- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

CRISPR-mediated Tagging with BirA Allows Proximity Labeling in Toxoplasma gondii
Published: Vol 8, Iss 6, Mar 20, 2018 DOI: 10.21769/BioProtoc.2768 Views: 12528
Reviewed by: David CisnerosAmit DeyAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Defining protein interaction networks can provide key insights into how protein complexes govern complex biological problems. Here we define a method for proximity based labeling using permissive biotin ligase to define protein networks in the intracellular parasite Toxoplasma gondii. When combined with CRISPR/Cas9 based tagging, this method provides a robust approach to defining protein networks. This approach detects interaction within intact cells, it is applicable to both soluble and insoluble components, including large proteins complexes that interact with the cytoskeleton and unique microtubule organizing center that comprises the apical complex in apicomplexan parasites.
Keywords: Mass spectrometryBackground
Analysis of protein-protein interactions is a key endeavor in addressing how proteins assemble and function as macromolecular complexes. Traditionally, protein complexes have been identified through co-immunoprecipitation (co-IP) with subsequent mass spectrometry analysis. However, some protein complex substituents can be artificially lost or gained during the lysis, pull-down, and washing steps of co-IP, which is especially problematic for insoluble membrane or structural proteins that require aggressive solubilization. As an alternative to co-IP, proximity-dependent biotin identification (BioID) provides a ‘snapshot’ of proteins in close proximity to a target protein of interest during normal cellular homeostasis (Roux et al., 2012). BioID utilizes a promiscuous Escherichia coli biotin protein ligase (BirA) fused to a target protein of interest. Biotin supplementation licenses the BirA fusion to biotinylate near-neighbors within 30 nm (Roux et al., 2012; Van Itallie et al., 2013), with a static labeling radius of ≤ 10 nm (Kim et al., 2014). Biotinylated proteins may be captured by affinity chromatography and identified by mass spectrometry (Roux et al., 2012).
Toxoplasma gondii belongs to the phylum Apicomplexa composed of thousands of obligate parasites. Due to ease of in vitro cultivation and genetic manipulation, T. gondii is considered a model organism for studying the biology of apicomplexans. Recently, Chen et al. (2015) adapted BioID for use in T. gondii, identifying several novel protein components of the inner membrane complex (IMC). BioID has since been employed in T. gondii research to identify interactors of kinases (Gaji et al., 2015), calmodulins (Long et al., 2017a), and to define the protein repertoire of other cellular compartments including the parasitophorous vacuole (Nadipuram et al., 2016), sutures of the IMC (Chen et al., 2017), and the apical complex (Long et al., 2017b). Here we will describe the protocol for generating a BirA gene fusions using CRISPR/Cas9 tagging (Shen et al., 2014; Shen et al., 2017), in vivo BirA biotin labeling and purification of biotinylated proteins from parasites, and identification of captured biotinylated proteins by mass-spectrometry. Since analysis of mass spectrometry datasets can be complicated by non-specific hits, we provide a method to filter out false-positive interactions and rank true-positives using Straightforward Filtering IndeX program (SFINX) (http://sfinx.ugent.be/) (Titeca et al., 2016). Candidate interactors that emerge from BioID/SFINX analysis should also be validated by secondary analyses. Therefore we also provide instructions for demonstrating co-localization by a complementary proximity ligation assay.
Materials and Reagents
- Pipette tips (Corning, catalog numbers: 4713 , 4712 )
- 1.7 ml Eppendorf tube (Corning, Costar®, catalog number: 3620 )
- T25, T175 flasks (Corning, catalog numbers: 430639 , 431080 )
- Syringes 10cc, 20cc (BD, catalog numbers: 302995 , 302830 )
- 22 G blunt needle (CML Supply, catalog number: 901-22-100M )
- D3 polycarbonate membrane (GE Healthcare, catalog number: 110612 )
- 4 mm electroporation cuvette (Harvard Apparatus, catalog number: 450126 )
- 24- and 96-well plates (MIDSCI, catalog numbers: TP92024 , TP92696 )
- Coverslips (Fisher Scientific, catalog number: 12-545-80 )
- 2 ml Eppendorf tubes (Fisher Scientific, catalog number: 054-08-138 )
- 1 L Stericup Filter Units (Merck, Millipore Sigma, catalog number: SCVPU11RE )
- Cryovials (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 5000-0050 )
- 5 ml, 10 ml, 25 ml pipettes (Fisher Scientific, catalog numbers: 13-676-10H , 13-676-10J , 13-676-10K )
- C18 CSH column (WATERS, catalog number: 186005295 )
- T. gondii RH∆ku80∆hxgprt strain (a gift from Dr. Vernon Carruthers, University of Michigan Medical School, Ann Arbor)
- Q5 Site-mutagenesis Kit with E. coli competent cells (New England Biolabs, catalog number: E0554S )
- Plasmids available at https://www.addgene.org/
- LB broth (BD, catalog number: 244610 )
- Ampicillin (Sigma-Aldrich, catalog number: A0166 )
- Plasmid Extraction Kit (Macherey-Nagel, catalog number: 740588.250 )
- M13 reverse universal primer
- Q5 DNA polymerase (New England Biolabs, catalog number: M0491S )
- PCR Cleanup Kit (Macherey-Nagel, catalog number: 740609.250 )
- Trypsin for tissue culture (Sigma-Aldrich, catalog number: T3924 )
- Mycophenolic acid (Sigma-Aldrich, catalog number: M3536 )
- Xanthine (Sigma-Aldrich, catalog number: X4002 )
