- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Reduced Representation Bisulfite Sequencing in Maize


Published: Vol 8, Iss 6, Mar 20, 2018 DOI: 10.21769/BioProtoc.2778 Views: 9309
Reviewed by: Renate WeizbauerAnnis Elizabeth RichardsonAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Oct 2013
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
DNA methylation is an epigenetic modification that regulates plant development (Law and Jacobsen, 2010). Whole genome bisulfite sequencing (WGBS) is a state-of-the-art method for profiling genome-wide methylation patterns with single-base resolution (Cokus et al., 2008). However, for an organism with a large genome, e.g., the 2.1 Gb genome of maize, WGBS may be very expensive. Reduced representation bisulfite sequencing (RRBS) has been developed in mammalian studies (Smith et al., 2009). By digesting the genome with MspI with a size selection range of approximately 40-220 bp, CG-rich regions covering only ~1% of the human genome can be specifically sequenced. However, unlike mammalian genomes, plant genomes do not exhibit clear CpG islands. Therefore the original RRBS protocol is not suitable for plants. Accordingly, we developed an in silico pipeline to select specific enzymes to generate a region of interest (ROI)-enriched, e.g., promoter-enriched, reduced representation genome in plants (Hsu et al., 2017). By digesting the maize genome with MseI and selecting 40-300 bp segments, we sequenced about one-fourth of the maize genome while preserving 84.3% of the promoter information. The protocol has been successfully established in maize and can be broadly used in any genome. Our in silico pipeline is combined with the RRBS library preparation protocol, allowing for the computational analysis and experimental validation.
Keywords: Bisulfite sequencingBackground
DNA methylation is a heritable epigenetic modification that plays an important role in many developmental processes of animals, plants and fungi by regulating gene expression and the chromatin structure (Law and Jacobsen, 2010). WGBS is a genome-wide scale method for profiling DNA methylation at single-base resolution, although high sequencing costs are required to achieve sufficient coverage (Cokus et al., 2008). In mammals, RRBS has been developed to specifically sequence CG-dense regions, e.g., CpG islands (Smith et al., 2009). In this protocol, we aimed to adapt RRBS for plants. To be specific, we developed an in silico pipeline (Figure 1) to performed enzyme selection by targeting specific genome regions to generate RRBS methylomes and provided an experimental validation protocol.
