- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Quantifying Symmetrically Methylated H4R3 on the Kaposi’s Sarcoma-associated Herpesvirus (KSHV) Genome by ChIP-Seq

Published: Vol 8, Iss 6, Mar 20, 2018 DOI: 10.21769/BioProtoc.2781 Views: 8324
Reviewed by: Alka MehraStefan de VriesPrashanth N Suravajhala
Original research article
The authors used this protocol in:

Jul 2017
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Post-translational modifications to histone tails contribute to the three-dimensional structure of chromatin and play an important role in determining the relative expression of nearby genes. One such modification is symmetric di-methylation of arginine residues, which may exhibit different effects on gene expression including blocking the binding of transcriptional activators, or recruiting repressive effector molecules. Recent ChIP-Seq studies have demonstrated the importance of cross-talk between different histone modifications in gene regulation. Thus, to acquire a comprehensive understanding of the combined efforts of these epigenetic marks, ChIP-Seq must be utilized for identifying specific enrichment on the chromatin. Tumorigenic herpesvirus KSHV, employs epigenetic mechanisms for gene regulation, and by evaluating relative abundance of multiple histone modifications in a thorough, unbiased way, using ChIP-Seq, we can get a superior insight concerning the complex mechanisms of viral replication and pathogenesis.
Keywords: KSHVBackground
Kaposi’s sarcoma-associated herpesvirus (KSHV) is an oncogenic human virus with two distinct phases during its lifecycle. After initial infection, KSHV establishes a persistent life-long infection in the host that is particularly problematic to the immune-compromised individuals. KSHV can cause various tumors in HIV/AIDS patients including Kaposi’s sarcoma, and multiple B-cell lymphomas (Chang et al., 1994; Cesarman et al., 1995; Russo et al., 1996; Soulier et al., 1995). With a large genome of about 165,000 bp, which encodes nearly 90 different open reading frames, KSHV has ample tools to evade the host immune surveillance system, alter host-cell growth pathways and produce infectious progeny virions.
During the latent phase only a fraction of the viral genes is expressed, which are oncogenic in nature and also help in replication and passaging of the viral episomes into the divided tumor cells (Uppal et al., 2014; Purushothaman et al., 2016). However, many factors including viral co-infection (HIV) and other stimulus such as, hypoxia, oxidative stress, or immune-suppressant medications can trigger the virus to shift into the active, lytic phase of the lifecycle leading to the production of infectious progeny virions, which egress from host cells surface to infect surrounding tissues (Purushothaman et al., 2015). These complex processes are carried out in a coordinated fashion with specific genes expressed in a sequential manner during the switch to a lytic (virus-producing) phase of the viral life cycle (Purushothaman et al., 2015; Aneja and Yuan, 2017).
KSHV employs epigenetic mechanisms to carefully regulate differential gene expression needed to maintain the virus in a specific phase of the lifecycle. Upon infection and entry into the human cells, viral genome acquires cellular histones, similar to the host genomes, and persists in euchromatin (transcription-permissive) or heterochromatin (transcription-repressive) states (Toth et al., 2013; Uppal et al., 2015). By these means, KSHV is able to restrict gene expression during latency to only a minimal subset of genes and is yet poised to rapidly shift into lytic replication phase. The development of KSHV-induced malignancies involves both phases of the lifecycle, thus it is essential for researchers to have a clear understanding of the mechanisms of how these epigenetic changes regulate viral genes expression.
Epigenetic regulation of gene expression relies on conformational changes to chromatin that alter the availability of certain proteins for transcription (Bernstein et al., 2007). One of the best-studied ways organisms induce conformational changes to the chromatin is through histone-tail modifications. Histone residues may be ‘modified’ by the addition of different molecules to specific residues on the histone proteins (Bannister and Kouzarides, 2011). While several modifications have been identified, two of the most-commonly studied types are lysine residue acetylation or lysine residue methylation and they can dictate the transcriptional state of the genes occupied by those modified histones (Zhang et al., 2015). For example, lysine acetylation is generally considered an ‘activating’ mark that upregulates gene expression. Another activating mark is histone 3, lysine 4 trimethylation (H3K4me3), yet another modification on histone H3 at lysine 27 in the same fashion (H3K27me3) is a ‘repressive’ mark, leads to transcriptional silencing (Bannister and Kouzarides, 2011).
Histone lysine acetylation (H3ac), H3K4me3, and H3K27me3 levels have been assessed on the KSHV genome but the landscape of other histone modifications during lytic reactivation had not yet been ascertained (Gunther and Grundhoff, 2010; Toth et al., 2010 and 2013). So, when proteomic interaction studies conducted in our lab suggested that a lytic viral protein, ORF59, could be involved in chromatin remodeling, particularly in regards to arginine methylation, we set out to study the histone arginine methylation. Our recent study, ‘KSHV encoded ORF59 modulates histone arginine methylation of the viral genome to promote viral reactivation’ examined the enrichment of a specific histone modification H4R3me2s (histone 4 arginine 3 symmetric di-methylation) across the viral genome (Strahan et al., 2017). H4R3me involves first the mono-methylation of the arginine followed by the addition of another methyl group in either a symmetric, or asymmetric fashion (H4R3me2s or H4R3me2a, respectively) (Di Lorenzo and Bedford, 2011). Arginine methylation is important for various several cellular processes including RNA processing, DNA repair, transcription, signal transduction, and chromatin remodeling (Pahlich et al., 2006). While the addition of methyl groups on arginine residues increases hydrophobicity that blocks hydrogen bonding but the overall charge is not altered so the binding between nucleic acids or other proteins remains undisturbed (Pahlich et al., 2006). Various protein arginine methyltransferases (PRMTs) are expressed in multiple subcellular locales to modify the protein arginine residues of nuclear as well as cytoplasmic proteins (Bedford and Clarke, 2009).