- Formaldehyde 10% ultrapure EM grade (Polysciences, catalog number: 04018-1 )
- Mouse anti-HA antibodies (BioLegend, catalog number: 901501 )
- Rabbit anti-GAP45 (a gift from Dr. Dominique Soldati-Favre, University of Geneva Medical School, Geneva, Switzerland)
- Goat anti-Mouse IgG (H+L) Secondary Antibody Conjugated with Alexa Fluor-488 (Thermo Fisher Scientific, InvitrogenTM, catalog number: A-11001 )
- IRDye 680CW Goat anti-Mouse IgG (H+L) (LI-COR, catalog number: 926-68070 )
- Streptavidin Alexa Fluor-488 conjugate (Thermo Fisher Scientific, catalog number: S32354 )
- IRDye 800CW streptavidin (LI-COR, catalog number: 925-32230 )
- IRDye 680CW Goat anti-rabbit IgG (H+L) (LI-COR, catalog number: 926-68071 )
- Goat anti-Rabbit IgG (H+L) Secondary Antibody Conjugated with Alexa Fluor-594 (Thermo Fisher Scientific, InvitrogenTM, catalog number: A-11037 )
- Liquid nitrogen
- FBS (GE Healthcare, catalog number: SH30071.03HI )
- 20% DMSO
- Streptavidin magnetic beads (Thermo Fisher Scientific, PierceTM, catalog number: 88816 )
- Ammonium bicarbonate (Sigma-Aldrich, catalog number: 11213 )
- DTT
- Iodoacetamide (IAM) (Sigma-Aldrich, catalog number: I1149 )
- DUOlink In Situ Red Starter Mouse/Rabbit (Sigma-Aldrich, catalog number: DUO92101 )
- DMEM (Thermo Fisher Scientific, GibcoTM, catalog number: 12800017 )
- Sodium bicarbonate (Sigma-Aldrich, catalog number: S5761 )
- HEPES (Sigma-Aldrich, catalog number: H3375 )
- L-glutamine (Sigma-Aldrich, catalog number: G7513 )
- Gentamicin (Sigma-Aldrich, catalog number: G1272 )
- Potassium phosphate dibasic (K2HPO4) (Sigma-Aldrich, catalog number: 1551128 )
- Potassium phosphate monobasic (KH2PO4) (Sigma-Aldrich, catalog number: 1551139 )
- Potassium chloride (KCl) (Sigma-Aldrich, catalog number: P9333 )
- Calcium chloride (CaCl2) (Sigma-Aldrich, catalog number: 793639 )
- Magnesium chloride (MgCl2) (Sigma-Aldrich, catalog number: M8266 )
- Ethylenediaminetetraacetic acid (EDTA) (Sigma-Aldrich, catalog number: E6758 )
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S7653 )
- D-biotin (Sigma-Aldrich, catalog number: 47868 )
Note: This product has been discontinued. - Tris (Sigma-Aldrich, catalog number: T1503 )
- EGTA (Sigma-Aldrich, catalog number: E3889 )
- Triton X-100 (Sigma-Aldrich, catalog number: T8787 )
- NP-40 (Sigma-Aldrich, catalog number: I8896 )
- Glycerol (Sigma-Aldrich, catalog number: G5516 )
- Sodium dodecyl sulfate (SDS) (Sigma-Aldrich, catalog number: L3771 )
- Deoxycholate (Sigma-Aldrich, catalog number: D6750 )
- Bromophenol blue (Sigma-Aldrich, catalog number: B0126 )
- DOC, deoxycholate (Sigma-Aldrich, catalog number: 30970 )
- Lithium chloride (LiCl) (Sigma-Aldrich, catalog number: 62476 )
- D10 medium (see Recipes)
- Cytomix buffer (see Recipes)
- Phosphate-buffered saline (PBS) (see Recipes)
- D-biotin stock (see Recipes)
- Cytoskeleton buffer (see Recipes)
- 5x SDS sample buffer (see Recipes)
- Buffer 1 (see Recipes)
- Buffer 2 (see Recipes)
- Buffer 3 (see Recipes)
- Buffer 4 (see Recipes)
Equipment
- Eppendorf centrifuge (Eppendorf, model: 5810 R )
- Incubator (Thermo Fisher Scientific, Thermo ScientificTM, model: Model 370 )
- Class II biological safety hood (Baker Co., model: SterilGARD® II, ClassII type A/B3 )
- BTX ECM-830 electroporator (Harvard Apparatus, model: ECM 830 )
- Hemocytometer (Hausser Scientific, catalog number: 3120 )
- Inverted phase contrast microscope (Nikon Instruments, model: Eclipse TS100 )
- 200 µl pipette
- Sonic Dismembrator 550 (Fisher Scientific, model: Model 550 )
- Magnetic stand (Thermo Fisher Scientific, catalog number: 12321D )
- Odyssey imaging system (LI-COR, model: Odyssey® CLx )
- Liquid nitrogen tank (Airgas, model: NI230LT22 )
- Q-exactive HF mass spectrometer (Thermo Fisher Scientific, Thermo ScientificTM, model: Q ExactiveTM HF )
Software
- sgRNA selection website: http://grna.ctegd.uga.edu/, Mascot, version 2.5.1 (Matrix Science)
- SFINX analysis website: http://sfinx.ugent.be/, Scaffold version 4.6.1 (Proteome Software Inc.)
Procedure
- Designing a Cas9/sgRNA plasmid targeting a specific gene for C-terminal tagging with BirA
Note: The protocol described here is adapted from Shen et al., 2017. We recommend reading this review for a background of CRISPR genome editing in T. gondii before attempting this protocol.- sgRNA selection
- In http://toxodb.org/toxo/, navigate to your gene of interest (GOI) webpage. This may be accomplished using a gene search based on expression or functional properties or simply by entering a known gene ID or keyword. Be careful to choose the correct T. gondii strain for your work. Download the genomic sequence from 500 bp upstream of the start codon to 500 bp downstream of the stop codon.
- Copy the first 200 bp following the stop codon and paste at http://grna.ctegd.uga.edu/. Enter a job name, select the T. gondii lineage database, and keep all other default settings.
- From the generated list of sgRNA sequences, select a 20 nt sgRNA sequence that has a 3’ NGG protospacer adjacent motif (PAM) motif. The sgRNA sequence can be either on the forward or reverse DNA strand.
- In http://toxodb.org/toxo/, navigate to your gene of interest (GOI) webpage. This may be accomplished using a gene search based on expression or functional properties or simply by entering a known gene ID or keyword. Be careful to choose the correct T. gondii strain for your work. Download the genomic sequence from 500 bp upstream of the start codon to 500 bp downstream of the stop codon.
