- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Detection and Analysis of Circular RNAs by RT-PCR

Published: Vol 8, Iss 6, Mar 20, 2018 DOI: 10.21769/BioProtoc.2775 Views: 28801
Reviewed by: Gal HaimovichOmar AkilWilliam C. W. Chen
Original research article
The authors used this protocol in:

Jul 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Gene expression in eukaryotic cells is tightly regulated at the transcriptional and posttranscriptional levels. Posttranscriptional processes, including pre-mRNA splicing, mRNA export, mRNA turnover, and mRNA translation, are controlled by RNA-binding proteins (RBPs) and noncoding (nc)RNAs. The vast family of ncRNAs comprises diverse regulatory RNAs, such as microRNAs and long noncoding (lnc)RNAs, but also the poorly explored class of circular (circ)RNAs. Although first discovered more than three decades ago by electron microscopy, only the advent of high-throughput RNA-sequencing (RNA-seq) and the development of innovative bioinformatic pipelines have begun to allow the systematic identification of circRNAs (Szabo and Salzman, 2016; Panda et al., 2017b; Panda et al., 2017c). However, the validation of true circRNAs identified by RNA sequencing requires other molecular biology techniques including reverse transcription (RT) followed by conventional or quantitative (q) polymerase chain reaction (PCR), and Northern blot analysis (Jeck and Sharpless, 2014). RT-qPCR analysis of circular RNAs using divergent primers has been widely used for the detection, validation, and sometimes quantification of circRNAs (Abdelmohsen et al., 2015 and 2017; Panda et al., 2017b). As detailed here, divergent primers designed to span the circRNA backsplice junction sequence can specifically amplify the circRNAs and not the counterpart linear RNA. In sum, RT-PCR analysis using divergent primers allows direct detection and quantification of circRNAs.
Keywords: Circular RNABackground
CircRNAs are covalently closed, single-stranded RNAs lacking 5’ or 3’ ends. Although their genesis is poorly understood, they can arise from pre-mRNAs by a process called backsplicing (Panda et al., 2017d; Jeck et al., 2013). CircRNAs have been reported to be abundant, ubiquitously expressed, and conserved across species (Jeck et al., 2013). A number of studies have established that circRNAs can regulate gene expression by acting as competitors of pre-mRNA splicing, as decoys for microRNAs, as sponges for RBPs, and possibly also as substrates for translation (Panda et al., 2017d). In recent years, more than one hundred thousand circRNAs have been reported bioinformatically from high-throughput RNA sequencing (RNA-seq) (Glazar et al., 2014). Unfortunately, there is little overlap among different bioinformatic pipelines and there is no ‘gold standard’ method to validate the accuracy of circRNAs identified by different bioinformatic tools (Szabo and Salzman, 2016). However, RT-PCR has been widely used for validation of circRNAs identified by RNA-seq. This protocol describes the design of divergent primers which face away from each other on the linear RNA, so that they can only amplify the circRNAs, and not the linear RNAs with the same sequence. The PCR amplicon for the detection of circRNAs using divergent primers spans the backsplice junction of circRNAs. This method has been successfully used in several studies for the detection and quantification of circRNAs.
