- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Sebinger Culture: A System Optimized for Morphological Maturation and Imaging of Cultured Mouse Metanephric Primordia

Published: Vol 8, Iss 4, Feb 20, 2018 DOI: 10.21769/BioProtoc.2730 Views: 7354
Reviewed by: Alessandro DidonnaXia WangAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2010
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Here, we present a detailed protocol on setting up embryonic renal organ cultures using a culture method that we have optimised for anatomical maturation and imaging. Our culture method places kidney rudiments on glass in a thin film of medium, which results in very flat cultures with all tubules in the same image plane. For reasons not yet understood, this technique results in improved renal maturation compared to traditional techniques. Typically, this protocol will result in an organ formed with distinct cortical and medullary regions as well as elongated, correctly positioned loops of Henle. This article describes our method and provides detailed advice. We have published qualitative and quantitative evaluations on the performance of the technique in Sebinger et al. (2010) and Chang and Davies (2012).
Keywords: Organ cultureBackground
The metanephric (permanent) kidneys of mammals develop from simple rudiments located at the caudal end of the intermediate mesoderm. In mice, at about embryonic day (‘E’) 10 these rudiments form and consist of two morphologically distinguishable components; an epithelial ureteric bud that arises as a diverticulum of the Wolffian (nephric) duct, and a metanephrogenic mesenchyme that forms next to the duct. As development progresses, the ureteric bud enters the metanephrogenic mesenchyme and undergoes many successive rounds of growth and branching to make a ‘tree’; this later remodels to produce a mature collecting duct system in which tubules radiate from a central cavity, the renal pelvis (Lindstrom et al., 2015). The renal pelvis drains to the ureter, which forms from the original stalk of the ureteric bud. As the ureteric bud develops, it induces cells from the metanephrogenic mesenchyme to condense around each of its tips to form a ‘cap mesenchyme’ (Schreiner, 1902; Reinhoff, 1922). The cap mesenchymes are stem cell populations that divide as the tips divide, so that each tip formed by bifurcation of an existing branch inherits its own cap (reviewed by Hendry et al., 2011). Cells at the more distal ends of the caps differentiate to form excretory nephrons, which connect to the ureteric bud branch from whose cap they formed (Georgas et al., 2009). Blood vessels invade from the base of the metanephros and follow the ureteric bud, making a network around (but never entering) the cap mesenchymes (Munro et al., 2017): later, these vessels will serve glomeruli and other parts of the kidney.
In vitro culture of metanephric kidney rudiments has a long history. Indeed, these were among the first embryonic organs to show continued development outside the body (Carrel and Burrows, 1910). The continued development of kidney rudiments outside of the body was a scientific, as well as a technical, advance: the autonomous development of isolated organs demonstrated that the information required to build them was ‘local’ and did not depend on the rest of the embryo. This observation added considerable support to the idea that architecture of the body is hierarchical, with modules (organs) that largely look after themselves and interact with the rest of the body only at specific functional interfaces. The earliest culture methods used rather complex media and culture supports, such as Grobstein’s use of clotted avian plasma and chick embryo extract (Grobstein, 1953). These systems were necessary because simply placing a kidney rudiment in a glass dish or flask resulted in its breakdown because cells adhered to the substrate and spread out to form a monolayer. Immersion in medium in non-adhesive dishes prevents cell dispersal but does not result in proper development (see ‘Data analysis’ section). Both imaging and reproducibility were greatly improved by Saxén’s adoption of a culture method developed by Trowell for culture of rat lymph nodes (Trowell, 1954). Saxén placed embryonic kidney rudiments, isolated from mice at E11, on filters that were supported by a stainless steel grid at the interface between gas and medium (Saxén et al., 1962).
The Trowell method has been a mainstay of research in kidney development for many decades. It allows for significant ureteric bud growth and branching, formation of nephrons, differentiation of their separate proximal and distal domains and connection of nephrons to ureteric bud branches. Rudiments grow flat enough to facilitate confocal microscopy of fixed specimens without the need for ‘clearing’ techniques, and even conventional epifluorescence microscopy is adequate for many purposes (e.g., Davies et al., 2014). Another advantage is that diffusion paths have proved to be short enough and open enough to enable mechanisms of development to be investigated in this method with drugs (e.g., Fisher et al., 2001), growth factors (e.g., Piscione et al., 1997), function-blocking antibodies (e.g., Falk et al., 1996) and, to a limited extent, siRNAs (e.g., Davies et al., 2004). The Trowell system, together with variants that place rudiments at the air-medium interface using use Transwell filters instead of stainless steel supports, remains very common in studies of kidney development.