- Generation of a Cas9/sgRNA plasmid containing the gene specific sgRNA
- Order DNA oligos for PCR mutagenesis of pSAG1::CAS9-U6::sgUPRT (Addgene #54467). The reverse primer (5’-AACTTGACATCCCCATTTAC-3’) is universal for all mutagenesis reactions with this Cas9/sgRNA plasmid. The forward primer (5’-[20 nt sgRNA sequence in 5’ to 3’ direction]–GTTTTAGAGCTAGAAATAGC-3’) will be unique for every sgRNA. For example, if the desired protospacer + PAM site is 5’-GCTGGTCTATCGCTAGCTCGAGG, the forward primer would be 5’-GCTGGTCTATCGCTAGCTCGGTTTTAGAGCTAGAAATAGC. Specific examples for individual genes can be found in these recent papers (Brown et al., 2017; Long et al., 2017a and 2017b).
- Use Q5 site-directed mutagenesis Kit (New England Biolabs) to mutate the sgRNA sequence of pSAG1::CAS9-U6::sgUPRT (Addgene #54467). We recommend reactions consisting of 6.25 µl 2x Q5 Hot-start enzyme mix (included in kit), 0.625 µl of each primer (working stock 10 µM), 1 µl (1 ng) pSAG1::CAS9-U6::sgUPRT, and 4 µl ddH2O. For thermal cycling, we recommend 25 cycles of 95 °C for 30 sec, 60 °C for 20 sec, and 72 °C for 5 min.
- Resolve 1 µl of the PCR reaction on a DNA gel to confirm the successful reaction with a DNA band at about 10 kb. Ligate 1 µl of the Q5 PCR reaction with the KLD mix (included in kit) according to the manufacturer’s instructions.
- Transform 2.5 µl of the KLD reaction into the supplied E. coli competent cells in the kit and grow on LB agar plates containing 100 µg/ml ampicillin 37 °C overnight.
- Grow three single colonies of E. coli overnight in 3 ml LB broth containing 100 µg/ml ampicillin at 37 °C. Pellet the bacterial cultures using centrifugation 3,000 x g for 10 min. Extract the plasmids from the bacterial pellets using a Plasmid Extraction Kit (Macherey-Nagel). Send the three plasmids for Sanger sequencing to confirm the successful insertion of sgRNA in the Cas9/sgRNA plasmid using M13 reverse universal primer. Select sequence-confirmed plasmid for further editing.
- Order DNA oligos for PCR mutagenesis of pSAG1::CAS9-U6::sgUPRT (Addgene #54467). The reverse primer (5’-AACTTGACATCCCCATTTAC-3’) is universal for all mutagenesis reactions with this Cas9/sgRNA plasmid. The forward primer (5’-[20 nt sgRNA sequence in 5’ to 3’ direction]–GTTTTAGAGCTAGAAATAGC-3’) will be unique for every sgRNA. For example, if the desired protospacer + PAM site is 5’-GCTGGTCTATCGCTAGCTCGAGG, the forward primer would be 5’-GCTGGTCTATCGCTAGCTCGGTTTTAGAGCTAGAAATAGC. Specific examples for individual genes can be found in these recent papers (Brown et al., 2017; Long et al., 2017a and 2017b).
- sgRNA selection
- Generation of a BirA tagging line using CRISPR/Cas9 tagging
- Generation of a gene-specific BirA tagging cassette
- Design forward and reverse primers for amplification of the BirA tagging cassette. The forward primer contains a 40 nt 5’ homology sitting right upstream of the stop codon (note that the stop codon was excluded), and follows with GCTAGCAAGGGCTCGGGC for anchoring at the linker in a tagging plasmid pLinker-BirA-HXGPRT-LoxP (Addgene #86668). The linker sits in front of the tag BirA and serves as a spacer between the tag and endogenous protein. It also works as an anchoring site for the forward primer. The reverse primer contains a 40 nt 3’ homology arm adjacent to the Cas9 cleavage site at the sgRNA sequence (3 bp upstream of the PAM site), and follows with ATAGGGCGAATTGGAGCTCC for anchoring at the end of the HXGPRT selection cassette in the tagging plasmid. Recommended scale of synthesis is 25 nmole DNA and oligos should be processed using a standard desalting procedure.
- Use the manufacturer’s protocol of Q5 DNA polymerase (New England Biolabs) to amplify a BirA cassette with the forward and reverse primers in a 90 µl PCR reaction (Figure 1). The tagging plasmid (1 ng in 90 µl of reaction) is used as a DNA template, and the annealing temperature is set at 60 °C for 2 min.
- Resolve 2 µl of the PCR reaction on a DNA gel to confirm the successful reaction to see if a PCR product of 3.5 kb is present.

Figure 1. Diagram of CRISPR/Cas9 tagging strategy. The efficiency of Cas9 site-specific cleavage combined with integration using short regions of homology allows for rapid tagging of genes of interest. Gene-specific homology regions consisting of 42 bp flanking regions (denoted as HR1 (purple) and HR2 (blue)) are combined with conserved linker regions (L (red) and T (black)) to generate amplicons. The conserved linkers flank the central tagging construct containing a BirA tag (green), a stop codon (s), and a generic 3’ UTR (yellow). Primers HR1 and HR2 are used to amplify the gene-specific BirA amplicon from the tagging plasmid. The BirA tagging cassette is then cloned into a gene-specific pCas9/sgRNA plasmid and transfected into the RH∆ku80 line, followed by drug selection. Diagram was adapted from (Long et al., 2017a).
- Design forward and reverse primers for amplification of the BirA tagging cassette. The forward primer contains a 40 nt 5’ homology sitting right upstream of the stop codon (note that the stop codon was excluded), and follows with GCTAGCAAGGGCTCGGGC for anchoring at the linker in a tagging plasmid pLinker-BirA-HXGPRT-LoxP (Addgene #86668). The linker sits in front of the tag BirA and serves as a spacer between the tag and endogenous protein. It also works as an anchoring site for the forward primer. The reverse primer contains a 40 nt 3’ homology arm adjacent to the Cas9 cleavage site at the sgRNA sequence (3 bp upstream of the PAM site), and follows with ATAGGGCGAATTGGAGCTCC for anchoring at the end of the HXGPRT selection cassette in the tagging plasmid. Recommended scale of synthesis is 25 nmole DNA and oligos should be processed using a standard desalting procedure.
