- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Adapting the Smart-seq2 Protocol for Robust Single Worm RNA-seq

Published: Vol 8, Iss 4, Feb 20, 2018 DOI: 10.21769/BioProtoc.2729 Views: 20840
Reviewed by: Pengpeng LiHillel SchwarzLokesh Kalekar
Original research article
The authors used this protocol in:

Apr 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Most nematodes are small worms that lack enough RNA for regular RNA-seq protocols without pooling hundred to thousand of individuals. We have adapted the Smart-seq2 protocol in order to sequence the transcriptome of an individual worm. While developed for individual Steinernema carpocapsae and Caenorhabditis elegans larvae as well as embryos, the protocol should be adaptable for other nematode species and small invertebrates. In addition, we describe how to analyze the RNA-seq results using the Galaxy online environment. We expect that this method will be useful for the studying gene expression variances of individual nematodes in wild type and mutant backgrounds.
Keywords: RNA-seqBackground
Low input RNA-seq protocols and amplification kits, such as Smart-seq (Takara Bio, USA, Inc) and SuperAmp (Miltenyl Biotec, Inc), have been increasingly developed and commercialized as a response to the growing prevalence of low input RNA-seq studies based on small tissues, single microorganisms, and single cells. These studies often explore and address heterogeneous gene expression among individuals of a certain population, such as a population of cells, a complex tissue, or a population of microscopic organisms. Improvements and adaptations of low input RNA-seq protocols for microscopic organisms, such as nematodes, will greatly benefit the field of nematology by allowing for the analysis of gene expression heterogeneity at the single nematode level. Here we have adapted the single cell RNA-seq protocol, Smart-seq2 (Picelli et al., 2013 and 2014; Trombetta et al., 2014), for single nematode RNA-sequencing. We successfully utilized adapted versions of this protocol in the transcriptomic analysis of the insect-parasitic nematode, Steinernema carpocapsae (Lu et al., 2017) as well as in the analysis of individual embryos and L1 larvae from two Steinernema and two Caenorhabditis species including C. elegans (Macchietto et al., 2017), but this protocol can be adapted for any species of nematode. While this protocol will work on nematodes without already sequenced genomes or transcriptomes, we limit our computational analysis to organisms with published genome annotations, such as S. carpocapsae (Dillman et al., 2015). Our need for single nematode RNA-sequencing arose as a method to circumvent the limitations of working with samples with low-inputs of RNA. For example, many of our in vivo experiments limited the number of nematodes we could utilize. Single nematode RNA-seq has allowed us to efficiently obtain high resolution gene expression data from these nematodes. The protocol has also enabled us to collect individual embryos to map out time courses of nematode embryonic development for comparative transcriptomics across multiple species. The development and advancement of low input RNA-seq protocols will aid investigators in circumventing issues related to using individual organisms and specialized/limited samples.
Materials and Reagents
- Gloves
- 8-strip, nuclease-free, 0.2-ml, thin-walled PCR tubes with caps (SARSTEDT, catalog numbers: 72.985.002 and 65.989.002 )
- Needle 25 G 1.5 inch regular (BD, PrecisionGlide, catalog number: 305127 )
- QubitTM assay tubes (Thermo Fisher Scientific, InvitrogenTM, catalog number: Q32856 )
- Pipette tips
- 1.5 ml Eppendorf tube
- Spatulas
- 70% ethanol or RNase away
- Proteinase K (QIAGEN, catalog number: 19131 )
- RNasin ribonuclease inhibitor (RNase inhibitor) (Promega, catalog number: N2611 )
- UltraPure DNase/RNase free distilled water (Thermo Fisher Scientific, GibcoTM, catalog number: 10977015 )
- Oligo-dT30VN primer (ordered from IDT (https://www.idtdna.com/site)): 5’-AAGCAGTGGTATCAACGCAGAGTACT30VN-3’
Note: This oligonucleotide anneals to all the RNAs containing a poly(A) tail. The 3’ end of this oligonucleotide contains ‘VN’, where ‘N’ is any base and ‘V’ is either A, C or G. The two terminal nucleotides are necessary for anchoring the oligonucleotide to the beginning of the poly(A) tail and for avoiding unnecessary amplification of long stretches of adenosines. Dissolve the oligonucleotide in TE buffer to a final concentration of 100 μM. Store this oligo at -20 °C for 6 months. - dNTP mix (10 mM each) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: R0192 )
- Superscript II reverse transcriptase kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: 18064014 )
