the ‘Recipes’ section.
- Move the seedlings from the ½ MS plates to the fluorescein 0.8% agar Petri dishes using the small tweezers. Do not squeeze the seedlings as their walls are very thin at this point, lift them instead by putting the tweezer prongs under the cotyledons with minimal pressure. Place their root onto the agar surface such that the seedling does not fall onto its side. Use the dissection microscope to ease handling of the seedlings. Roots can be gently manipulated into the agar to ensure the seedling remains upright.
- Incubate the seedlings in the growth room for 2.5 h under the light source to maximize fluorescein uptake.
- Prepare on the same day a batch of fluorescein-agar plates as described in the ‘Recipes’ section.
- Propidium iodide staining
- Create a 5 μg/ml propidium iodide solution in water and place it in as many 1.5 ml Eppendorf tubes as needed.
- Transfer the seedlings into these Eppendorf tubes (10-20 in each) using the same technique as before to avoid damaging them.
- Incubate for 15 min at RT, mix by gently inverting the tube.
- Move the seedlings from the Eppendorf tubes to imaging dishes (Cellview cell culture dishes) using a P1000 and a cut pipette tip. The dishes should contain as many non-overlapping samples as possible, usually between 3 and 5. The dissection microscope can be used at this stage to detect and remove damaged samples (cracks in the epidermis, snapped roots or squeezed sections).
- Wash once with 300 μl of sterile distilled water and remove as much water as possible by pippeting, in such a way that the seedlings still have a layer of liquid around them. When imaging, add sterile distilled water as needed if the samples dry out.
- Create a 5 μg/ml propidium iodide solution in water and place it in as many 1.5 ml Eppendorf tubes as needed.
- Imaging
The following list contains a typical list of settings to image these samples:- 25x oil immersion objective.
- Zoom 1 (this can be changed to better suit the size of the ROI, although systematic warps to the images appear < 0.7 zoom).
- Frame size: 2,048 x 2,048. This can be adjusted to the proportions of the ROI.
- Bit depth: 16 bits.
- Pinhole slice thickness: 1.9 μm (0.47 AU).
- Slice interval (z direction): 0.7 μm.
- Scan speed: 9.
- Averaging: 4-8.
- Fluorescein excitation: 488 nm.
- Propidium iodide excitation: 535 nm.
- 25x oil immersion objective.
Data analysis
The data obtained with this protocol should be processed using the same protocol described previously in Montenegro-Johnson et al. (2015) and Jackson et al. (2017a and 2017b). Namely, the propidium iodide channel is used to perform 3D segmentation of the cell boundaries as it stains cell walls, while the fluorescein signal within each cell is used to characterize bulk flow for each cell type in the hypocotyl. See Figure 1 for a typical example of the obtained confocal stacks before processing. The steps are as follows:
- Load the confocal stacks into Fiji using the Bio-formats plugin and export each channel to a TIFF image format stack.
- Load each TIFF stack onto MorphoGraphX.
- Apply a Gaussian blur (typically 0.3 μm smooth length in each direction) to the propidium iodide channel.
- Use the ITK watershed segmentation on the smoothed propidium iodide stack to find the cell boundaries. The threshold used varies depending on the staining and image acquisition settings.
- Edit the stacks by fusing oversegmeneted cells as needed.
- Create a mesh for the cells with ‘cube size: 2’ and no smooth passes as settings.
- Calculate fluorescein concentration using the heat map function with ‘volume’ and ‘internal signal’ settings.