- Transfection into a T. gondii recipient line and screening clonal lines
Note: Throughout this protocol, all manipulation of live host cells or parasites should be performed with aseptic technique in a BSL2 certified biosafety cabinet or hood.- Purify the BirA cassette with a Gel Extraction and PCR Cleanup Kit (Macherey-Nagel), according to the manufacturer’s protocol.
- Elute the DNA cassette by adding 10-15 µl of the Cas9/sgRNA plasmid (10-20 µg of plasmid), and elute the column again with 5 µl ddH2O, to make a cassette-Cas9/sgRNA mixture.
- Heat the cassette-Cas9/sgRNA mixture at 75 °C for 10 min in a 1.5 ml Eppendorf tube, and centrifuge to collect liquid on the wall.
- Culture human foreskin fibroblasts (HFF) in T25 flasks with D10 medium (Recipe 1) in a 37 °C, 5% CO2 incubator until they reach confluency. Trypsinize HFF cells using standard cell culture techniques to passage the cells by splitting 1:4.
- Infect completely confluent HFF monolayer in T25 flasks with the T. gondii line to be tagged at the density of 2.5 x 106 parasites per T25 flask. Here, T. gondii RH∆ku80∆hxgprt is used. Grow the T. gondii line for ~2 days until natural egress from HFF monolayer has occurred. Fresh HFF flasks will be needed for following transfection.
- To harvest the parasites, scrape the monolayer to remove from the flask and collect 5 ml of culture medium containing parasites. Release any remaining intracellular T. gondii by passing through a 22 G needle with a 10 ml syringe, carefully remove the needle and safely discard into a sharps container, and filter the parasites by passing through 3.0 μm polycarbonate membranes to remove host cell debris. Centrifuge the parasites in the flow-through at 800 x g, 18 °C for 10 min, and wash the parasite pellet with 10 ml cytomix buffer (Soldati and Boothroyd, 1993; Recipe 2) once and centrifuge the resuspension again. The typical yield from a single T25 flask of HFF is ~5-8 x 107 parasites in total.
- Re-suspend the parasite harvested from one T25 in 2 ml cytomix buffer for transfection.
- Combine 200 µl of the parasite suspension (5-8 x 106 parasites), and the cassette-Cas9/sgRNA mixture (Figure 1) in a 4 mm electroporation cuvette (BTX), and perform the electroporation with the following protocol: 1,700 V, 176 μsec of pulse length, two pulses with 100 msec interval with a BTX ECM-830 electroporator.
- Immediate after the electroporation, transfer the parasite suspension into fresh T25 flasks with confluent HFF and incubate them at 37 °C, 5% CO2.
- Twenty-four hours after electroporation and recovery, begin drug selection with 25 µg/ml mycophenolic acid supplemented with 25 µg/ml xanthine. Passage parasites as needed in drug selection medium until a drug-resistant population emerges (~2-3 passages).
- Following natural egress, count parasites with a hemocytometer, and dilute parasites to 3 parasites/150 µl, and add this dilution of parasites in 150 µl D10 medium into 96-well plates with confluent HFF, and grow parasites at 37 °C, 5% CO2 for 6 days without movement.
- Visually looking for wells containing only one plaque in the 96-well plates under an inverted-phase contrast microscope. For wells containing only one T. gondii plaque, mix the parasites and medium in wells with 200 µl pipette. Keep growing the parasites for another 3 days until parasite natural egress. Protocols for plaque formation and expansion of T. gondii in microtiter well plates have also been thoroughly described previously (Roos et al., 1994).
- Pick 10 clones from wells and inoculate half of the culture in wells with HFF in a 96-well plate and another half in 24-well plates with coverslips and HFF monolayer. Let the parasites on coverslips grow for 24 h, and fixed with 4% formaldehyde in PBS (Recipe 3), and proceed with membrane permeabilization using 0.25% Triton X-100 in PBS containing 10% FBS, and followed with indirect fluorescence microscopy using primary antibodies against HA (mouse) and GAP45 (rabbit), and then secondary antibodies against mouse and rabbit conjugated with Alexa Fluor 488 and 594 respectively. Parasites are then imaged under a fluorescent microscope to look clones that are positive for HA staining (green channel).
- Diagnostic PCR for testing the insertion of BirA at the targeted locus is used to confirm the purity of clones using one primer sitting 200 bp upstream of the translational stop codon, and another primer sitting 300 bp downstream of the stop codon. Correctly tagged T. gondii clones do not produce the endogenous PCR product, which is only detected from the parental line RH∆ku80∆hxgprt.
- Western blot can also be performed to confirm the expression of endogenous BirA-3HA tagging line using commercial anti-HA antibodies and secondary antibodies used for detection. For Western blotting, we typically load 106 cell equivalents per lane and resolve by separation on 10% PAGE gels prior to transfer to nitrocellulose. Our preferred method of detection with LI-COR IR dye secondary antibodies; however, optimal loading conditions may vary depending on detection method. Protocols for detection of epitope tagged proteins in T. gondii have been detailed previously (Brown et al., 2017; Long et al., 2017a and 2017b).
- Once positive clones are confirmed, they should be transferred to T25 flasks and preserved in cryo-vials in liquid nitrogen storage. Five million parasites should be inoculated in a T25 flask and grown for 1 day. The infected HFF monolayers should be washed twice with warm PBS and trypsinized with 1 ml trypsin solution to detach the monolayers (diluted in an equal volume of PBS). The trypsinization should be stopped by adding 1 ml of 50% FBS in D10 medium followed by addition of 1 ml 20% DMSO in the D10 medium for preservation. Transfer the mixture into cryovials and put in an isopropanol container for freezing at -80 °C. Transfer the cryovials into a box in liquid nitrogen on the next day.