- LNA-modified TSO (ordered from Exiqon (http://www.exiqon.com/))
5’-AAGCAGTGGTATCAACGCAGAGTACATrGrG+G-3’
Note: At the 5’ end, this TSO carries a common primer sequence, whereas, at the 3’ end, there are two riboguanosines (rG) and one LNA-modified guanosine (+G) to facilitate template switching. TSO dissolved in TE buffer can be stored in 100 μM aliquots at -80 °C for 6 months. Avoid repeated freeze-thaw cycles. - Betaine (BioUltra ≥ 99.0%) (Sigma-Aldrich, catalog number: 61962 )
- Magnesium chloride (MgCl2; anhydrous) (Sigma-Aldrich, catalog number: M8266 )
- Kapa HiFi HotStart ReadyMix (Kapa Biosystems, catalog number: KK2602 )
- IS PCR oligo (ordered from IDT (https://www.idtdna.com/site))
5’-AAGCAGTGGTATCAACGCAGAGT-3’
Note: This oligonucleotide acts as PCR primer in the amplification step after RT. Dissolve the oligonucleotide in TE buffer to a final concentration of 100 μM. This oligo can be stored at -20 °C for 6 months. - Agencourt Ampure XP beads (Beckman Coulter, catalog number: A63881 )
- Ethanol 99.5% (vol/vol) (Kemethyl, catalog number: SN366915-06 )
- QubitTM dsDNA HS assay kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: Q32854 )
- Agilent high sensitivity DNA kit (Agilent Technologies, catalog number: 5067-4626 )
- Buffer PM (QIAGEN, catalog number: 19083 )
- Nextera DNA library prep kit (24 samples) (Illumina, catalog number: FC-121-1030 )
- QIAquick PCR purification kit (50) (QIAGEN, catalog number: 28104 )
- Phusion high fidelity PCR master mix with HF buffer, 500 reactions (New England Biolabs, catalog number: M0531L )
- Tris-HCl pH 8.0 (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM9850G )
- Triton X-100 (Sigma-Aldrich, catalog number: T9284 )
- EDTA pH 8.0 (Mediatech, catalog number: 46-034-Cl )
- Polysorbate 20, Acros OrganicsTM (Tween 20) (Acros Organics, catalog number: 233362500 )
- RNaseZap (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM9780 )
- List of sequencing indexes (Buenrostro et al., 2015) (see Supplemental file)
Equipment
- Pipettes
- Mini-centrifuge with head for 8-strip PCR tubes
- Vortexer
- Thermocycler
- Stereo microscope
- Qubit® Fluorometer
- Agilent 2100 Bioanalyzer (Agilent Technologies, model: Agilent 2100 , catalog number: G2938C)
- Magnetic stand 96 (Thermo Fisher Scientific, catalog number: AM10027 )
Procedure
Warning: RNA is easily degradable by RNases and not very stable on ice! Therefore, clean all surfaces with 70% ethanol or RNase away, change gloves often and sterilize all materials used. Lysed samples should not be kept on ice for more than one hour as mRNA will degrade.
Notes:
- This protocol can be used for all larval stages, dauers and embryos of nematodes.
- A new sterile needle should be used for each worm.
- Isolating and lysing nematodes from cultured plates
Note: Prepare the buffers freshly on the day of worm collection.- Prepare lysis buffer (see Recipes in the end of the protocol) with Proteinase K by combining 46.8 µl of lysis buffer stock with 3.2 µl of Proteinase K for a total of 50 µl.
- From Step A1 combine 18 µl of lysis buffer with Proteinase K with 2 µl RNase inhibitor for a total of 20 µl of lysis buffer.
- Use the lysis buffer with Proteinase K and RNasin ribonuclease inhibitor for lysing worms.
- Set up a station for nematode collection (Figure 1A).
- Wash nematodes three times with deionized water:
- To wash nematodes from cultured plates, add 1 ml of water to plate.
- Swirl the plate three times to dislodge nematodes.
- Transfer worms with a pipette to a 1.5 ml Eppendorf tube.
- Briefly spin down.
- Remove supernatant and bring up to 1 ml with water.
- Repeat Steps A5d and A5e two more times or until the water becomes clear.
- To wash nematodes from cultured plates, add 1 ml of water to plate.
- Collect one worm using a pipette in 2 µl of water and transfer to the wall of PCR tube (Figure 1B). Cut the worm with 25 gauge needle (Figures 1C-1D) (Video 1).
Note: Cutting is suggested for all larval stages. Note that we did not cut the embryos.
 Video 1. A video demonstrating the procedure for cutting a single nematode in preparation for RNA extraction
Video 1. A video demonstrating the procedure for cutting a single nematode in preparation for RNA extraction - Add 2 µl lysis buffer with Proteinase K and RNasin ribonuclease inhibitor.
- Flick tube to mix, and spin down tubes in a mini-centrifuge.
- Incubate samples on the thermocycler using the following program on PCR cycler.
- 65 °C–10 min
- 85 °C–1 min
- 4 °C
- 65 °C–10 min
- Transfer samples to ice.
- Add 1 µl oligo-dT VN primer (10 μM) and 1 µl dNTP mix to each tube. Flick tubes to mix, and spin down tubes.
- Incubate samples on the thermocycler at 72 °C for 3 min.
- Take samples from the thermocycler and immediately place them on ice.