The three ecotypes used in the original study presented different average fluorescent readings, possibly due to innate differences in permeability and/or bulk uptake rates. Some ecotypes also displayed far greater variability than others. For this reason, in order to be pooled together all samples need to be normalized by within-sample mean fluorescence. Then to be comparable between ecotypes all pooled data has to be normalized by ecotype-wide mean corrected fluorescence.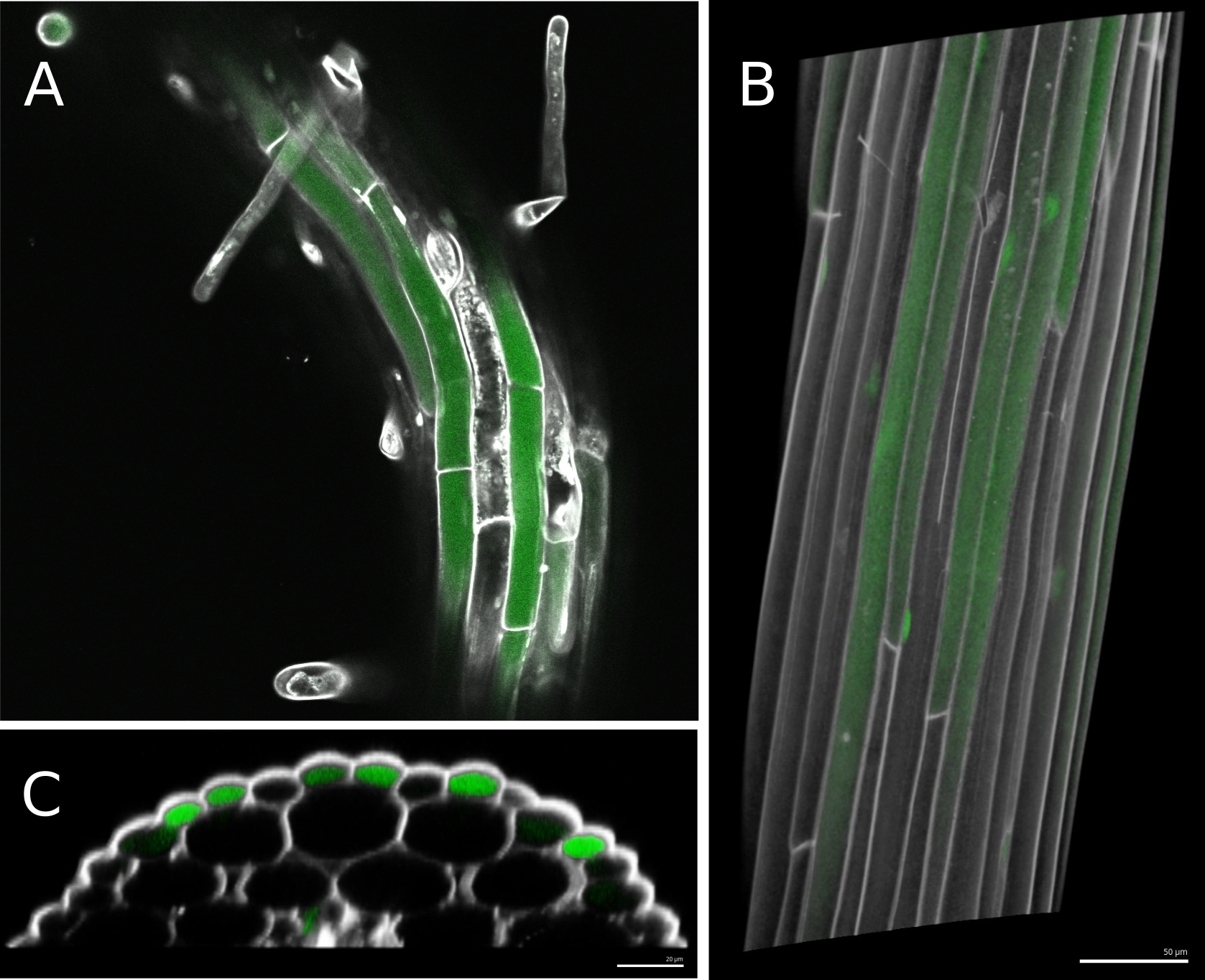
Figure 1. Typical results of imaging Arabidopsis hypocotyls after fluorescein exposure and propidium iodide staining. A. Single confocal stack of an Arabidopsis root, with root hairs visible. Two channels are shown, in grey scale propidium iodide, which stains cell walls, and in green fluorescein moving inside the living cells. B. 3D reconstruction of a hypocotyl from dozens of confocal stacks in MorphoGraphX. Transparency is used to show fluorescein signal accumulated inside the first layer of cells. C. A Transversal clip from the 3D reconstruction reveals a pattern of fluorescein concentration correlating with cell type spatial arrangements. This allows us to address which cells are involved in greater bulk flow through the epidermis.
Recipes
- ½ Murashige and Skoog medium (Murashige and Skoog, 1962)
2.3 g/L of Murashige and Skoog salt mixture with vitamins
0.8 g/L of agar-agar granulated powder
Add 80% of the final volume of filter-purified water (type I water, > 18.2 MΩ-cm)
Adjust pH with a 1 M KOH solution to 6.2
Top to selected final volume with filter-purified water (type I water, > 18.2 MΩ-cm)
Autoclave and pour into sterile Petri dishes (20 ml/dish) inside a tissue culture hood. Store poured Petri dishes at 4 °C before use (1 month shelf life) - Fluorescein plates
- Prepare in advance a 0.8 g/L agar-agar granulated powder mixture (follow the previous recipe but without adding the Murashige and Skoog salt mixture) and store at RT
- Prepare a 50 mM fluorescein solution (1,000x stock in 1:1 ethanol:sterile distilled water) and store it avoiding direct light sources (2 months shelf life)
- Melt the agar gel with a heated bath, steamer or by microwaving the gel thoroughly
- Wait until the solution cools off, reaching 50-60 °C
- Add the 1,000x fluorescein stock solution and pour the mix into Petri dishes (15 ml/dish). This need not be sterile and can be poured outside a tissue culture hood
- Prepare in advance a 0.8 g/L agar-agar granulated powder mixture (follow the previous recipe but without adding the Murashige and Skoog salt mixture) and store at RT
Acknowledgments
This work was supported by BBSRC grants BB/J017604/1, BB/L010232/1 and BB/N009754/1 to GWB, and by Leverhulme Trust Grant RPG-2016–049 to S.D.-N. and GWB. The authors declare no conflict of interest.