Our method has been successfully established in the maize genome, one of the major global crops, which has a 2.1 Gb genome (Hsu et al., 2017). In addition to its large size, 85% of the maize genome consists of various repetitive sequences (Schnable et al., 2009). This feature could cause multiple mapping, i.e., many short reads from sequencing, which need to be discarded. These characteristics make targeted bisulfite sequencing (BS-seq) more cost-effective. mCHH islands were found to be located upstream of transcription start sites (TSSs) with higher methylation level (Gent et al., 2013; Li et al., 2015). We therefore aimed to perform promoter-enriched RRBS in maize, and an in silico pipeline was developed for enzyme selection.
Our in silico pipeline currently has 85 pre-installed restriction enzymes. Users can easily append more enzymes. A genome FASTA file, refFlat annotation file, and repeat gff3 annotation file are the required input files to run this pipeline. ROIs, including promoters, exons, introns, splicing sites, repeats, UTRs and intergenic regions are pre-selected for enrichment analysis. As soon as the input files are prepared, users can run our pipeline to select ideal enzymes by typing simple commands. Users can also verify the prediction by performing RRBS library construction following the experimental protocol provided and performing sequencing.
Figure 1. Flowchart of maize RRBS in silico pipeline
Materials and Reagents
- Pipette tips
Note: Low retention tips are recommended. - 1.5 ml microcentrifuge tubes (Eppendorf, catalog number: 0030108051 )
- PCR tubes (Thermo Fisher Scientific, Applied BiosystemsTM, catalog number: A30588 )
- Clean razor blade
- Qubit dsDNA BR Assay Kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: Q32850 ) (used to quantify double strand DNA in combination with Qubit Fluorometer)
- MseI (New England Biolabs, catalog number: R0525S ) (used to digest maize genomic DNA, CutSmart Buffer is included)
Note: The enzyme could be replaced according to the in silico enzyme selection result in Procedure A. - Agarose
Note: For the gel size selection, we recommend low melting point agarose. - TBE buffer (Thermo Fisher Scientific, InvitrogenTM, catalog number: 15581044 )
- AMPure XP (Beckman Coulter, catalog number: A63881 )
- Absolute ethanol
Note: To dilute, use DNase/RNase-free distilled water. - 1 M Tris-HCl (Thermo Fisher Scientific, InvitrogenTM, catalog number: 15568025 )
Note: To dilute, use DNase/RNase-free distilled water. - NEBuffer 2 (New England Biolabs, catalog number: B7002S )
- Klenow Fragment (3’→5’ exo-) (New England Biolabs, catalog number: M0212S ) (used to end-repair and A-tail DNA)
- T4 DNA Ligase (New England Biolabs, catalog number: M0202S , T4 DNA ligation buffer is included) (used to ligate sequencing adaptors)
- 100-bp DNA ladder
- QIAquick Gel Extraction Kit (QIAGEN, catalog numbers: 28704 , 28706 )
- EpiTect Fast Bisulfite Conversion Kit (QIAGEN, catalog number: 59824 )