- Purify the BirA cassette with a Gel Extraction and PCR Cleanup Kit (Macherey-Nagel), according to the manufacturer’s protocol.
- Generation of a gene-specific BirA tagging cassette
- Purification of biotinylated proteins for Mass-spectrometry
- Confirmation of BirA activity in vivo
- Grow RH∆ku80∆hxgprt line and the BirA tagging line in T25 flasks containing HFF monolayer (containing 5 ml D10 medium) for 24 h at 37 °C, 5% CO2 in incubator and add 50 µl of 16 mM D-biotin solution (Recipe 4) in DMEM to make a final concentration at 160 µM. Grow the parasites for another 20 h.
- Harvest the parasites by passing through 22 G needles and D3 polycarbonate membrane to remove HFF debris, and collect the flow-through and centrifuge at 800 x g, 18 °C for 10 min to pellet the parasites. Wash parasites with cold PBS to remove excess biotin, and centrifuge and resuspend the parasite pellets in 80 µl PBS, and add 20 µl SDS sample buffer. Boil the samples at 100 °C for 5-10 min.
- Resolve the parasite lysate using SDS-PAGE, and perform Western blot using Streptavidin-800CW (LI-COR) to detect biotinylated proteins in the parasite lines. The BirA lane should exhibit additional bands not found in the parental RH∆ku80∆hxgprt line, as shown in Figure 2A with CaM1-BirA and CaM2-BirA.
- RH∆ku80∆hxgprt line and the BirA line can also be grown on coverslips with HFF monolayer in 24-well plates with addition of 160 µM D-biotin for 24 h, and used for testing by indirect immune-fluorescence microscopy using streptavidin Alexa Fluor 488, to detect biotinylated proteins at the localization of BirA protein (Figure 2B).
- Grow RH∆ku80∆hxgprt line and the BirA tagging line in T25 flasks containing HFF monolayer (containing 5 ml D10 medium) for 24 h at 37 °C, 5% CO2 in incubator and add 50 µl of 16 mM D-biotin solution (Recipe 4) in DMEM to make a final concentration at 160 µM. Grow the parasites for another 20 h.
- Purification of biotinylated proteins (Figure 2C)
- Inoculate the parental line RH∆ku80∆hxgprt line and the BirA tagging line in T175 flasks containing confluent HFF monolayer at the dosage of 2 x 107 parasites and grow for 24 h, and add D-biotin to the final concentration of 160 µM. Keep growing parasites in D-biotin for another 20 h until natural egress.
- Harvest parasites by passing through a 22 G needle and D3 polycarbonate membrane to remove debris, centrifuge and wash the parasite pellet with 50 ml cold PBS three times to remove excess D-biotin.
- Resuspend parasites in 2 ml cytoskeleton buffer (Recipe 5), and sonicate the resuspension with a microtip in Sonic Dismembrator 550 (Fisher Scientific) using a setting: 15 sec pulse, 15 sec interval time, and 2 min total pulse time.
- Incubate the parasite lysis on ice for 10 min, and transfer to 2 ml Eppendorf tubes, and centrifuge at 20,000 x g, 4 °C for 20 min. Transfer the supernatant to fresh tubes, and repeat the centrifugation to obtain clearer supernatant.
- Transfer the supernatant to 2 ml Eppendorf tubes containing 50 µl streptavidin magnetic beads (Pierce) that have been washed and balanced with cytoskeleton buffer for three times. Resuspend the supernatant and beads well and incubate at 4 °C overnight.
- Wash beads twice with 1.5 ml cytoskeleton buffer, and then wash once with 1.5 ml buffer 1 (Recipe 6), wash twice with 1.5 ml buffer 2 (Recipe 7), once with 1.5 ml buffer 3 (Recipe 8), twice with 1.5 ml buffer 4 (Recipe 9) and twice with 1.5 ml PBS. Each wash step needs to incubate for 5 min and the last two wash steps with buffer 4 and PBS should be performed to remove remaining SDS. The beads are pulled down by a magnetic stand, and supernatants are discarded.
- Transfer 10% of beads from the last wash step, and the beads are resuspended in 20 µl PBS and 5 µl 5x SDS sample buffer (Recipe 6) for SDS-PAGE and Western blot detection with streptavidin LI-COR C800. The remaining fraction of the beads (90%) can be stored temporarily at -80 °C.