Useful as it is, the Trowell culture system has a few problems. One is that the filter itself interferes with bright-field/phase contrast imaging because the filter pores appear, out of focus, in the image (though some Transwell systems do achieve good optical clarity). Another is that the tissue is too thick for reliable high-resolution, unattended time-lapse photography, because tubules leave the focal plane. A third problem is that some aspects of renal development, such as the formation of distinct cortex and medulla, and extension of nephrons’ loops of Henle from the cortex to the medulla, occur poorly if at all. In an attempt to address these issues, we developed an alternative culture system that uses the surface tension of very shallow medium to hold a kidney rudiment on to a clear glass substrate. To our surprise, the system not only solved the imaging problem, but it also allowed the organ rudiments to develop clear cortico-medullary zonation and nephron maturation proceeded as far as the production of clear and elongated loops of Henle (Sebinger et al., 2010) (Figure 1). The enhanced development is seen in cultures made from natural kidneys isolated from mouse embryos, and also in organoids engineered from suspensions of stem cells (Chang and Davies, 2012). 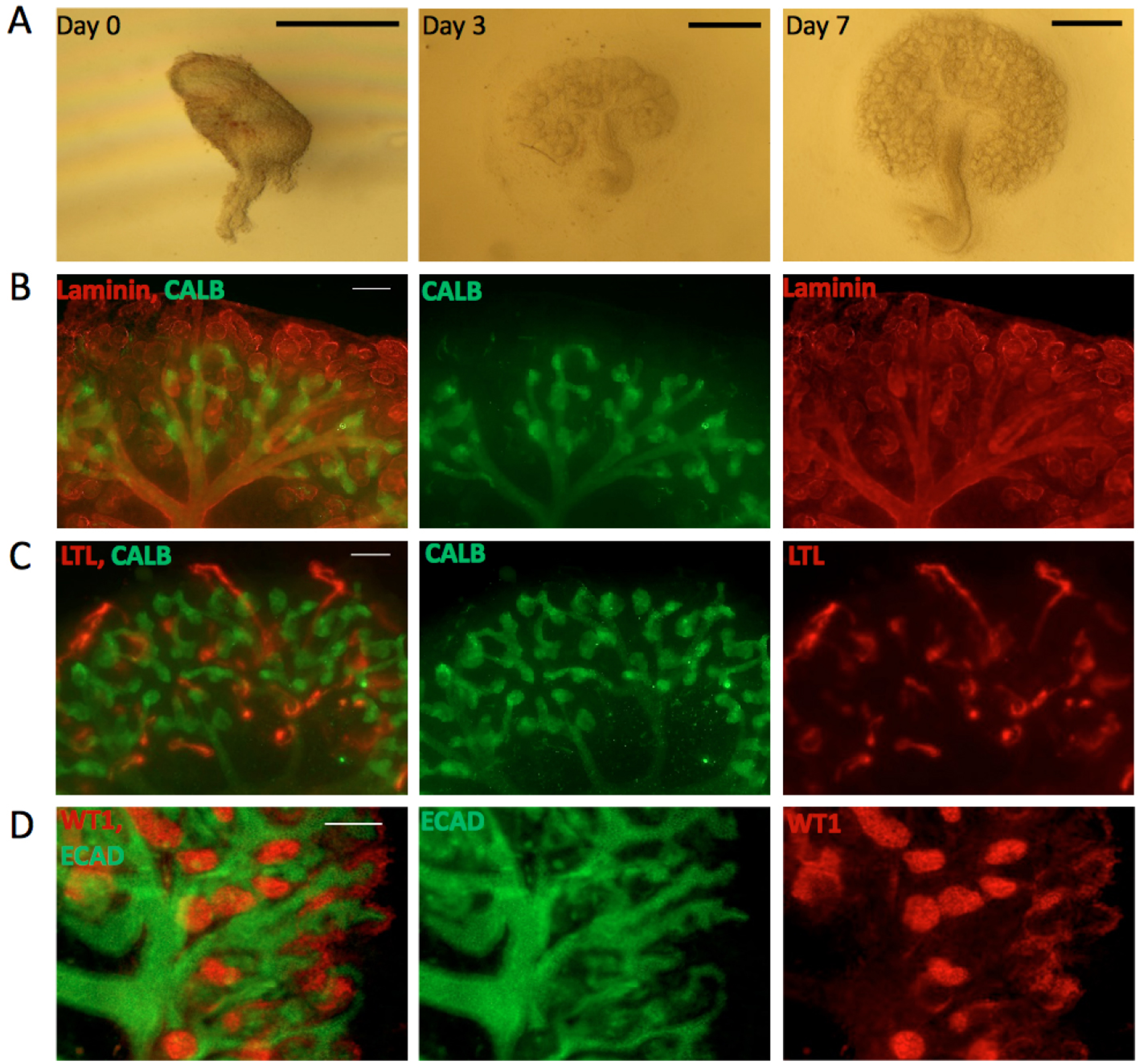
Figure 1. Kidneys cultured in the Sebinger system. A. Bright field images for E11.5 kidney grown in culture for 0, 3 and 7 days. Scale bars = 0.5 mm. B-D. E11.5 kidneys cultured for 7 days in the Sebinger system and stained for different renal markers to show maturation. B. Stained for the ureteric bud marker CALB (shown in green) and the basement membrane marker Laminin (shown in red). The red channel shows the presence of loops of Henle dipping into the medulla; C. Stained for CALB (green) and the proximal tubular marker LTL (red); D. Stained for ECAD (ureteric bud and distal tubular marker; shown in green) and WT1 (podocyte and cap mesenchyme marker; shown in red). Scale bars = 100 μm.