Interestingly, the conformational difference between asymmetrically modified H4R3me2 and symmetrically H4R3me2 affects transcriptional activation/repression very distinctly. Symmetrically modified H4R3me2 favors the repression of gene expression and transcriptional silencing, while in contrast asymmetrically modified H4R3me2 favors upregulation of gene transcription (Di Lorenzo and Bedford, 2011).
In order to specifically capture the symmetrically modified, H4R3me2s chromatin, it was first absolutely essential to verify that the antibody used for ChIP-Seq purposes would not cross-react with H4R3me2a. Anti-H4R3me2s antibody was obtained and the specificity to the symmetrically modified H4R3me2 was tested before using for immunoprecipitating chromatin from KSHV infected cells. Following the confirmation of its specificity, we proceeded to perform ChIP-Seq from latent and lytically reactivating KSHV positive TRExBCBL1-RTA cells. The advantage of using TRExBCBL1-RTA cells (KSHV infected cell line) was to induce the expression of Replication and Transcription Activator (RTA) by tetracycline/doxycycline, which is both necessary and sufficient to trigger lytic reactivation (Nakamura et al., 2003). The ChIP assay was performed similarly to previously done ChIP-Seq assays that include the following basic steps: harvest the cells and cross-link with formaldehyde, isolate and shear chromatin, immunoprecipitate DNA-protein complexes of interest, purify the DNA, and prepare sequencing libraries from the immunoprecipitated and respective inputs DNA (Figure 1). To obtain a more thorough understanding of the chromatin remodeling role of viral protein ORF59, we needed to test the relative enrichment of several different factors at the KSHV genome including H4R3me2s, ORF59, PRMT5, COPR5. Each of these ChIPs were done in triplicate and samples were combined for library preparation. ChIP-Seq experiments traditionally use approximately 20 million cells per sample; however, to quantify H4R3me2s (and enrichment of other factors as well) on the KSHV genome, we chose to use a modified Low-Cell ChIP protocol with 5 million cells per sample instead (Park, 2009). To accomplish this, the chromatin-shearing step of the ChIP assay was optimized using the Diagenode Bioruptor® Pico to improve the efficiency of the DNA immunoprecipitation. 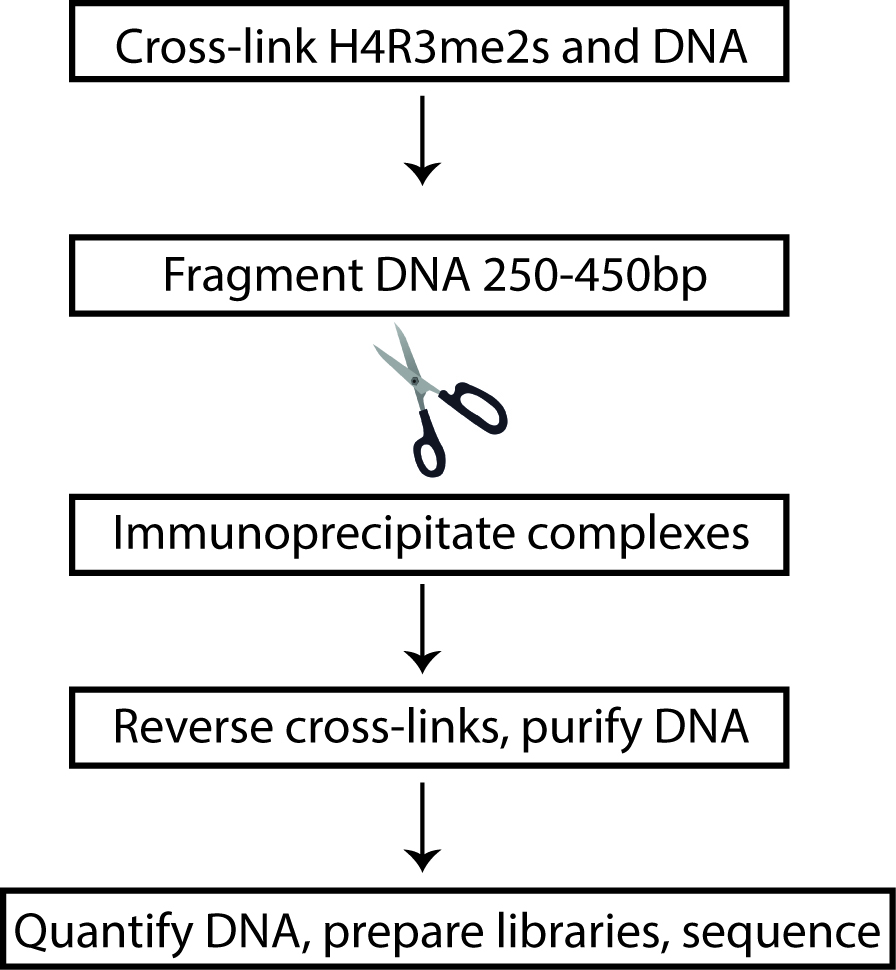
Figure 1. Flow-chart depiction of H4R3me2s ChIP. Cells from latent and lytic KSHV-positive cells were cross-linked to preserve DNA-protein interactions and the DNA was sheared into small fragments. DNA fragments were then immunoprecipitated, purified, and subjected to next-generation sequencing.