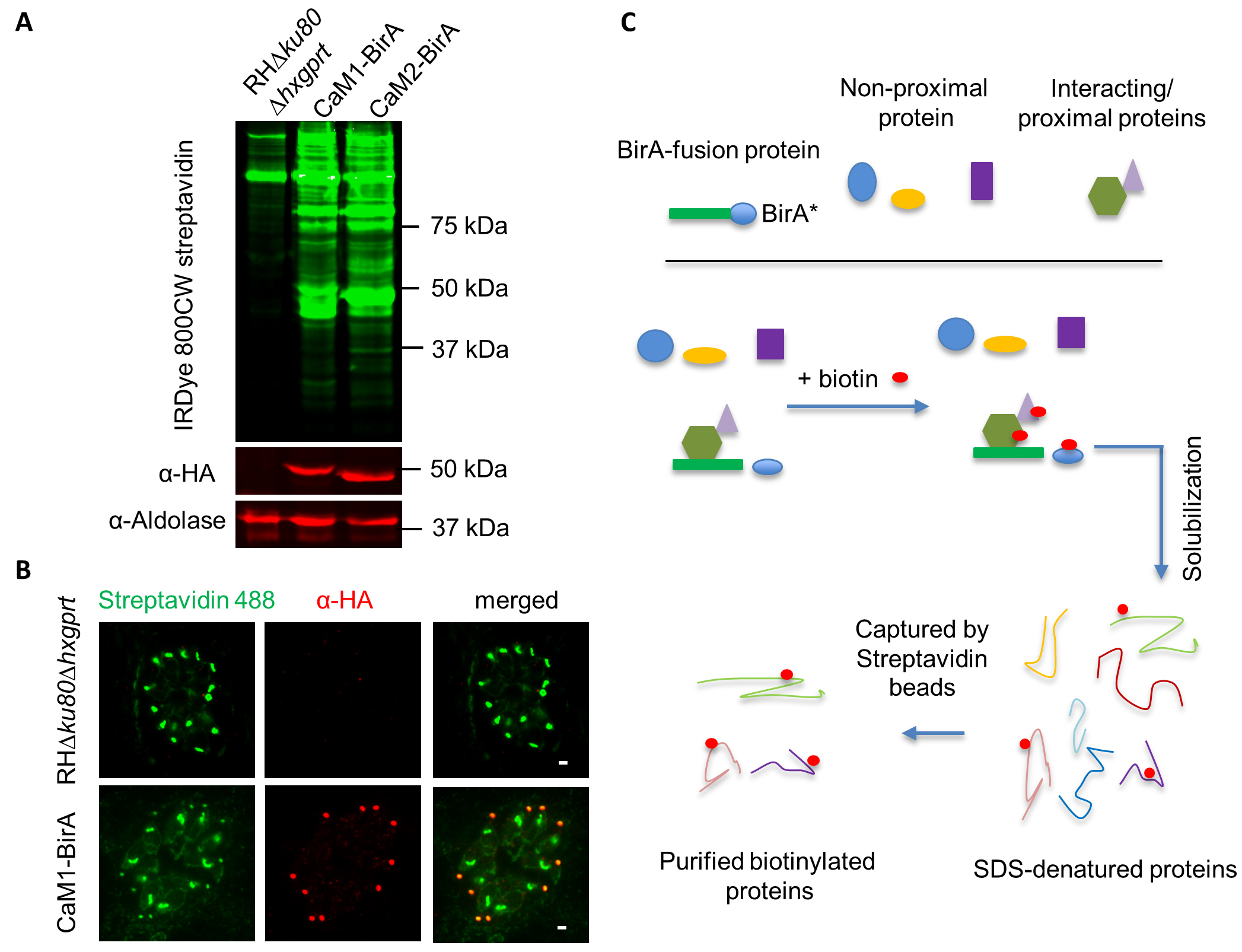
Figure 2. Strategy and examples of BirA tagging in T. gondii. A. Western blot detection of biotinylated proteins in the RH∆ku80∆hxgprt lines expressing CaM1-BirA or CaM2-BirA. IRDye 800CW streptavidin was used to detect biotinylated proteins. α-HA antibodies were used to detect BirA-3HA fusions, and α-aldolase was used as a loading control, and separately visualized by IRDye 680CW Goat anti-Mouse IgG and IRDye 680CW Goat anti-rabbit IgG. B. IFA detection of biotinylated proteins in the CaM1-BirA line. Streptavidin Alexa Fluor-488 was used to detect biotinylated proteins, and CaM1-BirA-3xHA detected by α-HA antibodies and visualized with anti-rabbit secondary antibodies conjugated with Alexa Fluor 599. Scale bar = 2 µm. C. Diagram of purification of biotinylated proteins from BirA-expressing T. gondii lines. Parasites were solubilized and biotinylated proteins were captured by streptavidin beads, as described in the Methods. This diagram is based on the original description in (Roux et al., 2012).
- Inoculate the parental line RH∆ku80∆hxgprt line and the BirA tagging line in T175 flasks containing confluent HFF monolayer at the dosage of 2 x 107 parasites and grow for 24 h, and add D-biotin to the final concentration of 160 µM. Keep growing parasites in D-biotin for another 20 h until natural egress.
- Confirmation of BirA activity in vivo
- Mass-spectrometry analysis of biotinylated proteins (performed by Dr. Michael Naldrett at the Proteomics & Metabolomics Facility, Center for Biotechnology, University of Nebraska, Lincoln (https://biotech.unl.edu/)
- Sample preparation
- Dissolve sample beads in 200 µl of 100 mM ammonium bicarbonate and reduce by adding 4 µl of 2 mM DTT for 1 h at 37 °C.
- Alkylate by adding 22 µl of 100 mM Iodoacetamide (IAM) for 20 min at 22 °C in the dark.
- Trypsin (5 µl of 0.1 mg/ml) is added per sample, and digestion is carried out overnight at 37 °C.
- Dissolve sample beads in 200 µl of 100 mM ammonium bicarbonate and reduce by adding 4 µl of 2 mM DTT for 1 h at 37 °C.
- LC-MS/MS
Run 5 µl tryptic digest sample on a NanoLC-MS/MS using a 2 h gradient on a 0.075 x 250 mm C18 Waters CSH column feeding into a Q-exactive HF mass spectrometer. - Spectral analysis
- Use Mascot to analyze results from the mass-spectrometry by searching the T. gondii ME49_20150114 (8322 sequences), cRAP_20150130 (117 sequences) and custom (containing a streptavidin sequence) databases assuming digestion by trypsin. Mascot is searched with a fragment ion mass tolerance of 0.060 Da and a parent ion tolerance of 10.0 PPM.
- Deamidated of asparagine and glutamine, oxidation of methionine, carbamidomethyl of cysteine and biotin of lysine and the N-terminus should be specified in Mascot as variable modifications.
- Use Scaffold version 4.6.1 to validate MS/MS based peptide and protein identifications. Peptide identifications are considered valid if they are supported by greater than 95.0% probability based on the Scaffold delta-mass correction (Keller et al., 2002). We generally accept protein identifications if they could be established at greater than 99.0% probability and contained at least 2 identified peptides. Lower cutoffs can also be used for exploratory work, but may lead to more false positive identifications.
- Use Mascot to analyze results from the mass-spectrometry by searching the T. gondii ME49_20150114 (8322 sequences), cRAP_20150130 (117 sequences) and custom (containing a streptavidin sequence) databases assuming digestion by trypsin. Mascot is searched with a fragment ion mass tolerance of 0.060 Da and a parent ion tolerance of 10.0 PPM.
- Sample preparation
- Validation of putative interactor proteins
Note: This step is suggested as a follow-up to BioID experiments but will likely take several months to complete. Some BioID experiments may only require a survey of bait-BirA ‘neighbors’ without validating specific interactors.- Localization of putative interactor proteins
- Interactors identified in the protein interactome may not have been previously localized. To verify the possible interactions at the right location, verifying their localization would be the first step.