Materials and Reagents
- 1 ml disposable syringes (Plastipak 1 ml, BD, catalog number: 303172 ) with fine needles (0.5 x 16 mm/25 G x 5/8”, Microlance 3, BD, catalog number: 300600 )
- 40 x 0.13 mm borosilicate glass coverslips (VWR, catalog number: 631-0177 )
- Silicone cones (flexiPERM Cone shape A, Greiner Bio One International)
These are at the time of writing available only on special order–phone Greiner–and delivery times can be long enough to make forward planning important. The cones can be re-used for years.
Note: We know of no suitable substitutes. - 100 mm sterile Petri dishes (for dissection–surface quality is irrelevant) (Cell Star®, Greiner Bio One International, catalog number: 664160 )
- 60 x 15 mm sterile Petri dishes (Cell Star®, Greiner Bio One International, catalog number: 628160 )
Note: We use Greiner but expect that others will also be suitable. - Glass Pasteur pipettes (150 mm, Volac, catalog number: D810 )
- Timed-mated mice at E11.5 (E10.5 is also suitable but is a more challenging dissection)
- Sterile distilled water
- 100% methanol
- Eagle’s minimal essential medium with Earle’s salts and non-essential amino acids (Sigma-Aldrich, catalog number: M5650 )
- Foetal bovine serum (FBS) (Biochrom, catalog number: S 0415 )
- Penicillin-streptomycin (Sigma-Aldrich, catalog number: P4333 )
- Phosphate buffered saline (PBS) (Sigma-Aldrich, catalog number: 79382 )
- Anti-laminin (Sigma-Aldrich, catalog number: L9393 )
- FITC anti-rabbit (Sigma-Aldrich, catalog number: F0382 )
- EmbryoMax® Penicillin-streptomycin solution, 100x (Sigma-Aldrich, catalog number: TMS-AB2-C )
- Hydrogen peroxide (Sigma-Aldrich, catalog number: H1009 )
- Ammonium hydroxide (Sigma-Aldrich, catalog number: A6899 )
- Cleaning solution (see Recipes)
- Dissecting medium (see Recipes)
- Culture medium (see Recipes)
- Hydration buffer (see Recipes)
Equipment
- Forceps
- Scalpel (curved blade, e.g., D-form, type 22, Swann Morton, catalog number: 0108 )
- 80 °C oven
Note: We use a Gallenkamp Hotbox size 1, but any 80 °C oven should be suitable. - Cell culture incubator at 37 °C, 5% CO2
Note: We use a NuAire 5500E (NuAire, model: NU-5500E ), but any stable incubator should work as well. - Dissecting microscopes
Note: We use ZEISS Stemi 2000C microscopes (ZEISS, model: Stemi 2000-C ) with transilluminating stages, but have demonstrated the technique in other laboratories with other models of dissecting microscope. The precise type of microscope is not important, but transillumination (rather than epi-illumination) is. - Clean area
Note: We do not use safety cabinets (for the non-pathogen-infected samples we use), because the vibration of the fans is a nuisance. We do use simple cabinets about the size of a safety cabinet, made from Perspex, to provide shelter from dust moved by Edinburgh winds blowing through ill-fitting antique lab windows).
Procedure
- Preparation
- Make the solutions listed in Recipes section.
- Sterilize 40 x 0.13 mm coverslips (depending on the manufacturer, they may need to be acid treated in 1 N HCl first; 10 min room temperature, followed by 3 rinses in sterile distilled water).
- Clean the flexiPERM cones in cleaning solution (50 ml or so–the exact volume does not matter) at 70 °C for 10 min. Rinse them 3 x and store them in sterile distilled water.
- Make the solutions listed in Recipes section.