References
- Barbier de Reuille, P., Routier-Kierzkowska, A. L., Kierzkowski, D., Bassel, G. W., Schupbach, T., Tauriello, G., Bajpai, N., Strauss, S., Weber, A., Kiss, A., Burian, A., Hofhuis, H., Sapala, A., Lipowczan, M., Heimlicher, M. B., Robinson, S., Bayer, E. M., Basler, K., Koumoutsakos, P., Roeder, A. H., Aegerter-Wilmsen, T., Nakayama, N., Tsiantis, M., Hay, A., Kwiatkowska, D., Xenarios, I., Kuhlemeier, C. and Smith, R. S. (2015). MorphoGraphX: A platform for quantifying morphogenesis in 4D. Elife 4: 05864.
- Christensen, N. M., Faulkner, C. and Oparka, K. (2009). Evidence for unidirectional flow through plasmodesmata. Plant Physiol 150(1): 96-104.
- De Bruyn, T., Fattah, S., Stieger, B., Augustijns, P. and Annaert, P. (2011). Sodium fluorescein is a probe substrate for hepatic drug transport mediated by OATP1B1 and OATP1B3. J Pharm Sci 100(11): 5018-5030.
- Duran-Nebreda, S. and Bassel, G. W. (2017). Bridging scales in plant biology using network science. Trends Plant Sci 22(12): 1001-1003.
- Eldredge, N. (1989). Macroevolutionary Dynamics: Species, Niches and Adaptive Peaks. McGraw Hill.
- Jackson, M. D., Xu, H., Duran-Nebreda, S., Stamm, P. and Bassel, G. W. (2017a). Topological analysis of multicellular complexity in the plant hypocotyl. Elife 6: e26023.
- Jackson, M. D. B., Duran-Nebreda, S. and Bassel, G. W. (2017b). Network-based approaches to quantify multicellular development. J R Soc Interface 14(135).
- Kobayashi, T., Urano, Y., Kamiya, M., Ueno, T., Kojima, H. and Nagano, T. (2007). Highly activatable and rapidly releasable caged fluorescein derivatives. J Am Chem Soc 129(21): 6696-6697.
- Konishi, Y., Hagiwara, K. and Shimizu, M. (2002). Transepithelial transport of fluorescein in Caco-2 cell monolayers and use of such transport in in vitro evaluation of phenolic acid availability. Biosci Biotechnol Biochem 66(11): 2449-2457.
- Montenegro-Johnson, T. D., Stamm, P., Strauss, S., Topham, A. T., Tsagris, M., Wood, A. T., Smith, R. S. and Bassel, G. W. (2015). Digital single-cell analysis of plant organ development using 3DCellAtlas. Plant Cell 27(4): 1018-1033.
- Morris, S. C. (2003). Life’s solution: inevitable humans in a lonely universe. Cambridge University Press.
- Murashige, T. and Skoog, F. (1962). A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiologia Plantarum 15(3): 473-497.
- Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B., Tinevez, J. Y., White, D. J., Hartenstein, V., Eliceiri, K., Tomancak, P. and Cardona, A. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9(7): 676-682.
- Tichauer, K. M., Guthrie, M., Hones, L., Sinha, L., St Lawrence, K. and Kang-Mieler, J. J. (2015). Quantitative retinal blood flow mapping from fluorescein videoangiography using tracer kinetic modeling. Opt Lett 40(10): 2169-2172.
- Wang, N. and Fisher, D. B. (1994). The use of fluorescent tracers to characterize the post-phloem transport pathway in maternal tissues of developing wheat grains. Plant Physiol 104(1): 17-27.
Article Information
Copyright
![]() Duran-Nebreda and Bassel. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Duran-Nebreda and Bassel. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Duran-Nebreda, S. and Bassel, G. W. (2018). Fluorescein Transport Assay to Assess Bulk Flow of Molecules Through the Hypocotyl in Arabidopsis thaliana. Bio-protocol 8(7): e2791. DOI: 10.21769/BioProtoc.2791.
- Jackson, M. D., Xu, H., Duran-Nebreda, S., Stamm, P. and Bassel, G. W. (2017a). Topological analysis of multicellular complexity in the plant hypocotyl. Elife 6: e26023.
Category
Plant Science > Plant physiology > Tissue analysis
Plant Science > Plant cell biology > Cell imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.