Another unique challenge faced in assessing chromatin structure of viral genomes is that H4R3me2s ChIP assay isolates both viral and cellular host DNA bound to H4R3me2s; and furthermore, upon lytic reactivation viral genomes are multiplied resulting in drastically different levels of viral DNA between two samples with an approximately identical number of cells (Figure 2). For this reason, we assessed H4R3me2s levels at a very early time point during lytic reactivation (12 h), before viral genome copies have had a chance to accumulate and possibly bias the downstream ChIP-Seq. As a result of these careful adjustments, we were able to successfully quantify relative enrichment of a repressive chromatin mark on the viral genome during two different phases of the lifecycle and demonstrate a novel chromatin-remodeling role for the early viral protein, ORF59.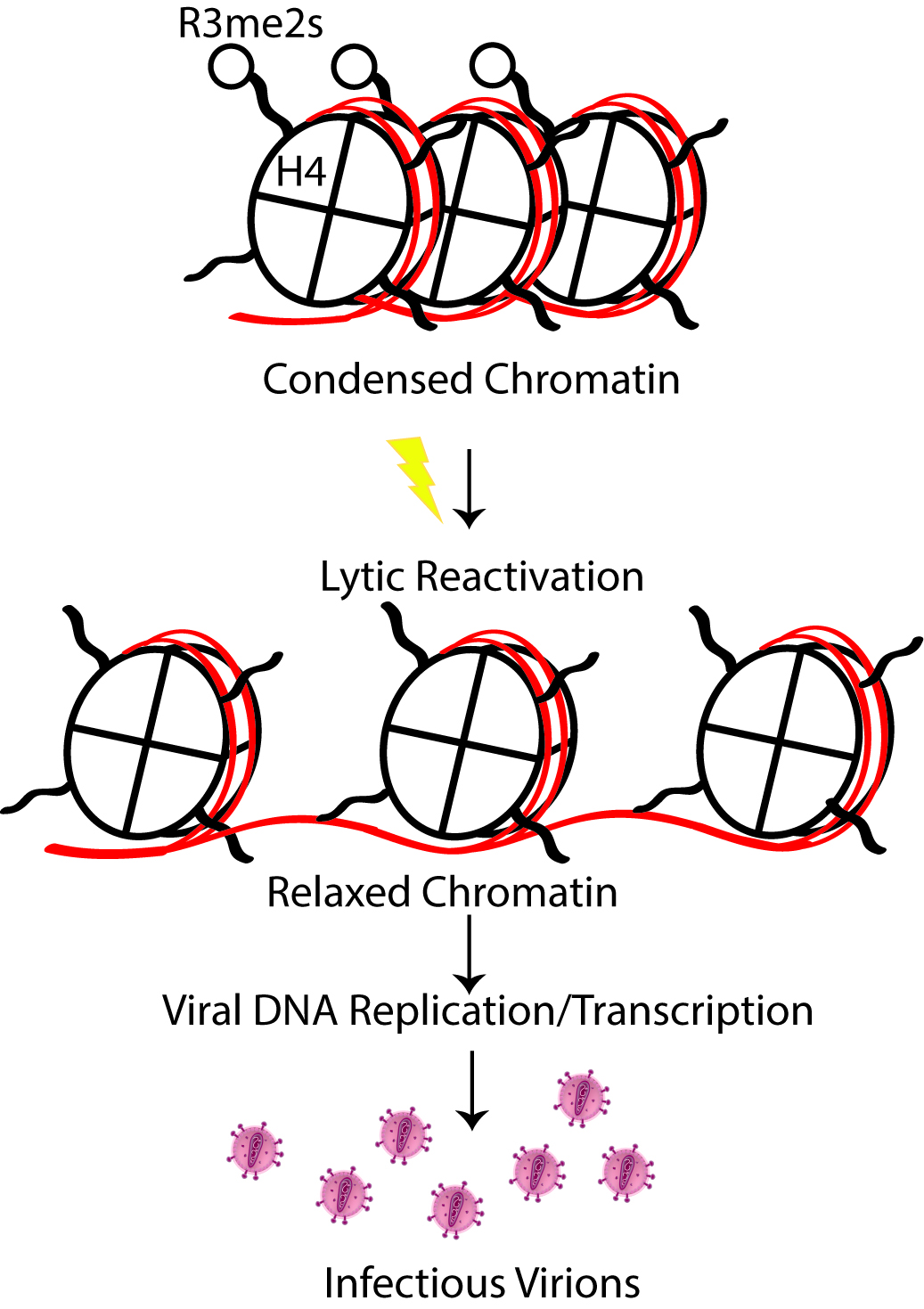
Figure 2. Schematic depiction of H4R3me2s role in KSHV lytic reactivation. H4R3me2s is an abundant hallmark of transcriptionally silent, heterochromatin and must be removed to favor active gene transcription.
Materials and Reagents
- Materials
- Pipette tips
- 1.5 ml Bioruptor® Pico Microtubes with Caps (Diagenode, catalog number: C30010016 )
- Illumina NextSeq 500 Mid Output KT v2 (150 cycles) (Illumina, catalog number: FC-404-2001 )
Note: Researchers should select the most appropriate flow cell for their experimental specifications. To sequence arginine methylation on the KSHV genome, this specific flow cell was sufficient.