Materials and Reagents
- Standard pipette tips with a volume capacity of 10 µl, 20 µl, 200 µl, and 1 ml
- Nuclease-free 1.7-ml microcentrifuge tubes (Denville Scientific, catalog number: C2171 )
- ThermoGridTM rigid strip 0.2-ml PCR tubes [(Denville Scientific, catalog number: C18064 (1000859) ]
- MicroAmp® optical 384-well reaction plate (Thermo Fisher Scientific, Applied BiosystemsTM, catalog number: 4309849 )
- Optical adhesive film (Thermo Fisher Scientific, Applied BiosystemsTM, catalog number: 4311971 )
- Dulbecco’s phosphate-buffered saline (DPBS) (Thermo Fisher Scientific, GibcoTM, catalog number: 14040-133 )
- Total RNA isolation-miRNeasy Mini Kit (QIAGEN, catalog number: 217004 )
- (Optional) TRIzol reagent (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 15596018 )
- Nuclease-free water (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM9930 )
- RiboLock RNase inhibitor (40 U/µl) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: EO0381 )
- RNase R (Lucigen, Epicentre, catalog number: RNR07250 )
- dNTP mix (10 mM each) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: R0193 )
- Random primers (150 ng/µl) (Sigma-Aldrich, Roche Diagnostics, catalog number: 11034731001 )
- Maxima reverse transcriptase (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: EP0741 )
- KAPA SYBR® FAST ABI prism 2x qPCR master mix (Kapa Biosystems, catalog number: KK4605 ), or SYBR Green from other vendors
- QIAquick Gel Extraction Kit (QIAGEN, catalog number: 28704 )
- TBE Buffer, 10x, Molecular Biology Grade (Sigma-Aldrich, catalog number: 574795-1L )
- 1 Kb Plus DNA Ladder (Thermo Fisher Scientific, InvitrogenTM, catalog number: 10787018 )
- UltraPureTM Agarose (Thermo Fisher Scientific, InvitrogenTM, catalog number: 16500500 )
- Ethidium bromide solution (Sigma-Aldrich, catalog number: E1510-10ML )
- 2% agarose gel (see Recipes)
Equipment
- Manual Pipettes set of 2 µl, 20 µl, 200 µl and 1,000 µl (Mettler-Toledo, Rainin, catalog number: 17014393 , 17014392 , 17014391 , and 17014382 )
- Cell scraper (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 179707PK )
- Vortex mixer (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 88880018 )
- UV transilluminator
- Refrigerated centrifuge (Eppendorf, model: 5430 R )
- NanoDropTM One/OneC Microvolume UV-Vis Spectrophotometer (Thermo Fisher Scientific, Thermo ScientificTM, model: NanoDrop OneTM , catalog number: ND-ONE-W)
- PCR strip tube rotor, mini centrifuge C1201 [Denville Scientific, catalog number: C1201-S (1000806) ]
- Eppendorf® Thermomixer® C (Eppendorf, model: Thermomixer® C , catalog number: 5382000015)
- Veriti® 96-well thermal cycler (Thermo Fisher Scientific, Applied BiosystemsTM, model: VeritiTM 96-Well, catalog number: 4375786 )
- OwlTM EasyCastTM B1 Mini Gel Electrophoresis Systems (Thermo Fisher Scientific, Thermo ScientificTM, model: OwlTM EasyCastTM B1 , catalog number: B1)
- Gel imaging system (ProteinSimple, catalog number: FluorChem E system )
- MPS 1000 mini plate spinner (Next Day Science, catalog number: C1000 )
- QuantStudio 5 Real-Time PCR System, 384-well (Thermo Fisher Scientific, Applied BiosystemsTM, model: QuantStudioTM 5, catalog number: A28140 )
Procedure
- Divergent primer design
- Get the mature sequence of circular RNA from the UCSC genome browser (https://genome.ucsc.edu/) using the genomic coordinates (Note 4).
- As shown in Figure 1, make the PCR amplicon template by joining the 100 nt sequence from the 3’ end to 100 nt sequence at the 5’ end of the circRNA (Note 5).

Figure 1. Schematic illustration of circRNA biogenesis from backsplicing of pre-mRNA (top) and schematic representation of the design of divergent primers using the circRNA junction as template for PCR amplification (bottom) - Use the above PCR amplicon template sequence to design PCR primers using the Primer 3 webtool (http://bioinfo.ut.ee/primer3/) or NCBI primer-BLAST (http://www.ncbi.nlm.nih.gov/tools/primer-blast/).
- Make sure the PCR amplicon is between 120-200 nt long (Note 6).
- If you know the CircBase ID of your circRNA (Glazar et al., 2014), you may design divergent primers using the CircInteractome webtool (Dudekula et al., 2016) (http://circinteractome.irp.nia.nih.gov/Divergent_Primers/divergent_primers.html).
- Get the mature sequence of circular RNA from the UCSC genome browser (https://genome.ucsc.edu/) using the genomic coordinates (Note 4).
- Total RNA isolation
- Take ~2 million cultured cells and remove the culture media.
- Wash the cells three times with cold PBS at 4 °C.
- Immediately scrape the cells and transfer them to a 1.7-ml tube using cold DPBS to rinse the plate.