- Isolation of E11.5 mouse kidney rudiments
Note: Takes about 20 min per pregnant mouse in skilled hands, a lot longer for beginners. Absolute beginners are advised to begin by isolating E12.5 kidneys before moving on to harder-to-see E11.5 kidneys. All steps should be done under sterile conditions.- Sacrifice the pregnant mother mouse by methods appropriate to the local animal licence and laws.
- Remove the uterus.
- In a large Petri dish, immerse the uteri in dissecting medium and use a scalpel to cut between bulges in the uterus: ‘rolling’ the curved edge of the scalpel over the tissue to be cut is easier than pulling it in the conventional way. Squeeze the embryo out of the cut end next to it with the forceps. Remove the heads of the embryos, and any placental/membrane material. (Figure 2)
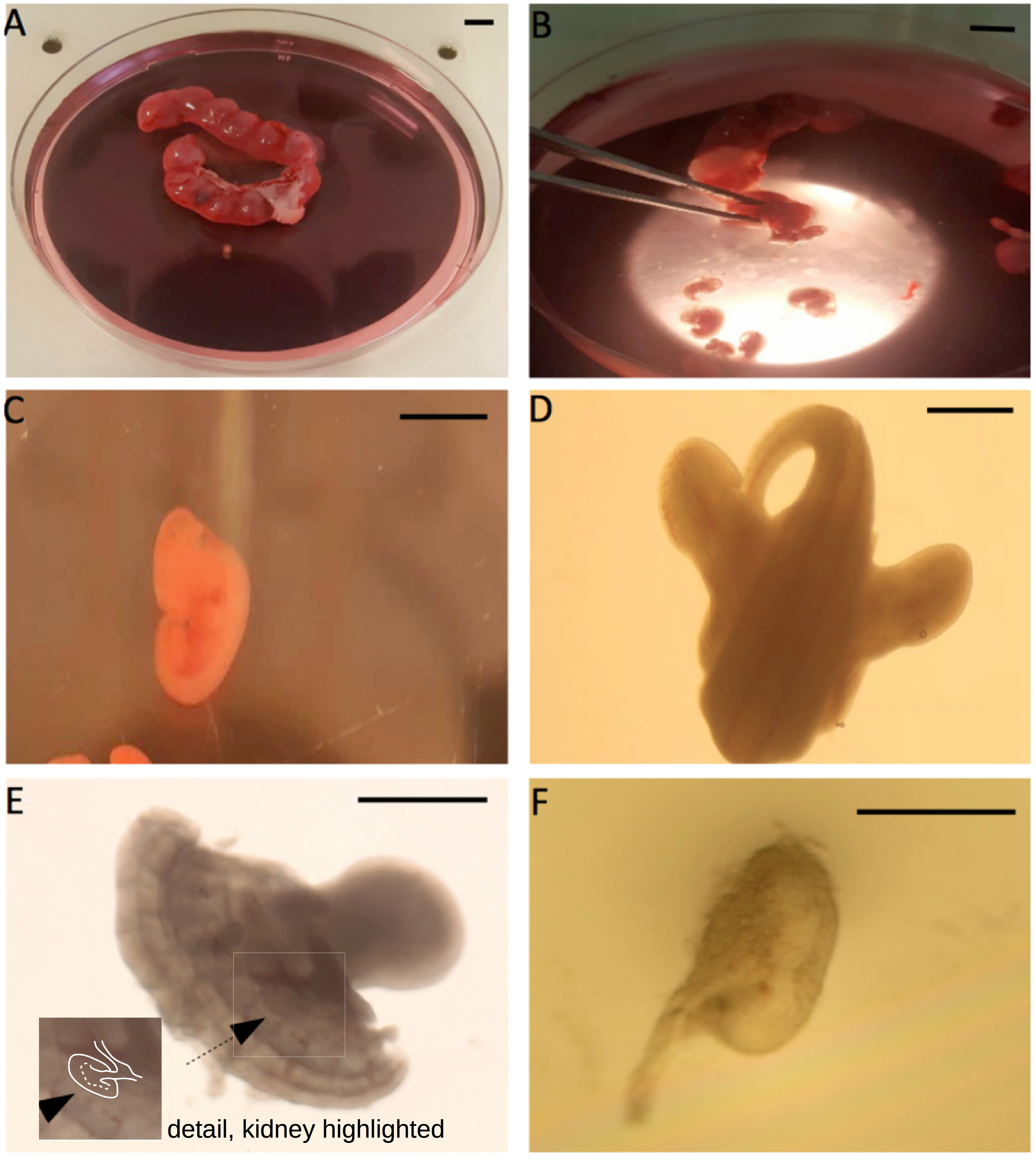
Figure 2. Illustration of the E11.5 metanephric kidney isolation. A. The pregnant uterus; B. Squeezing embryos out of the uterus with forceps; C. The extracted E11.5 embryos; D. The caudal part of the embryo; E. The rear of the embryo, after cutting it sagittally into two halves; the arrow points towards the metanephric kidney, edges of which are marked in white in the detail (seeing the kidney is the hardest part of the dissection, and takes practise). F. The isolated E11.5 metanephric kidney. Scale bar for A-C is 5 mm, for D and E is 1 mm, and 0.5 mm for F. - Transfer the embryo trunks to a 60 mm dish filled to about 5 mm depth with dissecting medium (the precise amount can be altered to suit individual preferences), which can be used straight from storage at 4 °C.