- Pipette tips
- Chemicals/stock solutions
- Protease inhibitors (leupeptin, aprotinin, sodium fluoride, pepstatin, and phenylmethylsulfonyl fluoride) (Sigma-Aldrich, catalog number: S8830 )
- Pierce 16% formaldehyde (w/v), Methanol-Free (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 28906 )
- 1 M glycine
- Bioruptor® Pico sonication beads (Diagenode, catalog number: C01020031 )
- RNase A, 20 mg/ml
- 1.5% agarose gel
- Specific ChIP grade antibodies
Note: To quantify H4R3me2s on the KSHV genome, following ChIP grade antibodies were used, rabbit anti-H4R3me2s (Active Motif, catalog number: 61187 ), rabbit anti-Histone H4 (Active Motif, catalog number: 61299 ), rabbit anti-control IgG (ChIP grade—Cell Signaling Technology, catalog number: 2729 ). - 1x TE
- 5 M NaCl
- 7.5 M ammonium acetate
- 100% ethanol
- GlycoBlue Coprecipitant (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM9516 )
- Proteinase K
- QIAGEN MinElute PCR purification Kit (QIAGEN, catalog number: 28004 )
- Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: Q32851 )
- NEXTflexTM ChIP Seq Kit (Illumina compatible) (Bioo Scientific, catalog number: NOVA-5143-02 )
- NEXTflexTM ChIP Seq Barcodes - 12 (Illumina compatible) (Bioo Scientific, catalog number: NOVA-514120 )
- KAPA Library Quantification Complete Kit (Universal), kit code KK4824 (Kapa Biosystems, catalog number: 07960140001 )
- Agilent Bioanalyzer High Sensitivity DNA chip Kit (Agilent Technologies, catalog number: 5067-4626 )
- 0.5 M PIPES, pH 8.0
- 1.7 M KCl
- 10% Nonidet-P40 (NP-40)
- 0.5 M EDTA, pH 8.0
- 1 M Tris-HCl pH 8.1
- Magnetic Protein A, and G beads
- Salmon sperm DNA, sheared 10 mg/ml (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM9680 )
- 10% Triton X-100
- 10% SDS
- 1 M NaHCO3
- Protease inhibitors (leupeptin, aprotinin, sodium fluoride, pepstatin, and phenylmethylsulfonyl fluoride) (Sigma-Aldrich, catalog number: S8830 )
- Buffers
- 1x PBS (137 mM NaCl, 10 mM phosphate, 2.7 mM KCl, and a pH of 7.4)
- Buffer D Chromatin Shearing Buffer (Diagenode, catalog number: C01020030 )
- Cell lysis buffer (see Recipes)
- ChIP dilution buffer (see Recipes)
- ChIP Magnetic A+G beads (see Recipes)
- ChIP Low Salt Wash (see Recipes)
- ChIP elution buffer (see Recipes)
- 1x PBS (137 mM NaCl, 10 mM phosphate, 2.7 mM KCl, and a pH of 7.4)
Equipment
- Pipettes
- Tabletop Eppendorf centrifuge, refrigerated and non-refrigerated
- Bioruptor® Pico Sonication device (Diagenode, catalog number: B01060001 )
- Magnetic stand
- Water bath
- Tube rotators
- Thermocycler
- Qubit Fluorometer (Thermo Fisher Scientific)
- Agilent 2100 Bioanalyzer (Agilent Technologies, model: Agilent 2100 , catalog number: G2939BA)
- Quantitative PCR machine
- Vortexer
- Illumina NextSeq 500 (Illumina, model: NextSeq 500 )
Note: The Illumina NextSeq machine used for these studies is the property of the Nevada Genomics Center, who performed all sequencing runs for this study.
Software
- CLC workbench 10.0.1 (Licensed from QIAGEN, Germantown, MD)
Procedure
- Chromatin immunoprecipitation assay
- Harvest approximately 20 million cells and centrifuge into a pellet. Aspirate culture medium, then resuspend in 5 ml ice-cold PBS containing protease inhibitors (1 μg/ml leupeptin, 1 μg/ml aprotinin, 1 μg/ml sodium fluoride, 1 μg/ml pepstatin, and 1 mM phenylmethylsulfonyl fluoride) and centrifuge again to wash cells.
- Resuspend cell pellet in 10 ml of 1% formaldehyde in PBS and incubate samples at room temperature for 10 min with gentle rocking.
Note: If using 16% methanol-free formaldehyde solution, mix 1 ml + 15 ml PBS to obtain 1% formaldehyde solution to cross-link cells in. - Immediately add glycine to samples at a final concentration of 125 mM to quench the cross-linking reaction and rock samples 5 min at room temperature.
Note: For 10 ml of cross-linking cell suspension, add 1.25 ml 1 M glycine to obtain a final concentration of 125 mM. - Centrifuge cells at 70 x g for 5 min, wash once more in 5 ml ice-cold PBS with protease inhibitors.
- Aspirate PBS from cell pellet and resuspend cells in 1 ml cell lysis buffer [5 mM piperazine-N,N’-bis (2-ethanesulfonic acid) (PIPES)-KOH (pH 8.0)-85 mM KCl-0.5% NP-40; Recipe 1] supplemented with protease inhibitors and incubate on ice for 10 min.
- Centrifuge at 800 x g for 5 min at 4 °C to collect the nuclei, aspirate supernatant from pellet.
- Meanwhile, prepare 1.5 ml sonication tubes by adding approximately 100 mg of Bioruptor® sonication beads and labeling tubes appropriately.
- Resuspend pellet in Buffer D Chromatin Shearing Buffer (Diagenode Inc.) in preparation for chromatin shearing using the Bioruptor® Pico instrument (Diagenode Inc.). For 20 million cells, resuspend nuclei in 1.8 ml Buffer D, and distribute 300 μl to 6 prepared sonication tubes.
- Sonicate using the Bioruptor® Pico sonication device using On/Off time for each cycle of 30 sec/30 sec for 60 cycles.
Note: Sonication parameters may take further optimization of conditions to obtain ideal chromatin shearing where fragments about 300 bp long are obtained. See Diagenode manuals for further suggestions on optimizing sonication efficiency. - Centrifuge sonication tubes in a tabletop centrifuge at 4,000 x g for 1 min to remove cell debris and transfer supernatant to clean tubes. Supernatant should be fairly clear after sonication. If desired, a small 15-20 μl aliquot may be taken from this step and treated with 1 μl RNase (20 mg/ml stock) for 30 min, then cross-links reversed by incubation at 65 °C for at least 4 h or overnight and resolved on a 1.5% agarose gel to check shearing efficiency (see Figure 3 for an example of shearing results).