- Collect the cell pellet by centrifugation at 500 x g for 5 min at 4 °C.
- Immediately add 700 µl of QIAzol Lysis Reagent provided in the miRNeasy Kit and disrupt the cell pellet by pipetting.
- Prepare the total RNA using the miRNeasy Kit following the manufacturer’s instructions (Note 1).
- The RNA in nuclease-free water can be stored for 6 months at -20 °C or -80 °C, or used immediately for RNase R digestion and cDNA synthesis.
- Take ~2 million cultured cells and remove the culture media.
- Degradation of linear RNA by digestion with RNase R and cDNA synthesis
- Measure RNA concentration with a NanoDrop spectrophotometer.
- Prepare an RNase R digestion reaction containing 2 µg of prepared RNA, 1 µl RiboLock, 2 µl 10x RNase R reaction buffer, and 1 µl of RNase R; adjust the volume to 20 µl with nuclease-free water (Note 2).
- Prepare a control reaction exactly the same as the RNase R reaction but without RNase R.
- Incubate the reactions at 37 °C for 30 min and immediately proceed to RNA isolation.
- Prepare the RNA from the RNase R and control treated samples using miRNeasy Kit following the protocol provided by the manufacturer and elute in 40 µl of nuclease-free water.
- Prepare the cDNA synthesis reaction containing 12 µl of prepared RNA, 1 µl RiboLock, 1 µl dNTP mix, 1 µl random primers, 4 µl 5x RT buffer, and 1 µl Maxima reverse transcriptase (Note 2).
- Prepare No-RT reaction containing everything except the Maxima reverse transcriptase (Note 3).
- Mix the reaction gently and centrifuge for 10 sec to settle the reaction at the bottom of the tube.
- Incubate the reaction at 25 °C for 10 min followed by 30 min incubation at 50 °C for cDNA synthesis.
- Inactivate the reverse transcriptase by incubating the reaction at 85 °C for 5 min.
- The prepared cDNA can be stored at -20 °C or used immediately for PCR analysis.
- Measure RNA concentration with a NanoDrop spectrophotometer.
- PCR and circRNA sequencing
- Prepare the forward and reverse divergent primer mix at a final concentration of 1 µM in nuclease-free water for the circRNA.
- Prepare the PCR reactions containing 25 µl of 2x SYBR Green mix, 0.1 µl cDNA, 12.5 µl divergent primer mix, and adjust the volume to 50 µl with nuclease-free water (Notes 2 and 10).
- Prepare another reaction same as above with 0.1 µl of no-RT instead of cDNA.
- Mix the reaction by tapping the tube with finger and centrifuge for a few seconds to settle the reactions at the bottom of the tube.
- Perform the PCR on a thermal cycler with a cycle setup of 3 min at 95 °C and 35 cycles of 5 sec at 95 °C plus 5 sec at 60 °C.
- Prepare ethidium bromide-containing 2% agarose gel (see Recipes) in 1x TBE buffer and resolve the whole 50 µl PCR product at 100 V until the loading dye reaches 3/4 of the gel.
- Visualize the PCR products on an ultraviolet transilluminator to confirm the size of the PCR product amplified (Note 7).
- Purify the PCR product from the agarose gel using the QIAquick Gel Extraction Kit following the manufacturer’s instructions (Figure 2).
- Quantify the PCR product concentration in the prepared DNA sample.
- Sequence the amplified PCR products with forward or reverse primers to find the backsplice junction sequence.