- The candidate protein coding genes can be endogenously tagged by HA or Ty epitope tags using a CRISPR tagging strategy, as described above for the BirA tagging. The only difference is that the BirA tag is replaced by HA or Ty tags in the PCR template for generation of tagging cassette. Here we recommend the plasmids stored in Addgene pLinker-6HA-HXGPRT-LoxP (#86552) and pLinker-2Ty-HXGPRT-LoxP (#86664).
- Once the HA or Ty tagging strain is generated, the strains can be grown on HFF monolayer on coverslips in a 24-well plate for indirect immune-fluorescence microscopy.
- The interactors co-localized with the bait proteins will go on for bioinformatic analysis in ToxoDB, such as cellular compartment, molecular function.
- The stability and function of the protein complex can be determined by conditional depletion of individual substituents using an auxin inducible degron system (Long et al., 2017a; Brown et al., 2017). A detailed protocol for using the auxin-inducible degron system is also available at Bio-protocol (Brown et al., 2018).
- Interactors identified in the protein interactome may not have been previously localized. To verify the possible interactions at the right location, verifying their localization would be the first step.
- Interaction confirmation by another proximity approach–proximity ligation assay (PLA)
- The protein interaction can be further confirmed by tagging one gene with HA and another gene with Ty in the same line using the CRISPR/Cas9 tagging strategy described above. In this situation, the Ty tag plasmid is recommended to harbor another resistance marker DHFR not HXGPRT.
- The HA tag and Ty tag in one T. gondii line can be recognized by rabbit HA and mouse Ty antibodies, and followed with corresponding secondary antibodies conjugated with PLA probes. The PLA procedures can follow the manufacturer’s protocol with DUOlink In Situ Red Start Kit (Sigma-Aldrich). The PLA approach detects protein interactions within 30 nm distance, which is similar to the distance of BirA labeling (≤ 30 nm) (Soderberg et al., 2006).
- The protein interaction can be further confirmed by tagging one gene with HA and another gene with Ty in the same line using the CRISPR/Cas9 tagging strategy described above. In this situation, the Ty tag plasmid is recommended to harbor another resistance marker DHFR not HXGPRT.
- Localization of putative interactor proteins
Data analysis
Analysis of protein interactome
Straightforward filtering index (SFINX) (http://sfinx.ugent.be/) is a program that provides a statistically robust method to filter false positives and identify bona fide interactions using peptide counts from proteomic studies (Titeca et al., 2016). Here we combine the proximity labeling approach with this program to build a proximity based protein-protein interaction network.
SFINX analysis:
- Datasets from two or more independent experiments should be combined into one Scaffold file. Set the program at ‘total unique peptide count’ with protein threshold 95% and a minimal number of peptides 2.
- Export the datasets with ‘current view’, and edit the excel file to remove unnecessary columns and rows, leave the gene accession number, the independent mass-spectrometry experiments (projects)’s name and peptide counts. Type in row names in the first row of the first column, as the SFINX website suggests in the info column. Save this basic file as Comma Separated Values file (.csv), or other file formats as suggested by the software.
- Create another .csv file (bait file) with the accession number of the gene of interest for BirA tagging in the experiments.
- Upload the basic file and bait file to the website and pick the correct file format. A protein-protein network will immediately be created with a strictness bar shown on the left. The strictness bar is based on the SFINX scores (derived from P scores), which is used to rank true positives. In our case, strictness 1 was taken, corresponding to P < 0.00004.
- The filtered data can be downloaded as an excel .tsc file by clicking ‘complete output’ or ‘cutoff output’. The downloaded file lists the interactors (preys), and scores and P scores for the interaction between interactors and BirA proteins (baits).
- To produce high quality protein-protein interaction networks with SFINX3, multiple bait proteins (i.e., BirA fusions) involved in the same pathway or location should be analyzed by mass spectrometry.
Notes
- We have also successfully used the statistical features provided within Scaffold to define meaningful interactions among replicates datasets. When running comparisons from replicate experiments, we analyze data using Student’s t-test either with or without correction for multiple comparisons. Corrected tests are much more stringent and hence less likely to identify false positives. However, they also greatly limit the number of potential interacting proteins. Where discovery is the main objective, it may be a better strategy to accept a higher false positive rate and validate potential interactions using some secondary analysis.
- Extra caution should be used when working with the DHFR resistance cassette as this confers resistance to pyrimethamine, the most commonly used drug to treat toxoplasmosis. The plasmids have been designed with loxP sites flanking the DHFR resistance cassette, making it possible to remove this region by transient transfection of Cre, flowed by cloning and screening by PCR.
- For optimal performance of SFINX, it is important to have two or more biological replicates for each BirA-labeled protein. The method is quite robust to differences in labeling efficiency between replicates, however, it is helpful to have similar overall peptide coverage between experimental runs. In our experience, SFINX is quite conservative in predicting interactions that exceed a statistical threshold for being unlikely to be due to chance. The output file gives posterior probabilities that can be used to set a threshold for what the user is comfortable accepting. In any cases, these outputs are only predictive of proximity interactions and follow up analysis using some other methods is necessary to confirm interactions.