- Using fine needles as dissecting instruments (see Materials and Reagents for details), transect the embryos just rostral to the hind limb buds. Retain the portions with the hind limb buds (these can be stored for a few days or transported on ice: see Davies, 2006, for details of the method).
- Remove the tails (which are otherwise a springy nuisance while dissecting).
- Lay each rear-end fragment of the embryo ventral-side-down, and cut it sagittally, directly down from the mid-line of the neural tube to its ventral surface.
- Inspect each half created in the last step, from the cut (anatomically medial) surface, and identify the kidney rudiment, which lies (at E11.5) next to the cranial limit of the hind limb bud. The ureteric bud diverticulum of the nephric duct is the most obvious spatial cue, and the extend of the metanephrogenic mesenchyme can be seen by a slight change in light-scattering properties (on some microscopes it appears lighter than the surrounding mesoderm, and on some it appears darker).
- Dissect the kidney rudiment free of the rest of the embryo, pinning tissue down with one needle and drawing the other needle against it to make accurate cuts, and transfer the kidney to a new dish in which rudiments can be pooled for later use (pulled glass pipettes provide one means of transferring rudiments easily: the heat of melting the glass to pull them will sterilize their ends).
Note: Throughout the dissection, care should be taken to ensure that media are not left in air for so long that they become alkaline (the pH indicator in the medium will indicate this by a colour change. We suggest 15 min as a maximum but air movement may mean shorter times are needed. In our experience, HEPES inhibits normal kidney development and we avoid its use).
- Sacrifice the pregnant mother mouse by methods appropriate to the local animal licence and laws.
- Sebinger culture
- Put the cover slips and the silicone cones in 80 °C oven for 20 min to dry (dryness is essential for proper adhesion between the silicone cone and the cover slips). They can be handled with gloved hands or forceps–use plastic forceps for cones to avoid damage that might result from the use of metal.
- Place a cone, narrow-end down, on to the coverslip and use a blunt-ended forceps to press the edge of the silicone cone against the cover slip to ensure tight adhesion.