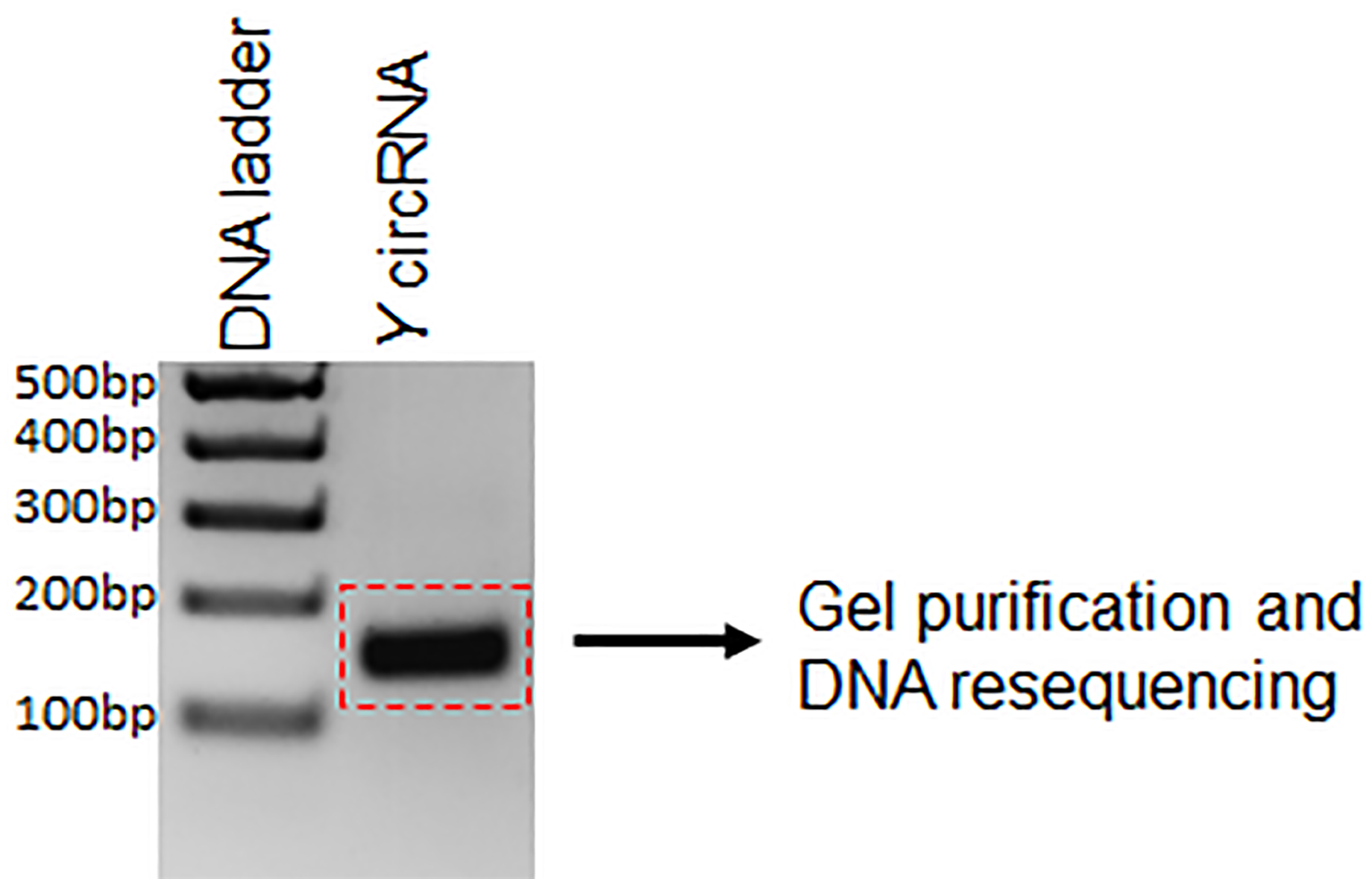
Figure 2. Example circRNA PCR product resolved and visualized on ethidium bromide-stained agarose gel. The PCR product is submitted for DNA sequencing after gel purification.
- Prepare the forward and reverse divergent primer mix at a final concentration of 1 µM in nuclease-free water for the circRNA.
- Quantitative PCR (qPCR) analysis of circRNA
- Prepare forward and reverse primer mixes for target mRNAs and circRNAs at a final concentration of 1 µM in nuclease-free water.
- Prepare the qPCR reactions in a 384-well plate containing 10 µl of 2x SYBR Green mix, 0.1 µl cDNA, and 5 µl primer mix. Adjust the volume to 20 µl with nuclease-free water (Notes 2 and 11).
- Vortex the reaction plate for few seconds after sealing the plate with optical adhesive film.
- Spin the plate for a few seconds to settle the reactions at the bottom of the wells.