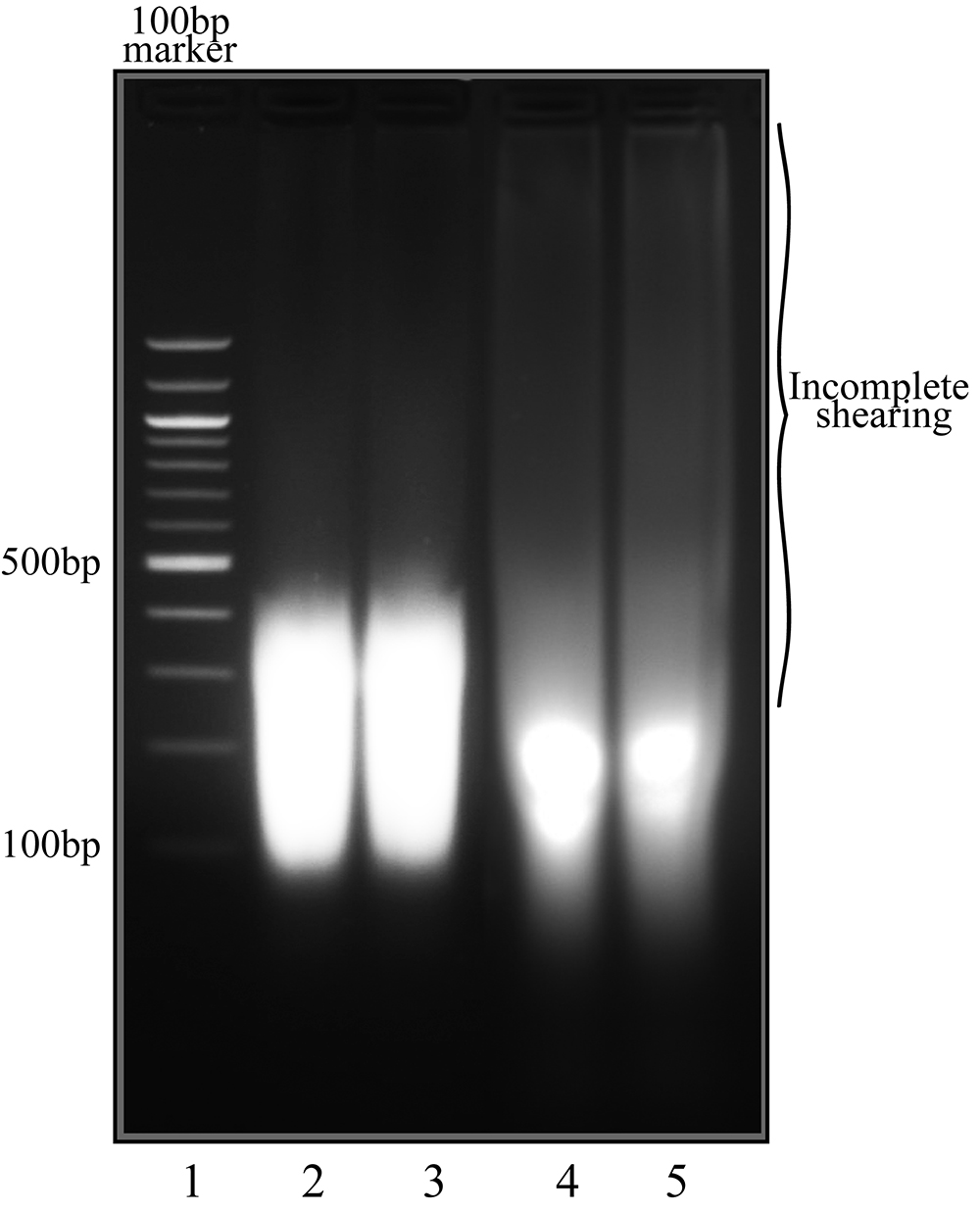
Figure 3. Chromatin shearing of KSHV positive cell lines. Lane 1, 100 bp marker. Lane 2-5 are shearing controls from TRExBCBL1-RTA cells, lanes 2 and 4 latent uninduced samples, lanes 3 and 5 are lytically reactivated cells. Lanes 2 and 3 demonstrate efficient shearing in preparation for ChIP assay, while lanes 4 and 5 show some residual DNA fragments (smearing) that are too large for ChIP-Seq assay. Lanes 2 and 3 were sheared using the Bioruptor® Pico for 30 sec on/off cycles for 60 min; lanes 4-5 were sheared using the identical parameters but the shorter time (30 min). - Add 1-3x volume of ChIP dilution buffer (1.2 mM EDTA, 16.7 mM Tris, pH 8.1, 167 mM NaCl, and protease inhibitors; Recipe 2) to sheared chromatin samples and divide volume in two, one set for IgG control ChIP, the other for specific antibody.
Note: For H4R3me2s ChIP from KSHV cells, we used 3x dilution of ChIP dilution buffer and 2 μg of specific antibody. Depending on antibody efficiency, a lesser dilution (1x or 2x volume of ChIP dilution buffer) of sheared chromatin can be used for ChIP assay. - Pre-clear samples with addition of 15 μl/ml of ChIP Magnetic A + G beads (Recipe 3) and rotate at 4 °C 30 min.
Note: If antibody specificity is excellent, the pre-clearing step may be omitted, however, to reduce background signal in the IgG samples when performing ChIP-Seq we recommend keeping this step. - Retain 10% of ChIP lysate for input fraction, store samples at 4 °C.
- Collect beads against a magnetic stand and transfer ChIP lysate to fresh tubes. Add IgG or specific antibody (2 μg or according to supplier’s instructions) and rotate ChIP samples overnight 4 °C.
- Next day, collect DNA-protein complexes by adding 25 μl/ml of ChIP Magnetic A + G beads and rotating 4 °C for 2 h.
- Wash ChIP samples 3 times each with 1 ml of ChIP Low Salt Wash buffer (0.1% SDS-1.0% Triton X-100-2 mM EDTA-20 mM Tris [pH 8.1]-150 mM NaCl; Recipe 4), and twice with 1x TE.