Note: Keep this kit at an appropriate temperature according to the manufacturer’s guide. - TruSeq DNA LT Sample Prep Kit (Illumina, catalog number: FC-121-2001 )
- PfuTurbo DNA Polymerase (Agilent Technologies, catalog number: 600250 )
- DNase/RNase-free distilled water (Thermo Fisher Scientific, InvitrogenTM, catalog number: 10977015 )
- Deoxynucleotide (dNTP) Solution Set (New England Biolabs, catalog number: N0446S )
- MinElute PCR Purification Kit (QIAGEN, catalog number: 28004 )
- 10 mM dNTP mix (see Recipes)
- 5x RRBS dNTP mix (see Recipes)
Equipment
- Pipettes:
- Thermo Fisher Scientific, Thermo ScientificTM, model: P5000, catalog number: 4641110N
- Thermo Fisher Scientific, Thermo ScientificTM, model: P1000, catalog number: 4641100N
- Thermo Fisher Scientific, Thermo ScientificTM, model: P300, catalog number: 4641090N
- Thermo Fisher Scientific, Thermo ScientificTM, model: P200, catalog number: 4641080N
- Thermo Fisher Scientific, Thermo ScientificTM, model: P100, catalog number: 4641070N
- Thermo Fisher Scientific, Thermo ScientificTM, model: P20, catalog number: 4641060N
- Thermo Fisher Scientific, Thermo ScientificTM, model: P10, catalog number: 4641030N
- Thermo Fisher Scientific, Thermo ScientificTM, model: P2, catalog number: 4641010N
- Thermo Fisher Scientific, Thermo ScientificTM, model: P5000, catalog number: 4641110N
- Qubit 2.0 Fluorometer (Thermo Fisher Scientific, Invitrogen, model: Qubit® 2.0 Fluorometer , catalog number: Q32866)
- Heating block (Eppendorf, model: ThermoMixer comfort , catalog number: 5355000011)
- Incubator (Only Science, Firstek, model: S300 )
- Thermocycler
- Thermo Fisher Scientific, Applied BiosystemsTM, model: GeneAmpTM PCR System 9700 , catalog number: 4413750
- Bio-Rad Laboratories, model: T100TM Thermal Cycler, catalog number: 1861096
- Thermo Fisher Scientific, Applied BiosystemsTM, model: GeneAmpTM PCR System 9700 , catalog number: 4413750
- Centrifuge
- Eppendorf, model: Centrifuge MiniSpin® plus , catalog number: 5453000011
- Eppendorf, model: Centrifuge 5424 , catalog number: 5424000410
- GYROZEN, model: 1730R
- Eppendorf, model: Centrifuge MiniSpin® plus , catalog number: 5453000011
- Magnetic stand
Software
- System requirements: Linux/Unix or Mac OS, python 2.7+, R 3.2.0
- Python and R scripts and a tutorial are available at: https://gitlab.com/fmhsu0114/maize_RRBS
Procedure
The complete pipeline contains 1. in slico enzyme selection, followed by 2. RRBS library preparation. The resulted library is to be sequenced to generate DNA methylation profiles at genomic regions of interest.
- In silico enzyme selection
- Clone the GitLab project from: https://gitlab.com/fmhsu0114/maize_RRBS.
- Download the genome FASTA file of your interest, i.e., maize_genome.fa, and use the following command to build the genome database:
$ python Build_Gnome.py AGPv3.fa
2 output files (genome.shelve and log-genome.txt) will be generated. - Download the annotation refFlat file, i.e., refFlat.txt, and use the following command to build the gene annotation database:
$ python Build_refgene_shelve.py refFlat.txt log-genome.txt
1 output file (refgenes.shelve) will be generated. - Download the annotation gff3 file and grep ‘repeats’ into an individual file, i.e., repeats.gff3, and use the following command to build the repeat annotation database:
$ python Build_repeats_Gnome.py repeats.gff3, log-genome.txt
1 output file (repeat.shelve) will be generated. - Build the genome CG, CHG and CHH sites database with the following command:
$ python Build_CG_CHG_CHH_Gnome.py genome.shelve log-genome.txt
1 output file (CG_CHG_CHH.shelve) will be generated. - Perform in silico digestion with the provided enzyme list in the GitLab project (enzyme_cutting_sites.txt) with the following command:
$ python Enzyme_digestione.py
fragment_info (per MSRE and per size range) for each enzyme and a Summary-enzyme-coverage.txt file will be generated. - Place the output files from 1.6 into one directory ./fragment/ and run the enrichment analysis with the following command:
$ python RRBS_fragments_enrichment.py repeats.shelve, refgenes.shelve
log2_fold_enrichment (per MSRE and per size range) files will be generated (see Table 1).
Table 1. Fold enrichment values in log2 scale of the MseI 40-300-bp fragments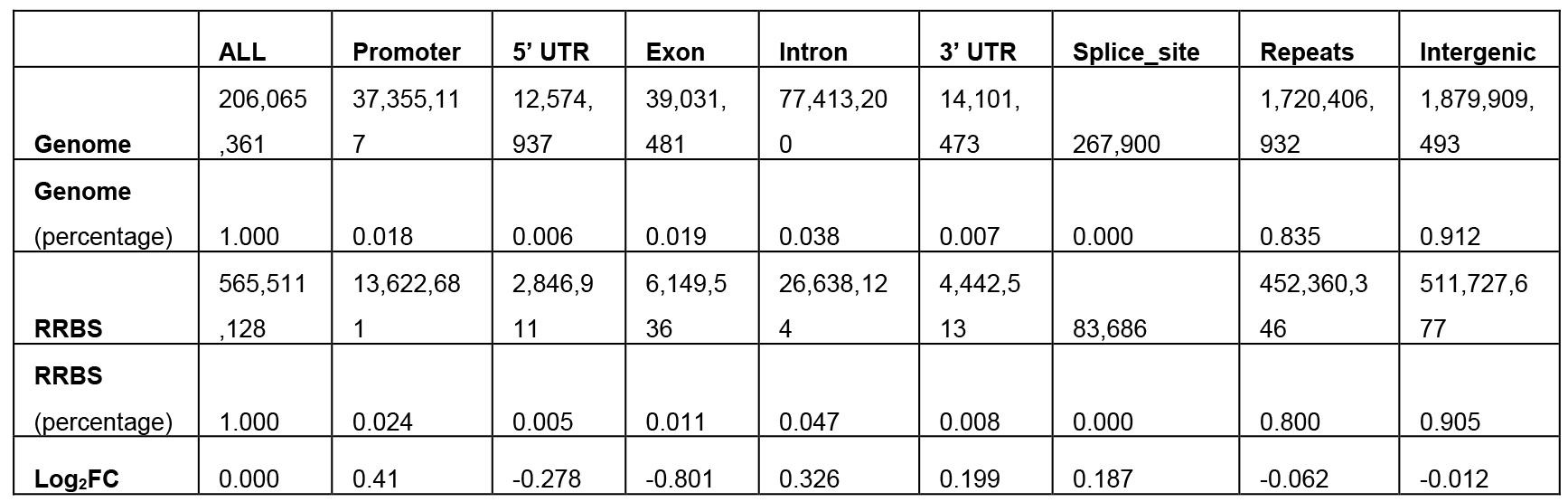
- Move all of the log2_fold_enrichment files into one directory ./output/ and plot the enrichment bar charts with the following command:
$ Rscript Barchart_enrichment.R
See Figure 2 to Figure 4 as examples.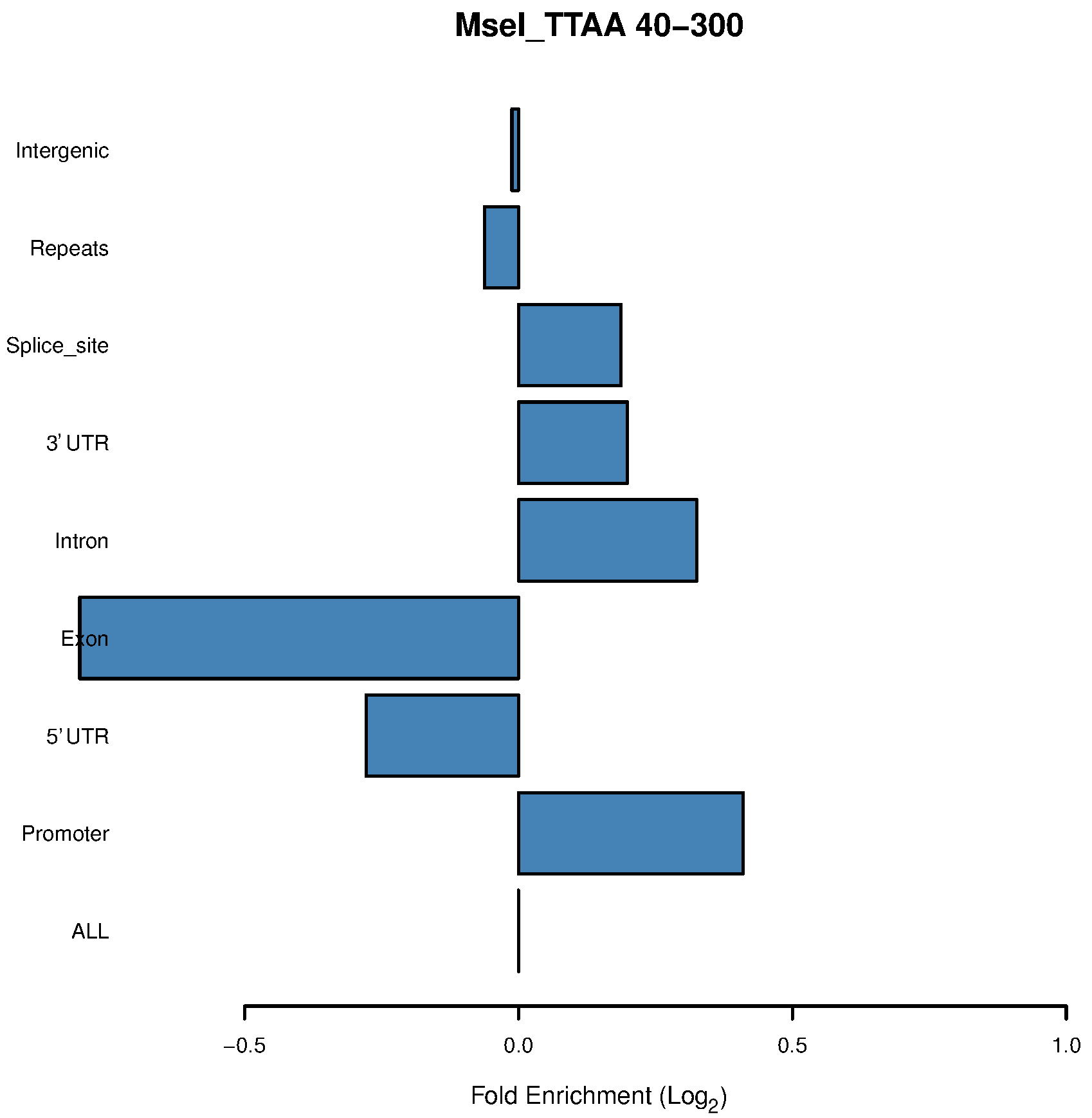
Figure 2. Bar chart of the MseI 40-300-bp fragments shows enrichment in promoters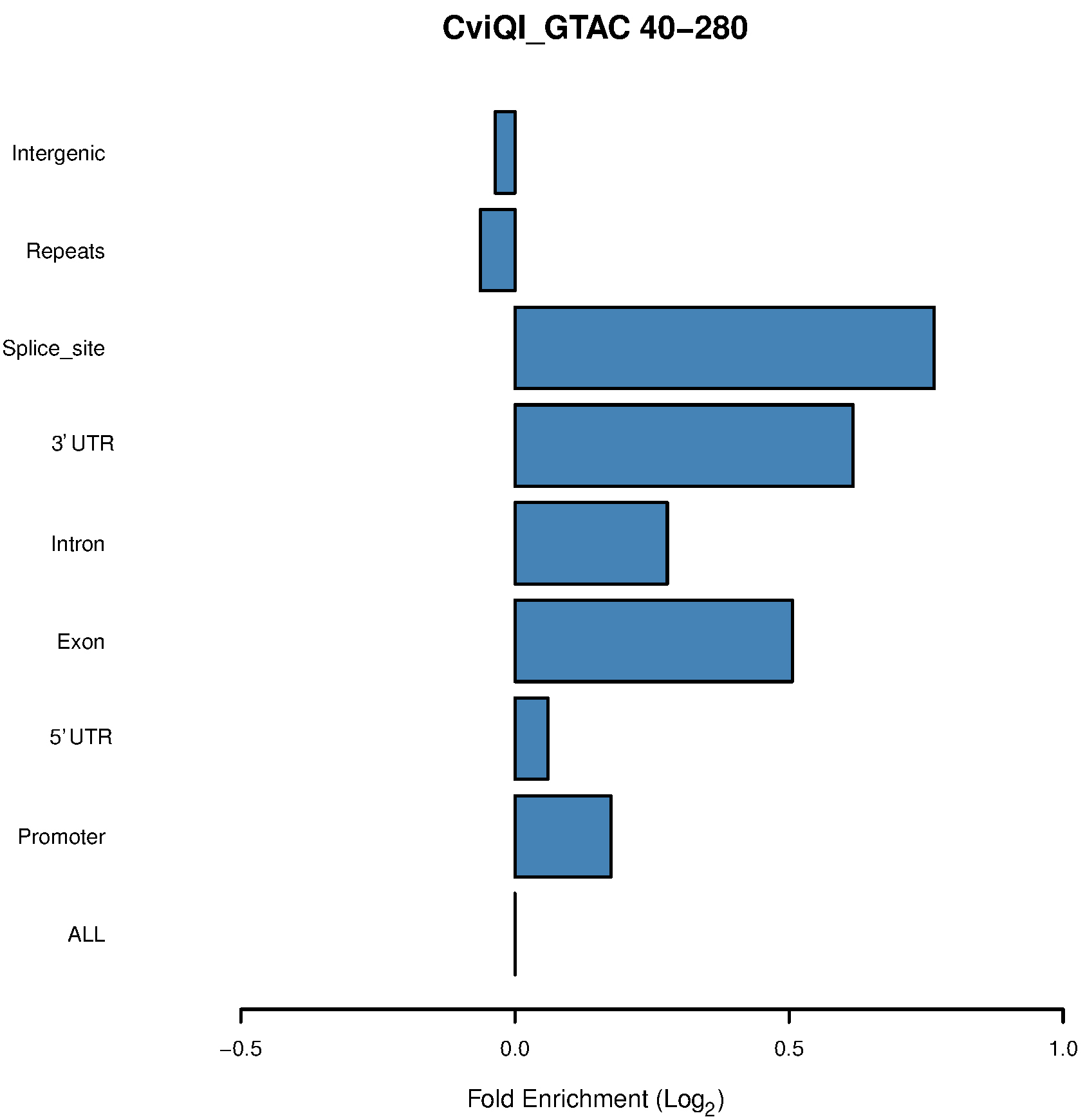
Figure 3. Bar chart of the CviQI 40-280-bp fragments shows enrichment in exons, introns, UTRs and splice sites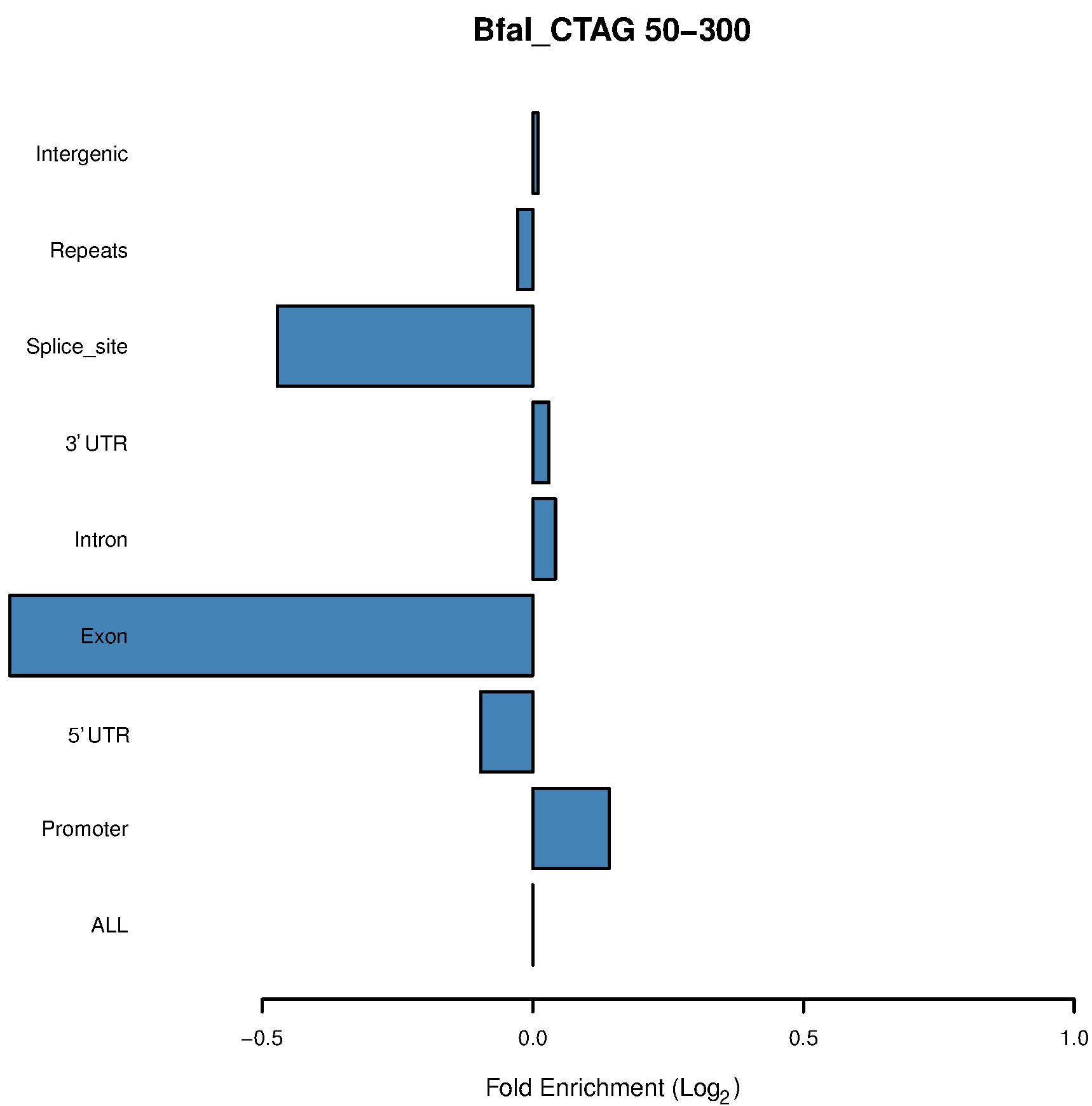
Figure 4. Bar chart of the BfaI 50-300-bp fragments shows enrichment in promoters - Use the following command to calculate the covered promoter and genebody region:
$ python RRBS_fragments_enrichment_gene_level_per_MRE_Size.py -i Enzyme -s size_range -c 1
This script has 3 parameters:
-i: restriction enzyme
-s: size selection range, e.g., 40-280
-c: number of cytosines per promoter/genebody to be included
An output file, Fragment-Enrichment-genebody-pmt-Enzyme-size_range-1.txt, will be generated (Table 2 and Table 3). This file contains the percentages of the promoters and genebody covered in the reduced representation (RR) genome.
Note: In Table 2 and Table 3, Pct refers to percentage; Pmt refers to promoter.