- Place the cover slip with the silicone cone attached to it on the base of a 60 mm Petri dish. (Figure 3)
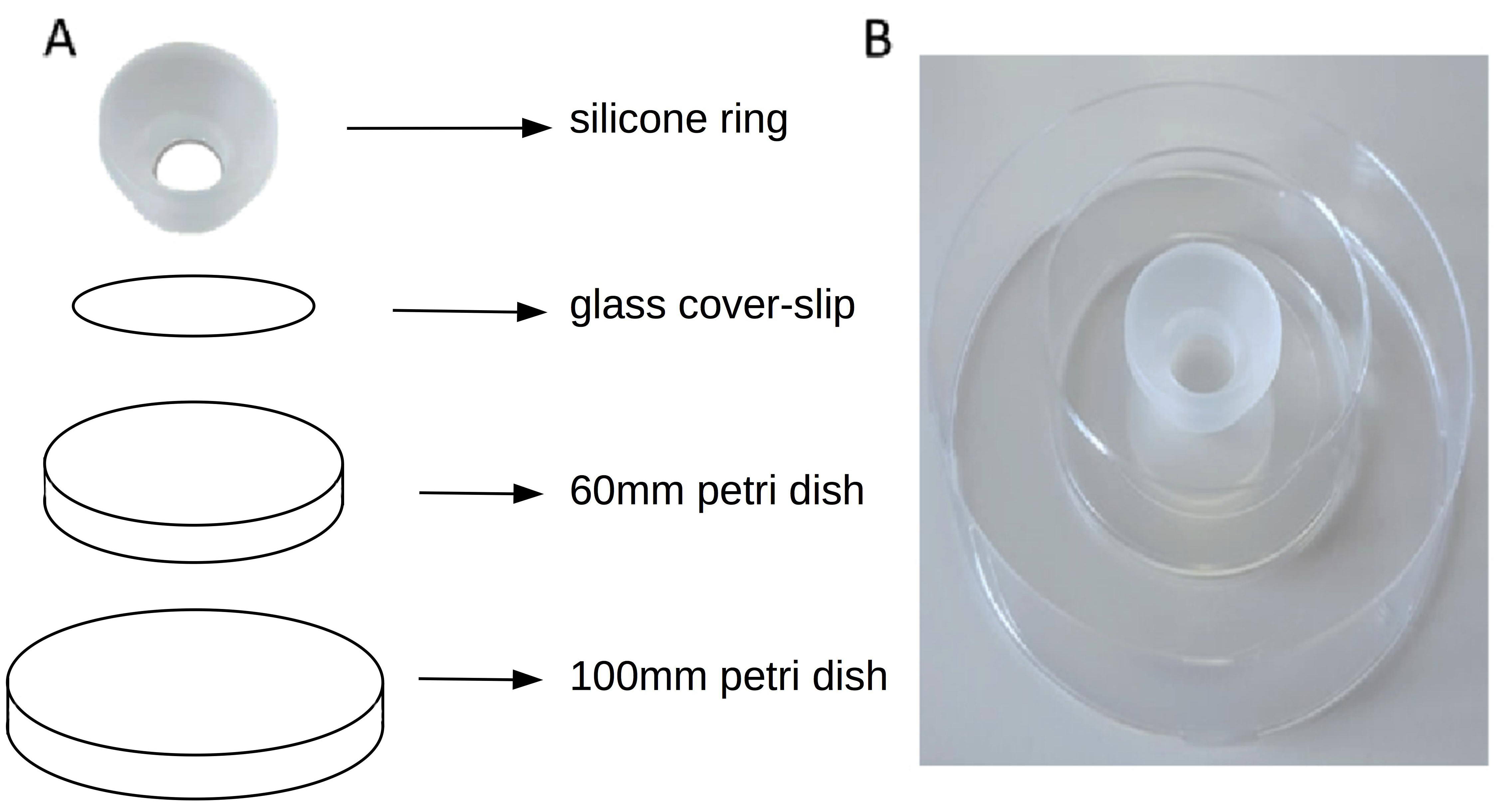
Figure 3. The Sebinger culture method. A. Shows the different component of the culture system. B. Shows the assembled Sebinger culture system. - Pipette a kidney rudiment into the approximate centre of the glass circle defined by the cone.
- Working quickly to avoid the risk of the tissue drying out, pipette away any medium carried over with the kidney rudiment, and place 85 μl of culture medium in the circle. It can be used straight from cold storage. Use a pipette tip to ensure that this medium spreads to cover the whole of the circle of glass bounded by the cone, including the kidney, and does not ‘bead’ into a single drop (Figure 4). Note that the optimal amount of medium varies slightly with batches of cones and with the glass. We therefore recommend that users try a series of volumes (e.g., 80, 82.5, 85. 97.5, 90 μl) with their own materials to determine the optimum.
- Place the 60 mm dish (with the cover slip, the silicone cone and the kidney rudiment now inside it) to the base of a 100 mm Petri dish and add 3 ml of hydration buffer into it (when the hydration buffer comes in contact with the silicone cone it detaches the cone from the cover slip and the system becomes leaky, this is why we use a separate dish to add the hydration buffer). (Figure 4)

Figure 4. The arrangement of the cone and dishes. A. Kidney rudiments are cultured in the centre of the silicone cone in a low volume of KCM medium and a humidifying buffer is added to the outer dish to prevent dryness. B. The KCM tends to bead as a single drop (left hand side image) and need to be distributed with a pipette tip to cover the glass circle enclosed by the silicone cone (right hand side image). - Place the lid on the 100 mm Petri dish then place the dish in a tissue culture incubator (37 °C, 5% CO2, 100% humidity). Kidneys can be cultured for 3 days; beyond that it is advisable to change the medium. A few cells at the periphery may egress and make a monolayer on the glass but the kidney as a whole should remain intact and coherent, as in Figure 1A. If it ‘rounds up’ but is still alive there is probably too much medium; if it rounds up as a dried mass, there is too little.