Recipes
- D10 medium
1 packet of DMEM powder
3.7 g sodium bicarbonate
2.38 g HEPES
10 ml of 200 mM L-glutamine
1 ml of 10 mg/ml gentamicin
Q.S. to 1 L with deionized H2O
Filter sterilize with 1 L Stericup Filter and store at 4 °C - Cytomix buffer
8.66 mM K2HPO4
1.36 KH2PO4
120 mM KCl
0.15 mM CaCl2
5 mM MgCl2
25 mM HEPES
2 mM EDTA
Adjust pH to 7.6
Filter sterilize with Stericup Filter and store at 4 °C - Phosphate-buffered saline (PBS)
137 mM NaCl
10 mM phosphate
2.7 mM KCl
Adjust pH to 7.4
Autoclave and store at 4 °C - D-biotin stock
160 mM D-biotin in DMEM - Cytoskeleton buffer
10 mM Tris, pH 7.4
100 mM NaCl
1 mM EDTA
1 mM EGTA
1% Triton X-100
1% NP-40
10% glycerol
0.2% SDS
0.5% deoxycholate - 5x SDS sample buffer
Bromophenol blue 0.25%
DTT 0.5 M
Glycerol 50%
SDS 10% - Buffer 1
20 mM Tris pH 7.5
2% SDS - Buffer 2
0.1% DOC
1% Triton X-100
500 mM NaCl
1 mM EDTA
50 mM HEPES
Adjust pH to pH 7.5 - Buffer 3
250 mM LiCl
0.5% NP-40
0.5% DOC
1 mM EDTA
10 mM Tris pH 8.1 - Buffer 4
50 mM Tris, pH 7.4
50 mM NaCl
Acknowledgments
This work was supported by a grant from the NIH (AI034036) to L.D.S. and adapted from protocols described in Long et al. (2017a and 2017b). The authors declare that there are no conflicts or competing interests.
References
- Brown, K. M., Long, S. and Sibley, L. D. (2017). Plasma membrane association by N-acylation governs PKG function in Toxoplasma gondii. MBio 8(3).
- Brown, K. M., Long, S. and Sibley, L. D. (2018). Conditional knockdown of proteins using auxin-inducible degron (AID) fusions in Toxoplasma gondii. Bio-protocol 8(4): e2728.
- Chen, A. L., Kim, E. W., Toh, J. Y., Vashisht, A. A., Rashoff, A. Q., Van, C., Huang, A. S., Moon, A. S., Bell, H. N., Bentolila, L. A., Wohlschlegel, J. A. and Bradley, P. J. (2015). Novel components of the Toxoplasma inner membrane complex revealed by BioID. MBio 6(1): e02357-02314.
- Chen, A. L., Moon, A. S., Bell, H. N., Huang, A. S., Vashisht, A. A., Toh, J. Y., Lin, A. H., Nadipuram, S. M., Kim, E. W., Choi, C. P., Wohlschlegel, J. A. and Bradley, P. J. (2017). Novel insights into the composition and function of the Toxoplasma IMC sutures. Cell Microbiol 19(4).
- Gaji, R. Y., Johnson, D. E., Treeck, M., Wang, M., Hudmon, A. and Arrizabalaga, G. (2015). Phosphorylation of a myosin motor by TgCDPK3 facilitates rapid initiation of motility during Toxoplasma gondii egress. PLoS Pathog 11(11): e1005268.
- Keller, A., Nesvizhskii, A. I., Kolker, E. and Aebersold, R. (2002). Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem 74(20): 5383-5392.
- Kim, D. I., Birendra, K. C., Zhu, W., Motamedchaboki, K., Doye, V. and Roux, K. J. (2014). Probing nuclear pore complex architecture with proximity-dependent biotinylation. Proc Natl Acad Sci U S A 111(24): E2453-2461.
- Long, S., Brown, K. M., Drewry, L. L., Anthony, B., Phan, I. Q. H. and Sibley, L. D. (2017a). Calmodulin-like proteins localized to the conoid regulate motility and cell invasion by Toxoplasma gondii. PLoS Pathog 13(5): e1006379.
- Long, S., Anthony, B., Drewry, L. L. and Sibley, L. D. (2017b). A conserved ankyrin repeat-containing protein regulates conoid stability, motility and cell invasion in Toxoplasma gondii. Nat Commun 8(1): 2236.
- Nadipuram, S. M., Kim, E. W., Vashisht, A. A., Lin, A. H., Bell, H. N., Coppens, I., Wohlschlegel, J. A. and Bradley, P. J. (2016). In vivo biotinylation of the Toxoplasma parasitophorous vacuole reveals novel dense granule proteins important for parasite growth and pathogenesis. MBio 7(4).
- Roos, D. S., Donald, R. G. K., Morrissette, N. S. and Moulton, A. L. (1994). Molecular tools for genetic dissection of the protozoan parasite Toxoplasma gondii. Methods Cell Biol 45: 28-61.
- Roux, K. J., Kim, D. I., Raida, M. and Burke, B. (2012). A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol 196(6): 801-810.
- Shen, B., Brown, K., Long, S. and Sibley, L. D. (2017). Development of CRISPR/Cas9 for efficient genome editing in Toxoplasma gondii. Methods Mol Biol 1498: 79-103.
- Shen, B., Brown, K. M., Lee, T. D. and Sibley, L. D. (2014). Efficient gene disruption in diverse strains of Toxoplasma gondii using CRISPR/Cas9. MBio 5(3): e01114-01114.
- Soderberg, O., Gullberg, M., Jarvius, M., Ridderstrale, K., Leuchowius, K. J., Jarvius, J., Wester, K., Hydbring, P., Bahram, F., Larsson, L. G. and Landegren, U. (2006). Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods 3(12): 995-1000.
- Soldati, D. and Boothroyd, J. C. (1993). Transient transfection and expression in the obligate intracellular parasite Toxoplasma gondii. Science 260(5106): 349-352.
- Titeca, K., Meysman, P., Gevaert, K., Tavernier, J., Laukens, K., Martens, L. and Eyckerman, S. (2016). SFINX: Straightforward filtering index for affinity purification-mass spectrometry data analysis. J Proteome Res 15(1): 332-338.
- Van Itallie, C. M., Aponte, A., Tietgens, A. J., Gucek, M., Fredriksson, K. and Anderson, J. M. (2013). The N and C termini of ZO-1 are surrounded by distinct proteins and functional protein networks. J Biol Chem 288(19): 13775-13788.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Long, S., Brown, K. M. and Sibley, L. D. (2018). CRISPR-mediated Tagging with BirA Allows Proximity Labeling in Toxoplasma gondii. Bio-protocol 8(6): e2768. DOI: 10.21769/BioProtoc.2768.
Category
Microbiology > Microbial proteomics > Whole organism
Molecular Biology > Protein > Protein-protein interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.