- Set up the qPCR reaction cycle for 2 min at 95 °C and 40 cycles of 2 sec at 95 °C and 10 sec at 60 °C on QuantStudio 5 Real-Time PCR System (Note 2).
- The percentage (%) RNA left after RNase R treatment using the delta CT method as described in Table 1 (Notes 8 and 9) (Figure 3).
Table 1. % of RNA left after RNase R treatment relative to control. The example CT values for linear and circRNAs in control and RNase R-treated samples, and calculation of RNA left after RNase R treatment.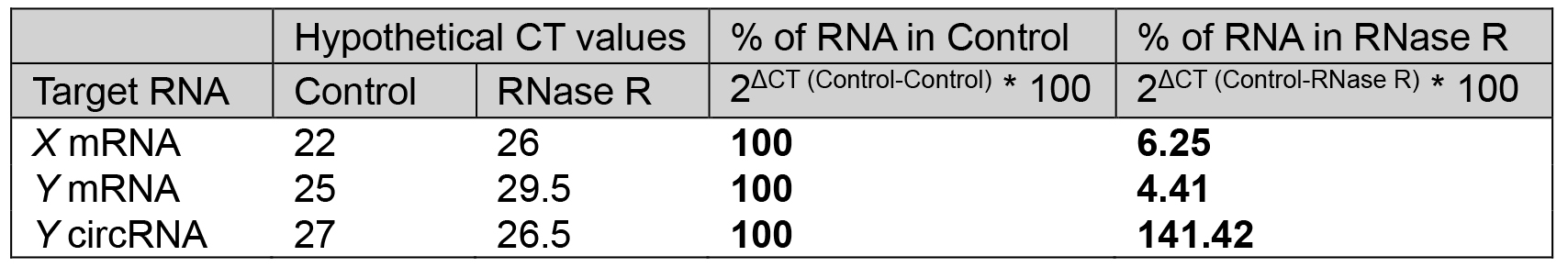
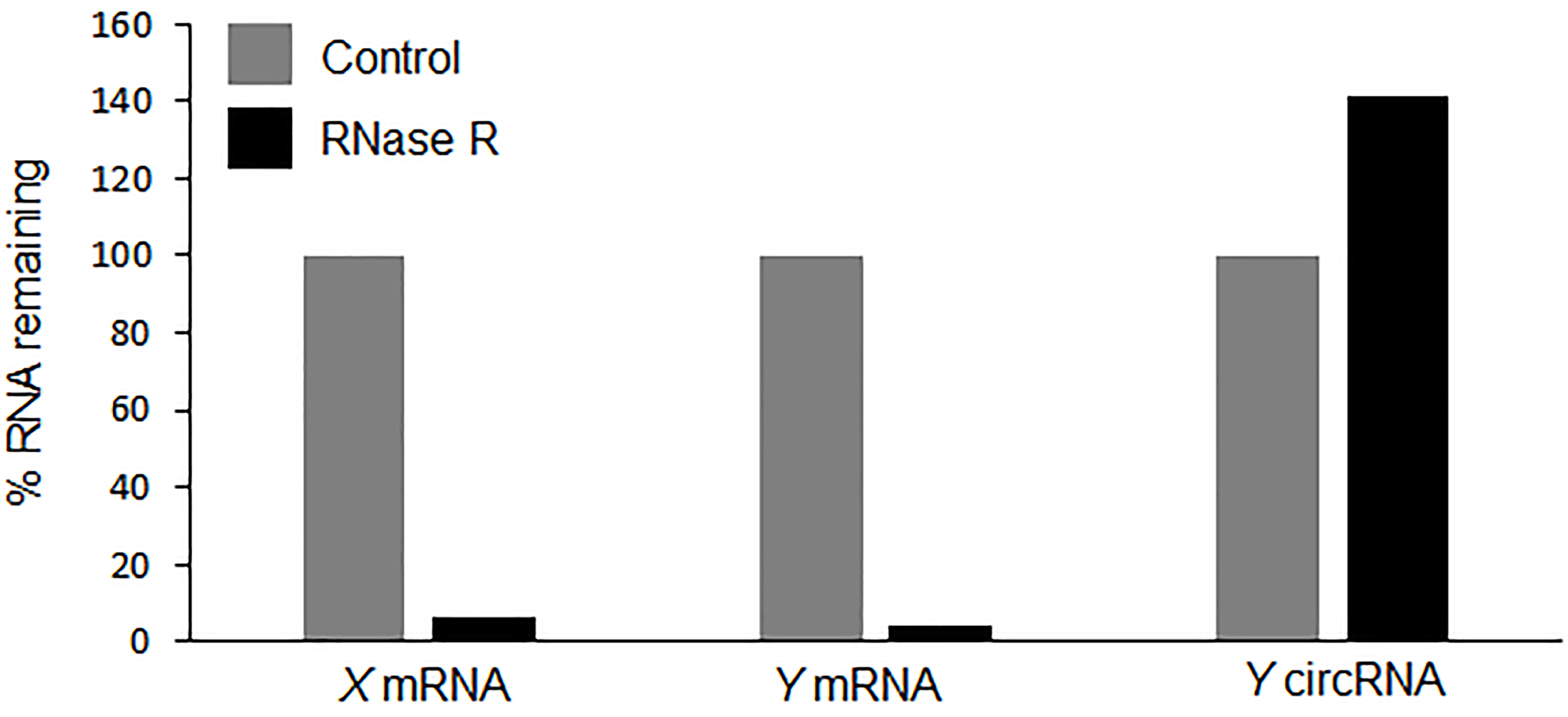
Figure 3. Hypothetical qPCR data showing the resistance of circRNA to RNase R treatment as calculated in Table 1. The qPCR results showing the levels of circRNAs and linear RNAs in RNase R (black) treated sample compared with the control treatment (grey).
- Prepare forward and reverse primer mixes for target mRNAs and circRNAs at a final concentration of 1 µM in nuclease-free water.
Data analysis
To validate the existence of a circRNA, Sanger sequencing is to be performed on the PCR product amplified with the divergent primers (Figure 2). The PCR product sequence should match exactly the expected circRNA junction sequence as predicted from the RNA-seq (Panda et al., 2017a and 2017c). However, this analysis does not inform on whether the backsplice junction sequence is coming from a scrambled exon linear transcript or a real backsplice junction. To study this possibility, RNA is digested with RNase R, a 5’ to 3’ exonuclease known to degrade linear RNAs. As shown in Figure 3, following RNase R treatment, the linear X mRNA and Y mRNA are depleted to a level lower than 10%, while Y circRNA was not degraded (Table 1). The fact that Y circRNA level did not show depletion while the counterpart linear Y mRNA depleted to a minimal level with RNase R treatment supports the notion that RNase R degrades linear RNAs specifically leading to enrichment of circRNA population (Figure 3) (Panda et al., 2017c).