Note: Refer to Figure 4 for visual representation of magnetic bead washing procedure.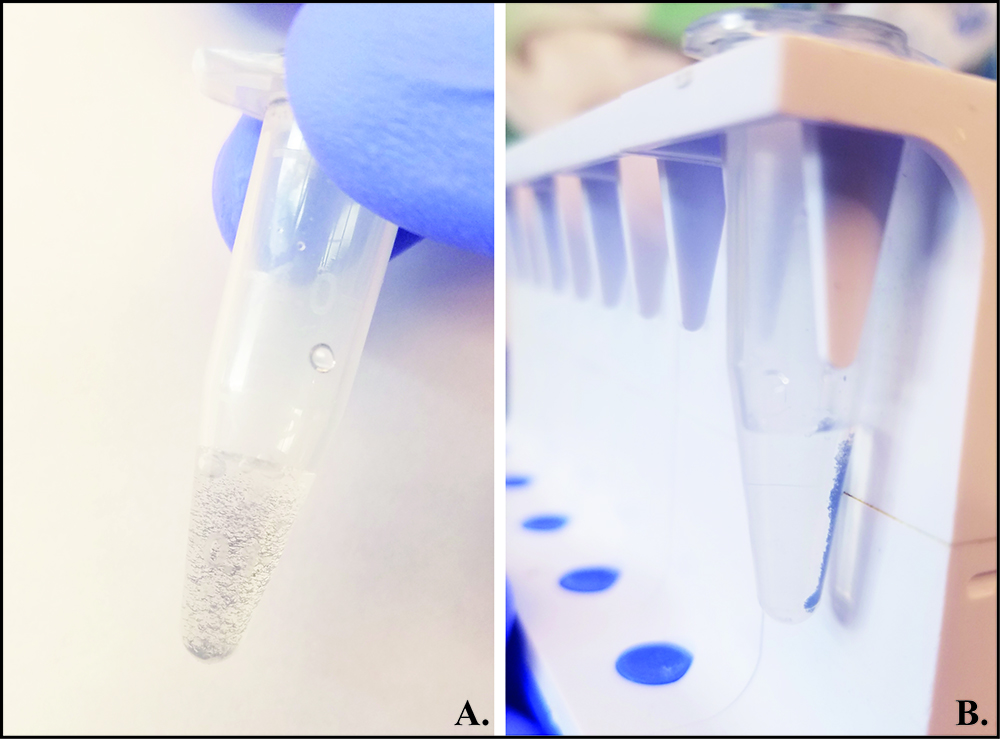
Figure 4. Magnetic bead washing procedure. A. Gently invert magnetic bead slurry with wash buffer to evenly mix and wash beads. B. Place the sample in magnetic rack and let stand until slurry appears clear because all magnetic beads have settled at the edge of the tube near the magnet. Carefully remove supernatant from beads and proceed with further washes or DNA elution step. This method removes loosely bound non-specific DNA fragments that contribute to background noise during sequencing. - Elute chromatin complexes from magnetic beads by adding 150 μl of freshly-prepared elution buffer (1% SDS-0.1 M NaHCO3; Recipe 5) and vortex very mildly for 15 min. Collect eluates by applying magnetic stand and transferring supernatant to clean tubes.
Note: To elute, set vortexer to a lowest speed and secure samples for continual vortexing for 15 min. Alternately, samples may be rotated for 15 min to achieve the same mild agitating effect. The elution step must be carried out at room temperature. - Repeat Step A17 for a total of 2 elution steps, and a total of 300 μl of eluted chromatin. Also, bring ChIP Input samples to a total volume of 300 μl, adding elution buffer if need be.
- Reverse cross-links by adding 18 μl of 5 M NaCl (0.3 M NaCl final concentration) and 1 μl RNase A (10 mg/ml), and incubating samples at 65 °C for at least 4 h or overnight.
Note: If the shorter reverse-crosslinking time is preferred (4 h), we recommend incubating samples in 65 °C water-bath to ensure efficient sample heating. If reversing crosslinks overnight, a hybridization oven set to 65 °C is sufficient. - After reverse-crosslinking is complete, precipitate samples by adding 1/15th volume 7.5 M ammonium acetate, and 2 volumes of 100% ethanol and incubating at -80 °C for at least 30 min.
Note: We recommend also adding a co-precipitant like GlycoBlue, 1 μl, to the samples at this stage to make visualizing the pellet easier. - Centrifuge at max speed in a 4 °C benchtop centrifuge for 15 min to pellet DNA. Semi-dry the pellet for approximately 5 min by leaving the tube open.
- Resuspend pellet in 100 μl 1x TE, and add 2 μl Proteinase K (10 mg/ml), and incubate samples at 45 °C for 2 h.
- Purify DNA using QIAGEN MinElute PCR Purification Kit, according to manufacturer’s instructions.
Note: Alternately, phenol-chloroform extraction may be performed at this step, and resulting DNA can be precipitated as in Step A20. Then add 1 ml 70% ethanol to each sample, centrifuging at max speed, 4 °C for 15 min, discarding ethanol, and repeating for a total of 3 washes. Let DNA pellets air dry briefly, then resuspend in 20-30 μl nuclease free water to proceed library preparation and sequencing. - Quantify DNA concentration using the Qubit High-Sensitivity DNA fluorometer, following manufacturer’s instructions. Briefly, Qubit dsDNA HS Reagent is mixed with an appropriate volume of Qubit dsDNA HS Buffer to make the Qubit dsDNA HS working solution. 1 μl of sample is mixed with 199 μl Qubit dsDNA HS working solution and the tube containing the sample is put into the machine for a reading that reveals the concentration of the original 1 μl sample. Once the concentration of DNA samples is determined, proceed to library preparation.
- Harvest approximately 20 million cells and centrifuge into a pellet. Aspirate culture medium, then resuspend in 5 ml ice-cold PBS containing protease inhibitors (1 μg/ml leupeptin, 1 μg/ml aprotinin, 1 μg/ml sodium fluoride, 1 μg/ml pepstatin, and 1 mM phenylmethylsulfonyl fluoride) and centrifuge again to wash cells.
- ChIP Library Preparation: NEXTflexTM ChIP-Seq Kit (Illumina Compatible) (Bioo Scientific Corp.)