Table 2. Promoter and genebody covered in the MseI 40-300-bp fragments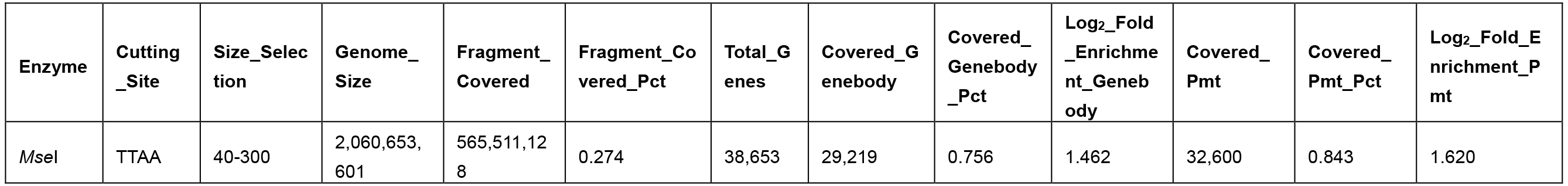
Table 3. Promoter and genebody covered in the BfaI 50-300-bp fragments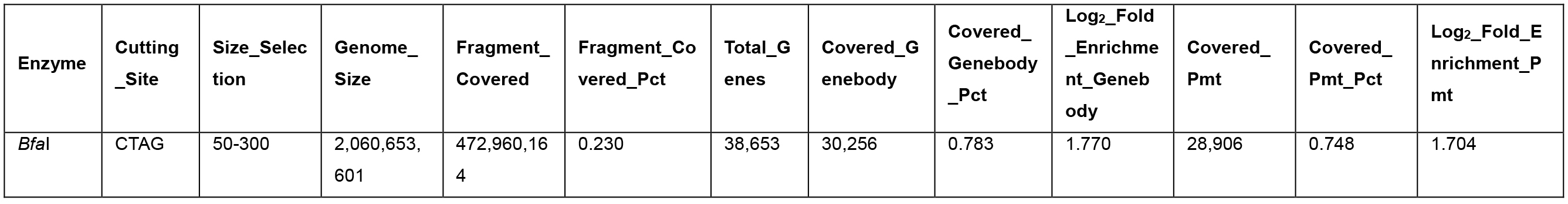
- Clone the GitLab project from: https://gitlab.com/fmhsu0114/maize_RRBS.
- RRBS library preparation
- Use Qubit Fluorometric Quantification with Qubit dsDNA BR assay to quantify maize genomic DNA according to the manufacturer’s instructions.
Note: To acquire high quality double strand genomic DNA, we recommend spin column-based DNA extraction kit, e.g., QIAGEN DNeasy Plant Mini kit. If Pheno-Chloroform method is used, we suggest another round of spin-column based purification. - Aliquot 1 μg of maize genomic DNA into a 1.5 ml tube, add 5 μl of 10x CutSmart Buffer, 1 μl of MseI and ddH2O to a final volume of 50 μl. Mix carefully with a pipette and incubate at 37 °C on the heating block for 4 h.
Note: MseI used here is designed for promoter-enriched RRBS in maize. For different genomes (organisms), researchers should conduct ‘in silico enzyme selection’ and replace MseI with the predicted enzyme, and modify the digestion time, buffer and temperature according to the manufacturer’s instructions. - [Optional] Take 5 μl for gel electrophoresis (100 ng, 1.5%, 0.5x TBE) and add 5 μl ddH2O to the enzyme digestion product.
Note: This step is to confirm the enzyme digestion efficiency. - Post-digestion AMPure beads purification
- Add 150 μl of well-inverted AMPure XP beads solution to 50 μl of DNA, vortex for 10 sec, briefly centrifuge and incubate at RT for 5 min.
Note: When vortexing solutions with AMPure XP beads, avoid high vortexing strength causing bubbles. After vortexing, use microcentrifuge to spin down quickly to remove liquids from the lid and the wall of the tube. - Place the tube on the magnetic stand for 3 min.
- Remove the supernatant.
- Add 200 μl of 80% EtOH without disturbing the beads and incubate at RT for 30 sec. Remove the EtOH.
Note: Do not remove the tube from the magnetic stand. - Repeat the washing Step B4d.
- Air-dry the beads for 5 min.
- Remove the tube from the magnetic stand.
- Add 32 μl of 10 mM Tris-HCl and pipet up and down to mix thoroughly. Incubate at RT for 2 min.
- Place the tube on the magnetic stand for 3 min.
- Transfer the supernatant to a PCR tube.
- Add 150 μl of well-inverted AMPure XP beads solution to 50 μl of DNA, vortex for 10 sec, briefly centrifuge and incubate at RT for 5 min.
- End repair and A-tailing
- Add 6 μl of 10x NEBuffer 2, 12 μl of 5x RRBS dNTP mix and 3 μl of 3’→5’ Klenow exo- to the purified DNA and add ddH2O to a final volume of 60 μl.
- Incubate using thermocycler at 30 °C for 20 min and 37 °C for 20 min, then place the tube on ice.
- Add 6 μl of 10x NEBuffer 2, 12 μl of 5x RRBS dNTP mix and 3 μl of 3’→5’ Klenow exo- to the purified DNA and add ddH2O to a final volume of 60 μl.
- DNA purification with AMPure XP beads
- Add 180 μl of well-inverted AMPure XP beads to the 60 μl of DNA, vortex for 10 sec, briefly centrifuge and incubate at RT for 5 min.
- Place the tube on the magnetic stand for 3 min.
- Remove the supernatant.
- Add 200 μl of 80% EtOH without disturbing the beads and incubate at RT for 30 sec. Remove the EtOH.
Note: Do not remove the tube from the magnetic stand. - Repeat the washing Step B6d.
- Air-dry the beads for 5 min.
- Remove the tube from the magnetic stand.
- Add 24 μl of 10 mM Tris-HCl and pipet up and down to mix thoroughly. Incubate at RT for 2 min.
- Place the tube on the magnetic stand for 3 min.
- Transfer the supernatant to a PCR tube.
- Add 180 μl of well-inverted AMPure XP beads to the 60 μl of DNA, vortex for 10 sec, briefly centrifuge and incubate at RT for 5 min.