Notes
- Total RNA can also be prepared with the TRIzol reagent (Thermo Fisher Scientific) or any other total RNA isolation kit.
- To avoid contamination, a PCR workstation may be used to prepare the reaction mixtures for RNase R treatment, cDNA synthesis, RT-PCR, and qPCR.
- A ‘No-RT’ cDNA reaction serves as a negative control for cDNA synthesis and only low-level background should be amplified in the RT-PCR using specific primer sets.
- The mature sequence of circRNA can be obtained by joining the exon sequence present between the backsplice site coordinates in the genome.
- If the circular RNA is shorter than 200 nt, then the mature circRNA sequence can be divided into two halves and the template for primer design can be generated by joining the 3’ half to the 5’ end of 5’ half.
- The PCR amplicon of the divergent primers should span the circRNA backsplice junction; care should be taken that no primer overlap with the junction sequence.
- The PCR product on agarose gels should show a single product of the expected size and the ‘No-RT’ reaction should not amplify a product.
- The dissociation curve analysis for each primer set should show a single peak.
- CT values should represent the average of triplicate reactions. No normalization is needed for the qPCR analysis. The actual level of linear RNA depletion in RNase R treatment can be estimated by normalizing it to the level of counterpart circRNA.
- Any Taq polymerase in place of SYBR green mix can be used for this PCR reaction.