For quantifying H4R3me2s enrichment on the viral KSHV genome, we used 10 ng of ChIP/Input DNA and chose to follow ‘Option 3’ of manufacturer’s instructions exactly for library preparation. A brief overview of the ChIP-Kit protocol is as follows:- End Repair of DNA samples, during this step, ends of DNA are blunted and 5’ phosphorylated.
- Gel-free size selection clean up, using magnetic beads DNA fragments below 300 bp and above 400 bp are discarded while fragments between 300-400 bp long are purified for the next step of library preparation.
- 3’ Adenylation, during this step 3’ ends are A-tailed.
- Adapter Ligation, during this step specific barcode DNA sequences are ligated to sequencing samples to enrich library concentration downstream and enable multiplexed sequencing runs.
- Clean up, during this step DNA sequences with barcodes ligated on are purified in preparation for the next step. This is important to remove excess adapter sequences or adapter dimers, which can unfavorably disrupt the next PCR step in the protocol.
- PCR amplification, during this step PCR is performed to enrich adapter-ligated products. To reduce PCR bias, the least number of cycles possible is recommended.
Note: For the library preparation PCR amplification, kit step F3, 16 repeat cycles were used for preparing the libraries used in our previous study, ‘KSHV encoded ORF59 modulates histone arginine methylation of the viral genome to promote viral reactivation’. As the kit recommends, the number of PCR cycles may be optimized depending on starting material concentration and quantity. - Clean up, this is the last clean-up and purification step of the library preparation protocol. We recommend using 23 μl of Resuspension Buffer for the final elution and collecting 20 μl of the final sample as opposed to the kit’s instructions (33 μl Resuspension Buffer, collecting 30 μl final sample) as this yields a more concentrated end product.
- End Repair of DNA samples, during this step, ends of DNA are blunted and 5’ phosphorylated.
- ChIP library validation and quantification
- To evaluate library quality, we suggest using the Agilent Bioanalyzer and running the High Sensitivity DNA chip. Properly prepared libraries yield one band of approximately 300-350 bp in size.
- To accurately quantify library sample concentration, we suggest using the KAPA Library Quantification Kit and following manufacturer’s instructions exactly. Accurate quantification of library concentration is essential for optimal clustering and maximizing the number of reads for each sample during the sequencing run.
- To evaluate library quality, we suggest using the Agilent Bioanalyzer and running the High Sensitivity DNA chip. Properly prepared libraries yield one band of approximately 300-350 bp in size.
- Sequencing
- High coverage sequencing means that each base is covered by a greater degree of aligned sequence reads, and thus the base calls assume a higher degree of confidence. According to Illumina’s instructions, coverage of approximately 100x is recommended for ChIP-Seq applications. Next-generation sequencing is a highly-standardized, validated process and we recommend following the prescribed procedures.
- For further information on Next Generation sequencing depth and coverage, manufacturer’s recommendations may be followed. Illumina’s recommendations can be accessed at https://www.illumina.com/science/education/sequencing-coverage.html for a detailed description of these important considerations.
- High coverage sequencing means that each base is covered by a greater degree of aligned sequence reads, and thus the base calls assume a higher degree of confidence. According to Illumina’s instructions, coverage of approximately 100x is recommended for ChIP-Seq applications. Next-generation sequencing is a highly-standardized, validated process and we recommend following the prescribed procedures.
Data analysis
- FastQ data generated from the NextSeq (Illumina, San Diego, CA) was annotated and the sequence reads were analyzed using CLC workbench 10.0.1 (QIAGEN, Germantown, MD). The sequence reads were downloaded from the Illumina Base Space repository that the Nevada Genomics Center uploaded. These files can be accessed through internet imported to the sequences analysis software, CLC Workbench in our case using the ‘Import Illumina Reads’ option.
- The reads obtained from input, control IgG antibody and specific antibody samples were mapped to the KSHV genome using ‘map reads to the reference’ operation of the CLC workbench.
- Mapped reads were subjected for identifying the enriched peaks in ChIP samples after comparing with the respective input samples peaks using the ‘ChIP-seq’ tool of the CLC Workbench. Relative enrichment is presented as a peak score calculated using minimum peak-calling P-value of 0.05. (Please refer to ‘KSHV encoded ORF59 modulates histone arginine methylation of the viral genome to promote viral reactivation’ by Strahan et al., 2017, publicly available at PLoS Pathogens).
- One challenge unique to assessing the relative abundance of this chromatin mark on the KSHV viral genome was that despite of high sequencing depth for each sample, only about 1% of the total ChIP-Seq reads mapped to the viral genome. This illustrates the importance of deep sequencing coverage for evaluating viral chromatin conformation.
Recipes
- Cell lysis buffer, 50 ml
500 μl 0.5 M PIPES, pH 8.0
2.5 ml 1.7 M KCl
2.5 ml 10% NP-40
Sterile ddH2O to 50 ml, store at room temperature
Add protease inhibitors fresh just before use - ChIP dilution buffer, 50 ml
125 μl 0.5 M EDTA pH 8.0
835 μl 1 M Tris-HCl pH 8.1
1.67 ml 5 M NaCl
Sterile ddH2O to 50 ml, store at room temperature
Add protease inhibitors fresh just before use - ChIP Magnetic A + G beads
- Combine 300 μl Protein A magnetic beads with 300 μl Protein G magnetic beads and wash with 1x TE 3 times
Note: Place tubes against a magnetic stand, remove supernatant, remove from magnetic stand, add 1 ml TE, repeat. - Resuspend bead slurry in 1x TE with 1 mg/ml sonicated salmon sperm DNA, and 1 mg/ml BSA and rotate overnight 4 °C
Note: Sonicated salmon sperm DNA is used as a blocking agent to reduce non-specific background DNA binding. - Next day, wash beads 1 time with TE, then resuspend in a final volume of 500 μl TE, beads are now ready for ChIP assay
- Combine 300 μl Protein A magnetic beads with 300 μl Protein G magnetic beads and wash with 1x TE 3 times
- ChIP Low Salt Wash, 50 ml
5 ml 10% Triton X-100
500 μl 10% SDS
200 μl 0.5 M EDTA pH 8.0
1 ml 1 M Tris-HCl pH 8.1
1.5 ml 5 M NaCl
Sterile ddH2O to 50 ml, store at room temperature
Add protease inhibitors fresh just before use and store on ice
Discard after washing steps - ChIP elution buffer
1 ml 10% SDS
1 ml 1 M NaHCO3 (freshly prepared)
Sterile ddH2O to 10 ml, store at room temperature and discard excess after use
Acknowledgments
This work was supported by the National Institute of Health (CA174459 and AI105000). This protocol was adapted to determine H4R3me2s enrichment on the KSHV genome from a combination of previous ChIP-Seq protocols and the LowCell number ChIP-protocol from Diagenode Inc. The authors would like to acknowledge the cooperation and help from the Nevada Genomics Center (University of Nevada, Reno, USA) in performing the Illumina sequencing runs.