- Ligate with a TruSeq methylated Illumina adaptor
- Add 3 μl of 10x T4 DNA ligation buffer, 2 μl of adaptor and 1 μl of T4 DNA ligase to the 24 μl end-repaired and A-tailed DNA to a final volume of 30 μl.
- Incubate at 16 °C overnight in a thermocycler.
- Add 3 μl of 10x T4 DNA ligation buffer, 2 μl of adaptor and 1 μl of T4 DNA ligase to the 24 μl end-repaired and A-tailed DNA to a final volume of 30 μl.
- Gel size selection (Figure 5)
- Load all the adapter-ligated DNA to 1.5% 0.5x TBE gel (15 μl per well, in total 2 wells) for gel electrophoresis with a 100-bp DNA ladder.
- Use a clean razor blade for each sample to cut 40-300-bp fragments and purify the DNA using the QIAGEN Qiaquick Gel Extraction Kit following the manufacturer’s instructions. Elute DNA twice with a total volume of 34 μl of EB buffer. Transfer the solution to a PCR tube. Take 1 μl for Qubit quantification (optional).
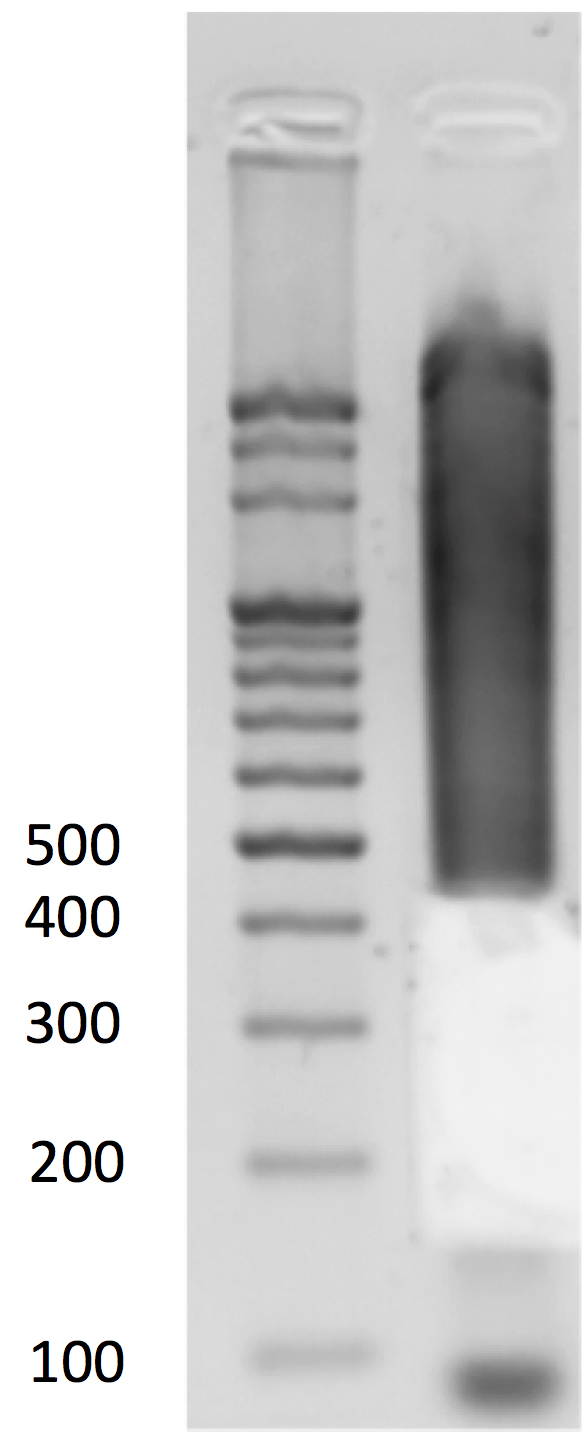
Figure 5. Gel size selection - Load all the adapter-ligated DNA to 1.5% 0.5x TBE gel (15 μl per well, in total 2 wells) for gel electrophoresis with a 100-bp DNA ladder.
- Bisulfite conversion
Note: Confirm that the reagents in the EpiTect Fast Bisulfite Conversion Kit are ready to use.- Add RNase-free water to the DNA solution to a final volume of 40 μl, mix with a pipette and aliquot 20 μl to another PCR tube.
- Add 35 μl of DNA Protect Buffer and 85 μl of a Bisulfite Solution to each tube to a final volume of 140 μl and mix thoroughly with gentle pipetting.
- Run in a thermocycler with the following program:
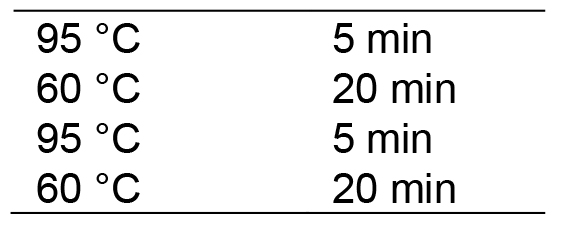
Then hold at 20 °C. Lid temperature: 100 °C.
- Add RNase-free water to the DNA solution to a final volume of 40 μl, mix with a pipette and aliquot 20 μl to another PCR tube.
- Briefly centrifuge the PCR tubes containing the bisulfite reaction with microcentrifuge to remove the liquid from the tube lid and transfer to clean 1.5 ml microcentrifuge tubes. For each tube, add 310 μl of freshly prepared Buffer BL containing 10 μg/ml carrier RNA, mix thoroughly with pipetting and then centrifuge briefly.
- Add 250 μl of 96-100% EtOH, gently pipet for 10 sec and then centrifuge briefly.
- Transfer all of the solution to MinElute DNA spin columns with collection tubes. Centrifuge at maximum speed for 1 min and discard the flow-through.
- Washing
Add 500 μl Buffer BW to each spin column, centrifuge at maximum speed for 1 min and discard the flow-through. - Desulfonation
- Add 500 μl of Buffer BD to each spin column and incubate for 15 min at RT.
- Centrifuge at maximum speed for 1 min and discard the flow-through.
- Add 500 μl Buffer BW to each spin column, centrifuge at maximum speed for 1 min and discard the flow-through.
- Repeat the washing Step B14c.
- Add 250 μl of 96-100% EtOH to each spin column and centrifuge at maximum speed for 1 min.
- Place each spin column into a clean 1.5 ml microcentrifuge tube, open the lid and incubate using a 60 °C heating block to evaporate the liquid.