Note: For time-lapse filming, we recommend in-incubator microscopes from Etaluma. - Specimens should be fixed on their cover-slip, according to the protocol appropriate for any antibody staining to be used. We generally apply 100% methanol at -20 °C immediately after removing the culture medium, and allow it to warm to room temperature over 15 min, then wash in phosphate-buffered saline. If alternative fixation techniques involve detergents (Tween, Triton etc.), great care should be taken to change solutions very gently to avoid detaching the culture from the glass. Incubation in antibody is usually overnight at 4 °C in Bijou (5 ml) bottles. An example protocol, used for the laminin stain in Figure 1, is overnight incubation in 200 μl 1/100 anti-laminin (Sigma-Aldrich) in PBS at 4 °C, a 7 h wash in PBS at 4 °C, overnight incubation in 200 μl 1/100 FITC anti-rabbit (Sigma-Aldrich) in PBS at 4 °C, two 2 h washes in PBS at 4 °C, followed by mounting, still on the filter, on a slide in PBS or 1:1 PBS-glycerol, with the cover-slip being held away from the main slide with fragments a broken coverslip used as spacers. This prevents crushing of the tissue. We do not embed or section.
- Put the cover slips and the silicone cones in 80 °C oven for 20 min to dry (dryness is essential for proper adhesion between the silicone cone and the cover slips). They can be handled with gloved hands or forceps–use plastic forceps for cones to avoid damage that might result from the use of metal.
Data analysis
- This is a method of culture, rather than a method to make a specific measurement, and the method will support a range of questions and analyses. Its features do, however, make it better suited for some questions than others. For avoidance of false-positive data caused by individual variation, if embryos from more than one mother mouse are to be used we recommend pooling kidneys from all the embryos, followed by random allocation to experimental and control groups.
- The extent of manual manipulation involved in the method makes the final size and shape of the organ rudiment quite variable, and some do not grow at all, usually because the film of medium has become a drop as in Figure 4B, left panel. At the very least, experimenters need to settle some exclusion criteria so that failed cultures can be excluded fairly from analyses (ideally, before any analyses are made except for simple observation of the medium film). Variation in growth between samples, probably arising from tiny differences in placement of specimens in the film, meaning that measurements of features such as total organ area will generate large error bars and require many replicates before any meaningful comparisons of experimental and control kidneys can be made. We advise against using such a measure. Better quantitative measurements include the number of branch tips in the collecting duct tree, and the number of nephrons. Examples of branch tip counting analysis can be found in Fisher et al. (2001) and Michael et al. (2005). Even for this, there is always an ambiguity about how ‘T-shaped’ a branch end must be for it to be counted as two tips not one, and there is always some ambiguity about what is mature enough to be called a nephron. We therefore strongly advise that all samples are blind-coded or, if this is not practical, images of them are blind-coded and counted by someone who does not know whether samples come from experiment or control.
- Much information gained from observation of organ culture, whether normal or subject to experimental manipulation, is qualitative rather than quantitative: tubular morphology is an example. Again, researchers are encouraged to use blind-coding, perhaps using images viewed by multiple independent people, and perhaps to use a semi-quantitative scoring system even for morphology (for example, classification of a nephron as ‘normal’ or ‘abnormal’). Some data are about directions, for example the direction in which a Loop of Henle grows. Directions require a coordinate system. A simple coordinate system can be made by extending lines radially from, for example, the centre of the kidney (determined, for example, using ImageJ’s Centre of Mass function), or from the first branching point. Directions of loops of Henle, for example, can be measured in terms of the angle between the tubule of the loop and the radial line passing through its tip. Again, having multiple biological replicates and multiple people making measurements on the same blind-coded samples will provide good measurements of inter-observer variability and of inter-sample variability.
Recipes
- Cleaning solution
This consists of 5:1:1 (by volume) H2O:H2O2:NH4OH
Make this by adding the hydrogen peroxide and ammonium hydroxide to the water, not the other way round, and use personal protective equipment (gloves, eye protection) when handling the concentrated solutions - Dissecting medium
Dissolve Eagle’s minimal essential medium with Earle’s salts and non-essential amino acids in ddH2O as per manufacturer’s instruction - Culture medium: KCM
Note: Simply add the serum and antibiotics to the Eagle’s medium (Dissecting medium).
Dissecting medium
10% fetal bovine serum
1/100 penicillin/streptomycin stock (this stock contains 10.000 U/ml penicillin and 10 mg/ml streptomycin) - Hydration buffer
PBS (from Sigma-Aldrich tablets: allow an hour for the tablets to dissolve, and mix well, then add penicillin/streptomycin diluted from the 100x stock mentioned in Recipe 3 above)
Acknowledgments
Development and use of the technique described here has been supported by funding from the following; European Commission FP6 grant MRTN-CT-2006-036097, Medical Research Council grant MR/K010735/1, Kidney Research UK grant RP_002_20160223, and a PhD scholarship from the Egyptian Educational and Cultural Bureau. The protocol was first described in the methods section of Sebinger, 2010. The authors have no competing interests.
References
- Carrel, A. and Burrows, M. T. (1910). Cultivation of adult tissues and organs outside the body. J Am Med Ass 55: 1379-1381.