Conflict of Interest: Authors declare no conflict of interests and competing interests, which may impact design and implementation of the protocol described above.
References
- Aneja, K. K. and Yuan, Y. (2017). Reactivation and lytic replication of Kaposi's sarcoma-associated herpesvirus: an update. Front Microbiol 8: 613.
- Bannister, A. J. and Kouzarides, T. (2011). Regulation of chromatin by histone modifications. Cell Res 21(3): 381-395.
- Bedford, M. T. and Clarke, S. G. (2009). Protein arginine methylation in mammals: who, what, and why. Mol Cell 33: 1-13.
- Bernstein, B. E., Meissner, A. and Lander, E. S. (2007). The mammalian epigenome. Cell 128(4): 669-681.
- Cesarman, E., Chang, Y., Moore, P. S., Said, J. W. and Knowles, D. M. (1995). Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med 332(18): 1186-1191.
- Chang, Y., Cesarman, E., Pessin, M. S., Lee, F., Culpepper, J., Knowles, D. M. and Moore, P. S. (1994). Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266(5192): 1865-1869.
- Di Lorenzo, A. and Bedford, M. T. (2011). Histone arginine methylation. FEBS Lett 585(13): 2024-2031.
- Gunther, T. and Grundhoff, A. (2010). The epigenetic landscape of latent Kaposi sarcoma-associated herpesvirus genomes. PLoS Pathog 6(6): e1000935.
- Nakamura, H., Lu, M., Gwack, Y., Souvlis, J., Zeichner, S. L. and Jung, J. U. (2003). Global changes in Kaposi’s sarcoma-associated virus gene expression patterns following expression of a tetracycline-inducible Rta transactivator. J Virol 77: 4205-4220.
- Pahlich, S., Zakaryan, R. P. and Gehring, H. (2006). Protein arginine methylation: Cellular functions and methods of analysis. Biochim Biophys Acta 1764:1890-1903.
- Park, P. J. (2009). ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet 10(10): 669-680.
- Purushothaman, P., Dabral, P., Gupta, N., Sarkar, R. and Verma, S. C. (2016). KSHV genome replication and maintenance. Front Microbiol 7: 54.
- Purushothaman, P., Uppal, T. and Verma, S. C. (2015). Molecular biology of KSHV lytic reactivation. Viruses 7(1): 116-153.
- Russo, J. J., Bohenzky, R. A., Chien, M. C., Chen, J., Yan, M., Maddalena, D., Parry, J. P., Peruzzi, D., Edelman, I. S., Chang, Y. and Moore, P. S. (1996). Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc Natl Acad Sci U S A 93: 14862-14867.
- Soulier, J., Grollet, L., Oksenhendler, E., Cacoub, P., Cazals-Hatem, D., Babinet, P., d'Agay, M. F., Clauvel, J. P., Raphael, M., Degos, L. and et al. (1995). Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 86(4): 1276-1280.
- Strahan, R. C., McDowell-Sargent, M., Uppal, T., Purushothaman, P. and Verma, S. C. (2017). KSHV encoded ORF59 modulates histone arginine methylation of the viral genome to promote viral reactivation. PLoS Pathog 13(7): e1006482.
- Toth, Z., Brulois, K. and Jung, J. U. (2013). The chromatin landscape of Kaposi's sarcoma-associated herpesvirus. Viruses 5(5): 1346-1373.
- Toth, Z., Brulois, K., Lee, H. R., Izumiya, Y., Tepper, C., Kung, H. J. and Jung, J. U. (2013). Biphasic euchromatin-to-heterochromatin transition on the KSHV genome following de novo infection. PLoS Pathog 9(12): e1003813.
- Toth, Z., Maglinte, D. T., Lee, S. H., Lee, H. R., Wong, L. Y., Brulois, K. F., Lee, S., Buckley, J. D., Laird, P. W., Marquez, V. E. and Jung, J. U. (2010). Epigenetic analysis of KSHV latent and lytic genomes. PLoS Pathog 6(7): e1001013.
- Uppal, T., Banerjee, S., Sun, Z., Verma, S. C. and Robertson, E. S. (2014). KSHV LANA--the master regulator of KSHV latency. Viruses 6(12): 4961-4998.
- Uppal, T., Jha, H. C., Verma, S. C. and Robertson, E. S. (2015). Chromatinization of the KSHV genome during the KSHV life cycle. Cancers (Basel) 7(1): 112-142.
- Zhang, T., Cooper, S. and Brockdorff, N. (2015). The interplay of histone modifications - writers that read. EMBO Rep 16(11): 1467-1481.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Strahan, R. C., Hiura, K. S. and Verma, S. C. (2018). Quantifying Symmetrically Methylated H4R3 on the Kaposi’s Sarcoma-associated Herpesvirus (KSHV) Genome by ChIP-Seq. Bio-protocol 8(6): e2781. DOI: 10.21769/BioProtoc.2781.
Category
Systems Biology > Epigenomics > Sequencing > ChIP-seq
Microbiology > Microbe-host interactions > Virus
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.