- Elute the DNA by adding 15 μl of Buffer EB onto the center of the spin column membrane, close the lid and incubate at RT for 1 min.
- Centrifuge at 15,000 x g for 1 min.
- Add 500 μl of Buffer BD to each spin column and incubate for 15 min at RT.
- PCR amplification
- Add 2.5 μl Illumina TruSeq PCR primer cocktail, 0.5 μl of 10 mM dNTP, 5 μl of 10x Pfu Turbo Buffer, 0.5 μl of Pfu Turbo DNA polymerase and 26.5 μl of ddH2O to the eluted DNA solution to a final volume of 50 μl. Mix thoroughly and centrifuge briefly.
- Run in a thermocycler with the following program:
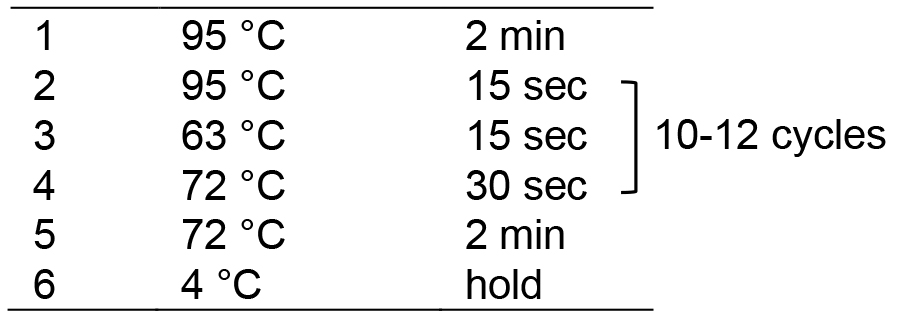
- Add 2.5 μl Illumina TruSeq PCR primer cocktail, 0.5 μl of 10 mM dNTP, 5 μl of 10x Pfu Turbo Buffer, 0.5 μl of Pfu Turbo DNA polymerase and 26.5 μl of ddH2O to the eluted DNA solution to a final volume of 50 μl. Mix thoroughly and centrifuge briefly.
- DNA purification with AMPure XP beads
- Add 150 μl of well-inverted AMPure XP beads to the 50 μl of DNA, vortex for 10 sec, briefly centrifuge and incubate at RT for 5 min.
- Place the tube on the magnetic stand for 3 min.
- Remove the supernatant.
- Add 200 μl of 80% EtOH without disturbing the beads and incubate at RT for 30 sec. Remove the EtOH.
Note: Do not remove the tube from the magnetic stand. - Repeat the washing Step B16d.
- Air-dry the beads for 5 min.
- Remove the tube from the magnetic stand.
- Add 22 μl of 10 mM Tris-HCl and pipet up and down to mix thoroughly. Incubate at RT for 2 min.
- Place the tube on the magnetic stand for 3 min.
- Transfer the supernatant to a 1.5 ml microcentrifuge tube.
- Add 150 μl of well-inverted AMPure XP beads to the 50 μl of DNA, vortex for 10 sec, briefly centrifuge and incubate at RT for 5 min.
- Quantify the final library concentration with the Qubit assay.
- Sequence the RRBS library with Illumina paired-end sequencing. Any regular length of pair-end reads, e.g., 100-250 bp, is fine.
- Use Qubit Fluorometric Quantification with Qubit dsDNA BR assay to quantify maize genomic DNA according to the manufacturer’s instructions.
Data analysis
- Our in silico pipeline creates a table and bar chart for each enzyme with its associated range of size selection. Figure 1 is a flowchart of this pipeline, and users could see input and output files of each step. Figure 2 is a bar chart showing the enrichment of each potential ROI from approximately 40-300 bp size-selected MseI-digested DNA. The X-axis is in log2 scale, so values > 0 can be considered to be an enrichment of these MseI-digested DNA fragments. Table 1 includes the values used to plot Figure 2. From Figure 2 and Table 1, we can see that the MseI-digested 40-300-bp fragments are enriched in promoters. As an alternative, Figure 3 shows the result from another enzyme (CviQI) 40-280 bp, with fragments more enriched in exons, splice sites, introns and UTRs.
- Different enzymes produce similar enrichment values. Figure 2 and Figure 4 suggest that MseI 40-300-bp and BfaI 50-300-bp fragments are enriched in promoters. Table 2 and Table 3 show the statistics of the promoters and genebody percentages covered. For the MseI RR genome, by sequencing 566 Mbp of the 2.1 Gb genome, 84.3% of promoters are included. As for the BfaI RR genome, sequencing 23.0% of the genome includes 74.8% of the promoters. Users need to determine the tradeoff between the cost of 4.4% of the genome and 8.5% of the promoters.
Conclusion
- We present an in silico pipeline to perform ROI-directed RRBS in maize with an experimentally verifiable prediction. The most critical steps for users to run our pipeline are: 1) Preparing the input files; 2) Choosing an optimal enzyme to show clear enrichment of the selected ROIs (see Figure 2).
- Input files can often be downloaded from public databases, such as Ensembl (Zerbino et al., 2017). It is important to note that our pipeline requires an individual repeat annotation file in .gff3 format. In addition to the bar charts and table (Figures 2 and 3, Table 1), when judging which size-selection region is optimal, the total RR genome size and read length of the sequencing platform should also be considered. For example, if two RR genomes show 2x enrichment in promoters and one covers 50% of the whole genome while the other covers 25%, the tradeoff between sequencing cost and quantity of information should be considered.
- For experimental validation, size selection and bisulfite conversion are the most critical steps. In this protocol, we used gel size selection to ensure that the fragment length could be visualized. As an alternative, AMPure XP beads can also be used. To ensure efficient bisulfite conversion, 0.01% sheared lambda phage DNA could be pooled into original genomic DNA prior to enzyme digestion. Since lambda phage DNA lacks methylated cytosine, non-conversion rate can be estimated by calculating the methylation level of lambda phage DNA to evaluate bisulfite conversion efficiency.
Recipes
- 10 mM dNTP mix
10 mM dATP
10 mM dTTP
10 mM dCTP
10 mM dGTP - 5x RRBS dNTP mix
200 μM dATP
200 μM dTTP
Acknowledgments
This study was supported by funding from the Institute of Plant and Microbial Biology, Academia Sinica and grants obtained from Taiwan Ministry of Science and Technology (106-2633-B-001-001) to P.-Y.C. We thank Yi-Jing Lee for providing the equipment information. The authors declare that they have no competing financial interests.