- As pipetting 0.1 µl of cDNA is difficult and error-prone, a master mix of cDNA and water is prepared depending on the final number of reactions required for each cDNA sample. Alternatively, the 20 µl prepared cDNA can be diluted to 1 ml with nuclease-free water and 5 µl of the diluted cDNA can be used in the qPCR reaction.
Recipes
- 2% agarose gel
2 g of agarose in 100 ml of 1x TBE
Ethidium bromide (EtBr) at a final concentration of approximately 0.2 μg/ml
Acknowledgments
ACP was supported by the Science & Engineering Research Board, a statutory body of the Department of Science &Technology (DST), Government of India (SERB/F/6890/2017-18). ACP and MG were supported by the National Institute on Aging, Intramural Research Program, National Institutes of Health.
This protocol was adapted from the previously published papers (Dudekula et al., 2016 and Panda et al., 2017b). The protocol was tested and optimized by different researchers in the Gorospe laboratory, National Institute on Aging, NIH (Panda et al., 2017c and Abdelmohsen et al., 2017). The authors have no conflicts of interest or competing interests to declare.
References
- Abdelmohsen, K., Panda, A. C., De, S., Grammatikakis, I., Kim, J., Ding, J., Noh, J. H., Kim, K. M., Mattison, J. A., de Cabo, R. and Gorospe, M. (2015). Circular RNAs in monkey muscle: age-dependent changes. Aging (Albany NY) 7(11): 903-910.
- Abdelmohsen, K., Panda, A. C., Munk, R., Grammatikakis, I., Dudekula, D. B., De, S., Kim, J., Noh, J. H., Kim, K. M., Martindale, J. L. and Gorospe, M. (2017). Identification of HuR target circular RNAs uncovers suppression of PABPN1 translation by CircPABPN1. RNA Biol 14(3): 361-369.
- Dudekula, D. B., Panda, A. C., Grammatikakis, I., De, S., Abdelmohsen, K. and Gorospe, M. (2016). CircInteractome: A web tool for exploring circular RNAs and their interacting proteins and microRNAs. RNA Biol 13(1): 34-42.
- Glazar, P., Papavasileiou, P. and Rajewsky, N. (2014). circBase: a database for circular RNAs. RNA 20(11): 1666-1670.
- Jeck, W. R. and Sharpless, N. E. (2014). Detecting and characterizing circular RNAs. Nat Biotechnol 32(5): 453-461.
- Jeck, W. R., Sorrentino, J. A., Wang, K., Slevin, M. K., Burd, C. E., Liu, J., Marzluff, W. F. and Sharpless, N. E. (2013). Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 19(2): 141-157.
- Panda, A. C., Abdelmohsen, K. and Gorospe, M. (2017a). RT-qPCR detection of senescence-associated circular RNAs. Methods Mol Biol 1534: 79-87.
- Panda, A. C., De, S., Grammatikakis, I., Munk, R., Yang, X., Piao, Y., Dudekula, D. B., Abdelmohsen, K. and Gorospe, M. (2017b). High-purity circular RNA isolation method (RPAD) reveals vast collection of intronic circRNAs. Nucleic Acids Res 45(12): e116.
- Panda, A. C., Grammatikakis, I., Kim, K. M., De, S., Martindale, J. L., Munk, R., Yang, X., Abdelmohsen, K. and Gorospe, M. (2017c). Identification of senescence-associated circular RNAs (SAC-RNAs) reveals senescence suppressor CircPVT1. Nucleic Acids Res 45(7): 4021-4035.
- Panda, A. C., Grammatikakis, I., Munk, R., Gorospe, M. and Abdelmohsen, K. (2017d). Emerging roles and context of circular RNAs. Wiley Interdiscip Rev RNA 8(2).
- Szabo, L. and Salzman, J. (2016). Detecting circular RNAs: bioinformatic and experimental challenges. Nat Rev Genet 17(11): 679-692.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Panda, A. C. and Gorospe, M. (2018). Detection and Analysis of Circular RNAs by RT-PCR. Bio-protocol 8(6): e2775. DOI: 10.21769/BioProtoc.2775.
Category
Cancer Biology > General technique > Molecular biology technique
Molecular Biology > RNA > RNA detection
Molecular Biology > RNA > qRT-PCR
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.