- Chang, C. H. and Davies, J. A. (2012). An improved method of renal tissue engineering, by combining renal dissociation and reaggregation with a low-volume culture technique, results in development of engineered kidneys complete with loops of Henle. Nephron Exp Nephrol 121(3-4): e79-85.
- Davies, J. A. (2006). A method for cold storage and transport of viable embryonic kidney rudiments. Kidney Int 70(11): 2031-2034.
- Davies, J. A., Hohenstein, P., Chang, C. H. and Berry, R. (2014). A self-avoidance mechanism in patterning of the urinary collecting duct tree. BMC Dev Biol 14: 35.
- Davies, J. A., Ladomery, M., Hohenstein, P., Michael, L., Shafe, A., Spraggon, L. and Hastie, N. (2012). Development of an siRNA-based method for repressing specific genes in renal organ culture and its use to show that the Wt1 tumour suppressor is required for nephron differentiation. Hum Mol Genet 2004: 235-46.
- Falk, M., Salmivirta, K., Durbeej, M., Larsson, E., Ekblom, M., Vestweber, D. and Ekblom, P. (1996). Integrin alpha 6B beta 1 is involved in kidney tubulogenesis in vitro. J Cell Sci 109 (Pt 12): 2801-2810.
- Fisher, C. E., Michael, L., Barnett, M. W. and Davies, J. A. (2001). Erk MAP kinase regulates branching morphogenesis in the developing mouse kidney. Development 128(21): 4329-4338.
- Georgas, K., Rumballe, B., Valerius, M. T., Chiu, H. S., Thiagarajan, R. D., Lesieur, E., Aronow, B. J., Brunskill, E. W., Combes, A. N., Tang, D., Taylor, D., Grimmond, S. M., Potter, S. S., McMahon, A. P. and Little, M. H. (2009). Analysis of early nephron patterning reveals a role for distal RV proliferation in fusion to the ureteric tip via a cap mesenchyme-derived connecting segment. Dev Biol 332(2): 273-286.
- Grobstein, C. (1953). Inductive epithelio-mesenchymal interaction in cultured organ rudiments in the mouse. Science 118: 52-55.
- Hendry, C., Rumballe, B., Moritz, K. and Little, M. H. (2011). Defining and redefining the nephron progenitor population. Pediatr Nephrol 26(9): 1395-1406.
- Lindstrom, N. O., Chang, C. H., Valerius, M. T., Hohenstein, P. and Davies, J. A. (2015). Node retraction during patterning of the urinary collecting duct system. J Anat 226(1): 13-21.
- Michael, L., Sweeney, D. E. and Davies, J. A. (2005). A role for microfilament-based contraction in branching morphogenesis of the ureteric bud. Kidney Int 68(5): 2010-2018.
- Munro, D. A. D., Hohenstein, P. and Davies, J. A. (2017). Cycles of vascular plexus formation within the nephrogenic zone of the developing mouse kidney. Sci Rep 7(1): 3273.
- Piscione, T. D., Yager, T. D., Gupta, I. R., Grinfeld, B., Pei, Y., Attisano, L., Wrana, J. L. and Rosenblum, N. D. (1997). BMP-2 and OP-1 exert direct and opposite effects on renal branching morphogenesis. Am J Physiol 273(6 Pt 2): F961-975.
- Reinhoff, W. F. (1922). Development and growth of the metanephros or permanent kidney in chick embryos. Johns Hopkins Hospital Bulletin 33: 392-406.
- Saxén, L., Vainio, T. and Toivonen, S. (1962). Effect of polyoma virus on mouse kidney rudiment in vitro. J Natl Cancer Inst 29: 597-631.
- Schreiner, K. E. (1902). Ueber die entwicklung der amniotenniere. Zeitsch. f. wiss. Zool. 71: 1-188.
- Sebinger, D. D., Unbekandt, M., Ganeva, V. V., Ofenbauer, A., Werner, C. and Davies, J. A. (2010). A novel, low-volume method for organ culture of embryonic kidneys that allows development of cortico-medullary anatomical organization. PLoS One 5(5): e10550.
- Trowell, O. A. (1954). A modified technique for organ culture in vitro. Exp Cell Res 6(1): 246-248.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Elhendawi, M. and Davies, J. A. (2018). Sebinger Culture: A System Optimized for Morphological Maturation and Imaging of Cultured Mouse Metanephric Primordia. Bio-protocol 8(4): e2730. DOI: 10.21769/BioProtoc.2730.
Category
Cell Biology > Cell isolation and culture > Organ culture
Developmental Biology > Morphogenesis > Organogenesis
Cell Biology > Cell imaging > Fluorescence
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.