References
- Cokus, S. J., Feng, S., Zhang, X., Chen, Z., Merriman, B., Haudenschild, C. D., Pradhan, S., Nelson, S. F., Pellegrini, M. and Jacobsen, S. E. (2008). Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452(7184): 215-219.
- Gent, J. I., Ellis, N. A., Guo, L., Harkess, A. E., Yao, Y., Zhang, X. and Dawe, R. K. (2013). CHH islands: de novo DNA methylation in near-gene chromatin regulation in maize. Genome Res 23(4): 628-637.
- Hsu, F. M., Yen, M. R., Wang, C. T., Lin, C. Y., Wang, C. R. and Chen, P. Y. (2017). Optimized reduced representation bisulfite sequencing reveals tissue-specific mCHH islands in maize. Epigenetics Chromatin 10(1): 42.
- Law, J. A. and Jacobsen, S. E. (2010). Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet 11(3): 204-220.
- Li, Q., Gent, J. I., Zynda, G., Song, J., Makarevitch, I., Hirsch, C. D., Hirsch, C. N., Dawe, R. K., Madzima, T. F., McGinnis, K. M., Lisch, D., Schmitz, R. J., Vaughn, M. W. and Springer, N. M. (2015). RNA-directed DNA methylation enforces boundaries between heterochromatin and euchromatin in the maize genome. Proc Natl Acad Sci U S A 112(47): 14728-14733.
- Schnable, P. S., Ware, D., Fulton, R. S., Stein, J. C., Wei, F., Pasternak, S., Liang, C., Zhang, J., Fulton, L., Graves, T. A., Minx, P., Reily, A. D., Courtney, L., Kruchowski, S. S., Tomlinson, C., Strong, C., Delehaunty, K., Fronick, C., Courtney, B., Rock, S. M., Belter, E., Du, F., Kim, K., Abbott, R. M., Cotton, M., Levy, A., Marchetto, P., Ochoa, K., Jackson, S. M., Gillam, B., Chen, W., Yan, L., Higginbotham, J., Cardenas, M., Waligorski, J., Applebaum, E., Phelps, L., Falcone, J., Kanchi, K., Thane, T., Scimone, A., Thane, N., Henke, J., Wang, T., Ruppert, J., Shah, N., Rotter, K., Hodges, J., Ingenthron, E., Cordes, M., Kohlberg, S., Sgro, J., Delgado, B., Mead, K., Chinwalla, A., Leonard, S., Crouse, K., Collura, K., Kudrna, D., Currie, J., He, R., Angelova, A., Rajasekar, S., Mueller, T., Lomeli, R., Scara, G., Ko, A., Delaney, K., Wissotski, M., Lopez, G., Campos, D., Braidotti, M., Ashley, E., Golser, W., Kim, H., Lee, S., Lin, J., Dujmic, Z., Kim, W., Talag, J., Zuccolo, A., Fan, C., Sebastian, A., Kramer, M., Spiegel, L., Nascimento, L., Zutavern, T., Miller, B., Ambroise, C., Muller, S., Spooner, W., Narechania, A., Ren, L., Wei, S., Kumari, S., Faga, B., Levy, M. J., McMahan, L., Van Buren, P., Vaughn, M. W., Ying, K., Yeh, C. T., Emrich, S. J., Jia, Y., Kalyanaraman, A., Hsia, A. P., Barbazuk, W. B., Baucom, R. S., Brutnell, T. P., Carpita, N. C., Chaparro, C., Chia, J. M., Deragon, J. M., Estill, J. C., Fu, Y., Jeddeloh, J. A., Han, Y., Lee, H., Li, P., Lisch, D. R., Liu, S., Liu, Z., Nagel, D. H., McCann, M. C., SanMiguel, P., Myers, A. M., Nettleton, D., Nguyen, J., Penning, B. W., Ponnala, L., Schneider, K. L., Schwartz, D. C., Sharma, A., Soderlund, C., Springer, N. M., Sun, Q., Wang, H., Waterman, M., Westerman, R., Wolfgruber, T. K., Yang, L., Yu, Y., Zhang, L., Zhou, S., Zhu, Q., Bennetzen, J. L., Dawe, R. K., Jiang, J., Jiang, N., Presting, G. G., Wessler, S. R., Aluru, S., Martienssen, R. A., Clifton, S. W., McCombie, W. R., Wing, R. A. and Wilson, R. K. (2009). The B73 maize genome: complexity, diversity, and dynamics. Science 326(5956): 1112-1115.
- Smith, Z. D., Gu, H., Bock, C., Gnirke, A. and Meissner, A. (2009). High-throughput bisulfite sequencing in mammalian genomes. Methods 48(3): 226-232.
- Zerbino, D. R., Achuthan, P., Akanni, W., Amode, M. R., Barrell, D., Bhai, J., Billis, K., Cummins, C., Gall, A., Giron, C. G., Gil, L., Gordon, L., Haggerty, L., Haskell, E., Hourlier, T., Izuogu, O. G., Janacek, S. H., Juettemann, T., To, J. K., Laird, M. R., Lavidas, I., Liu, Z., Loveland, J. E., Maurel, T., McLaren, W., Moore, B., Mudge, J., Murphy, D. N., Newman, V., Nuhn, M., Ogeh, D., Ong, C. K., Parker, A., Patricio, M., Riat, H. S., Schuilenburg, H., Sheppard, D., Sparrow, H., Taylor, K., Thormann, A., Vullo, A., Walts, B., Zadissa, A., Frankish, A., Hunt, S. E., Kostadima, M., Langridge, N., Martin, F. J., Muffato, M., Perry, E., Ruffier, M., Staines, D. M., Trevanion, S. J., Aken, B. L., Cunningham, F., Yates, A. and Flicek, P. (2017). Ensembl 2018. Nucleic Acids Res.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Hsu, F., Wang, C. R. and Chen, P. Y. (2018). Reduced Representation Bisulfite Sequencing in Maize. Bio-protocol 8(6): e2778. DOI: 10.21769/BioProtoc.2778.
Category
Systems Biology > Epigenomics > DNA methylation
Plant Science > Plant molecular biology > DNA
Molecular Biology > DNA > DNA sequencing
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.