Figure 1. Preparing and cutting an individual nematode for lysis. A. A picture of the setup used to isolate, cut, and prepare individual nematodes for RNA-seq. B. A picture of an individual nematode isolated in a pipette tip, being transferred to a PCR tube to be cut. C. A picture of a single nematode in 2 µl of DEPC-treated water. D. A picture of a single nematode in a PCR tube that has been cut in half using a 25 gauge needle. In panels B-D the individual nematode is circled in red.
- Prepare lysis buffer (see Recipes in the end of the protocol) with Proteinase K by combining 46.8 µl of lysis buffer stock with 3.2 µl of Proteinase K for a total of 50 µl.
- Smart-seq2 reverse transcription (RT) of mRNA
- Prepare reverse transcription master mix (RT MM). (No DNase required.)

- Add 5.7 μl of RT MM to each PCR tube.
- Quickly vortex and spin down in a mini-centrifuge.
- Put on thermocycler and run the following program:

Note: Stop point. Store samples -20 °C.
- Prepare reverse transcription master mix (RT MM). (No DNase required.)
- Smart-seq2 PCR amplification (Figure 2)
- Place samples on ice. Prepare KAPA HiFi master mix.

- Add 15 μl of KAPA HiFi master mix to each sample.
- Put on thermocycler and run the following program:

Note: Stopping point. Samples can be stored at -20 °C indefinitely.
Figure 2. Overview of the Smart-seq2 protocol adapted for individual nematodes. Modified from Picelli et al. (2014). 1. A nematode is cut and lysed to release total RNA. 2. The Oligo(dT) binds the poly(A) tail at the 3’ end of mRNA (section A of Procedure). 3. The 1st strand of cDNA is synthesized by MMLV reverse transcriptase (RT) which adds non-template guided cytosines at the 3’ end of the cDNA. These cytosines are used to anchor the LNA-modified TSO. Reverse transcriptase then uses the TSO as a template to complete the 1st strand (section B of Procedure). 4. The ISPCR primer anneals to the 3’ end of the 1st strand allowing DNA polymerase to bind and to synthesize the 2nd strand of cDNA and subsequently amplify the cDNA (section C of Procedure). 5. The double-stranded (ds) cDNA is purified and checked for quality (sections D and E of Procedure). 6. Transposomes tagment the ds cDNA by fragmenting it and ligating adapter sequences (section F of Procedure). 7. The tagmented ds cDNA is cleaned and 8. Ligated to sample specific indexes (section G of Procedure).
- Place samples on ice. Prepare KAPA HiFi master mix.
- cDNA bead clean-up
- Take samples out of -20 °C and let them sit at room temperature for 10 min.
- Take Ampure XP beads from fridge and vortex at high speed for 1 min. The solution should be homogenous.
- Aliquot 26 μl of Ampure XP beads into a 1.5 ml Eppendorf tube. Beads need to be warmed up to room temperature approximately 8 min.
- Add the sample (~26 μl) to the Ampure XP beads (1:1). Pipette up and down 10 times until thoroughly mixed.
- Incubate the sample with the Ampure XP beads for 8 min at room temperature on a tube rack.
- Make 600 μl of fresh 80% nuclease-free ethanol.
- After incubation, place sample on a magnetic bead stand for 5 min.
Note: The beads will be bound to cDNA of interest. - Remove supernatant using pipette without disturbing the beads.
Note: Place the supernatant into the old sample PCR tube and keep it until the cDNA concentration is checked. - Leave tubes on magnetic bead stand, add 200 μl of 80% ethanol to the beads. Wait 30 sec.
- Remove supernatant using a needle attached to a vacuum line or a pipette, being careful not to disturb the beads.
Note: Do not let beads dry out. - Repeat Steps D9-D10 two more times.
- Let beads dry on the magnetic stand for ~5 min with the cap open. Monitor the drying.
- When the beads begin to crack, remove the tube from the magnet and immediately add 17.5 μl of EB buffer to the beads. Pipette up and down 10 times to thoroughly mix. The solution should be homogenous and brown.
- Incubate the sample on a tube rack at room temperature for 2 min.
- Put the sample back on the magnetic bead stand for 2 min or until the liquid turns clear. Collect 15 μl of supernatant from the tube while on the magnetic stand. Be careful not to disturb beads. Place the supernatant into a clean labeled Eppendorf tube and place on ice.
Note: There should be no brown specs in the liquid collected. If you aspirate any beads, return the sample to the bead tube and repeat Steps D14-D15. - Discard tubes left in the magnetic stand.
- Take samples out of -20 °C and let them sit at room temperature for 10 min.
- Measurement of cDNA concentration and sample quality check
Note: Calibrate the Qubit Fluorometer according to manufacturer’s guide.- Qubit reagents for one sample:
198 μl of dsDNA HS Buffer
1 μl of dsDNA HS reagent 200x
1 μl of cDNA
Add the dsDNA HS reagent, dsDNA HS buffer and cDNA to a Qubit tube. Quickly vortex and spin down in a mini-centrifuge. - Incubate the tube for 2 min in the dark.
- Place cDNA sample into the Qubit fluorometer. On the Qubit screen, select ‘DNA’, select ‘High Sensitivity’, and select ‘read sample’. Select ‘read stock concentration’ and change the volume to ‘1 μl’. Record the cDNA concentration (ng/μl).
- Run BioAnalyzer (BA) on the cDNA following the protocol for the Agilent High Sensitivity DNA Kit.
Samples should be run on a BioAnalyzer machine using an Agilent High Sensitivity DNA Kit to generate profiles of the distribution of cDNA lengths (representative of the mRNA transcript lengths) in each sample. If the sample is not degraded, you will see a clear peak in the fragment sizes in the 500-10,000 bp range with no peaks at smaller fragment sizes indicative of full length cDNA resulting from full length mRNA transcripts (Figure 3A). The profile will show no peaks or many small peaks if the mRNA transcripts were degraded (likely due to RNase contamination or mishandling of the RNA) (Figure 3B).
Figure 3. Examples of Smart-seq2 BioAnalyzer (BA) profiles. A. An ideal BA profile of cDNA showing full length transcripts with large peaks around 1,000 bp and no PCR primer contamination (70 bp). B. A BA profile of cDNA made from highly degraded RNA resulting in an abundance of small fragments shown by multiple peaks between 35 and 300 bp.
- Qubit reagents for one sample:
- Tagmentation of cDNA (Figure 2)
Notes:- All reagents and samples should be kept on ice.
- This protocol uses the standard Nextera DNA library prep kit rather than Nextera XT DNA sample prep kit.
- Buffer PM is used because it has acetic acid and consequentially addresses the pH value of samples.
- Set a heating block to 55 °C and warm 30 μl of EB buffer.
- Calculate how to get 20 ng of cDNA from sample in 8 μl (see example below).

- In a PCR tube place 20 ng of Ampure-purified PCR product. Add EB to bring volume to 8 μl.
- In a NEW PCR tube, add 10 μl of Tagment DNA buffer from Nextera DNA Library Prep Kit.
- Add 2.2 μl Tagment DNA enzyme 1 from Nextera DNA Library prep Kit.
- Set a new P20 pipette to 15 μl and mix the transposase enzyme and transposase buffer by pipetting up and down 10 times.
- Transfer all of the mixed transposase enzyme and buffer into your cDNA and IMMEDIATELY pipette up and down at least 10 times to mix very well.
- Place sample on the PCR block for 5 min at 55 °C to initiate the tagmentation reaction.
- While sample is on PCR block, obtain a QIAquick DNA cleanup column, buffer PM and PE. The tagmented cDNA will be passed through this column to remove the enzyme from the DNA fragments.
- Add 60 μl of buffer PM to the 20 μl of tagmented cDNA. Mix well by pipetting up and down until the solution becomes clear.
- Pipette the entire volume into the QIAGEN spin column.
- Spin the QIAGEN column down in a centrifuge at 12,470 x g for 1 min. The cDNA will stick to the white filter in the column, and the liquid will pass through into the collection tube.
- Dump the flow-through into the trash. Place the column back inside the collection tube.
- Add 750 μl of buffer PE to the column.
- Spin the QIAGEN column down in a centrifuge at 12,470 x g for 1 min.
- Dump the flow-through into the trash. Place the column back inside the collection tube.
- Spin the QIAGEN column down in a centrifuge at 12,470 x g for 1 min.
- Place the column into a new, clean the Eppendorf tube that is labelled with the sample name.
- Add 30 μl of warmed EB buffer to the center of the white filter inside the column, being careful to not touch the filter with the pipette tip or to pipette the buffer onto the walls of the column.
- Let the column sit for 1 min.
- Spin down the column in a centrifuge at 12,470 x g for 1 min.
- Remove the column and keep the flow-through.
- The flow-through is the tagmented cDNA.
Note: At this step samples can be handed to the facility at which they will be sequenced. If barcodes are to be added by researcher, follow the next steps.
- All reagents and samples should be kept on ice.
- Amplification of adapter-ligated fragments (Figure 2)
- In a PCR tube add the following reagents:

- Put samples on PCR cycler and run the following program (Buenrostro et al., 2015):

Note: After the stopping point, samples can be stored at -20 °C indefinitely.
- In a PCR tube add the following reagents:
- DNA library bead clean-up
Follow steps described in Procedure D with the following changes:- Step D3, use 70 μl of Ampure XP beads (1:1).
- Step D4, add the sample (~70 μl).
- Step D14, add 30 μl of EB.
- Step D16, collect 27.5 μl of supernatant.
- Step D3, use 70 μl of Ampure XP beads (1:1).
- Measurement of DNA library concentration and sample quality check
- Follow steps in Procedure E to acquire DNA library concentration with Qubit.
- Run BioAnalyzer to check quality of library. (Optional, but highly recommended!)
Samples should be run on a BioAnalyzer to check for quality of tagmentation from Procedure F. A fully tagmented library will have a BioAnalyzer profile with a clear peak around 200 bp (Figure 4A). A partially tagmented library will have a BioAnalyzer profile with a uniform plateau between 150 to 1,000 bp and possibly some smaller peaks below 150 bp or above 1,000 bp (Figure 4B).
Figure 4. Examples of Smart-seq2 BioAnalyzer profiles after tagmentation and library preparation. A. An example of a sample that has been fully tagmented shows one peak around 200 bp. B. An example of a sample that was not fully tagmented shows a broad distribution of fragments ranging from 150-1,000 bp.
- Follow steps in Procedure E to acquire DNA library concentration with Qubit.
- Library sequencing
The libraries can be handed to a sequencing facility. Samples are sequenced using pair-end, 43 base pair reads. Pair-end sequencing improves the quality of the dataset enabling reads to more accurately align to the reference genome. Libraries can also be sequenced with single-end reads. Each sample should be sequenced to a depth of at least 10 million reads to reliably detect one transcript per million (TPM).
Data analysis
Analysis of Smart-seq2 data
RNA-seq libraries are typically sequenced to a depth of 10-20 million reads to reliably profile gene expression levels in samples of interest. Although fast and reliable software such as STAR (Dobin et al., 2013), Kallisto (Bray et al., 2016), and Salmon (Patro et al., 2017) have been developed for analyzing reads and obtaining gene/isoform expression levels, analysis of high throughput data usually requires computing and data storage resources beyond the capabilities of a typical desktop or laptop machine. Such tasks require access to a high performance computing cluster or online resources such as Galaxy (Afgan et al., 2016). Galaxy provides free access to analysis tools and computing/storage resources through a user-friendly online interface. Sequencing data generated using any of the various sequencing protocols can be uploaded to Galaxy that has appropriate analysis tools available to obtain interpretable results. Here we present a detailed tutorial for uploading Steinernema carpocapsae RNA-seq data to Galaxy and obtaining gene expression levels using Salmon.
- Creating a user account
Use of Galaxy resources requires a user account. A free user account can be obtained by registering at Galaxy. Log on to usegalaxy.org using any web browser of choice.- At the webpage menu click on ‘Login or Register’ and then ‘Register’.
- Enter a valid email address and fill in the required information.
- Log in to Galaxy when prompted.
- Open the email sent to the email address you provided in the step ‘2’ and click on the link provided to activate your account.
- At the webpage menu click on ‘Login or Register’ and then ‘Register’.
- Uploading data to Galaxy (Figure 5)
- RNA-Seq analysis using Salmon requires RNA-Seq data (fastq file format) and the transcriptome (fasta file format). The transcriptome for S. carpocapsae can be downloaded from WormBase ParaSite website as follows.
- Log on to parasite.wormbase.org/index.html.
- Click on ‘Downloads’.
- Search for ‘Steinernema carpocapsae’ on the list.
- Under ‘Full-length Transcripts’, click on ‘FASTA’ to download the gene sequences of S. carpocapsae in fasta format.
- Log on to parasite.wormbase.org/index.html.
- Paired-end S. carpocapsae RNA-seq data from Lu et al. (2017) can be downloaded at GEO under accession GSE89961.
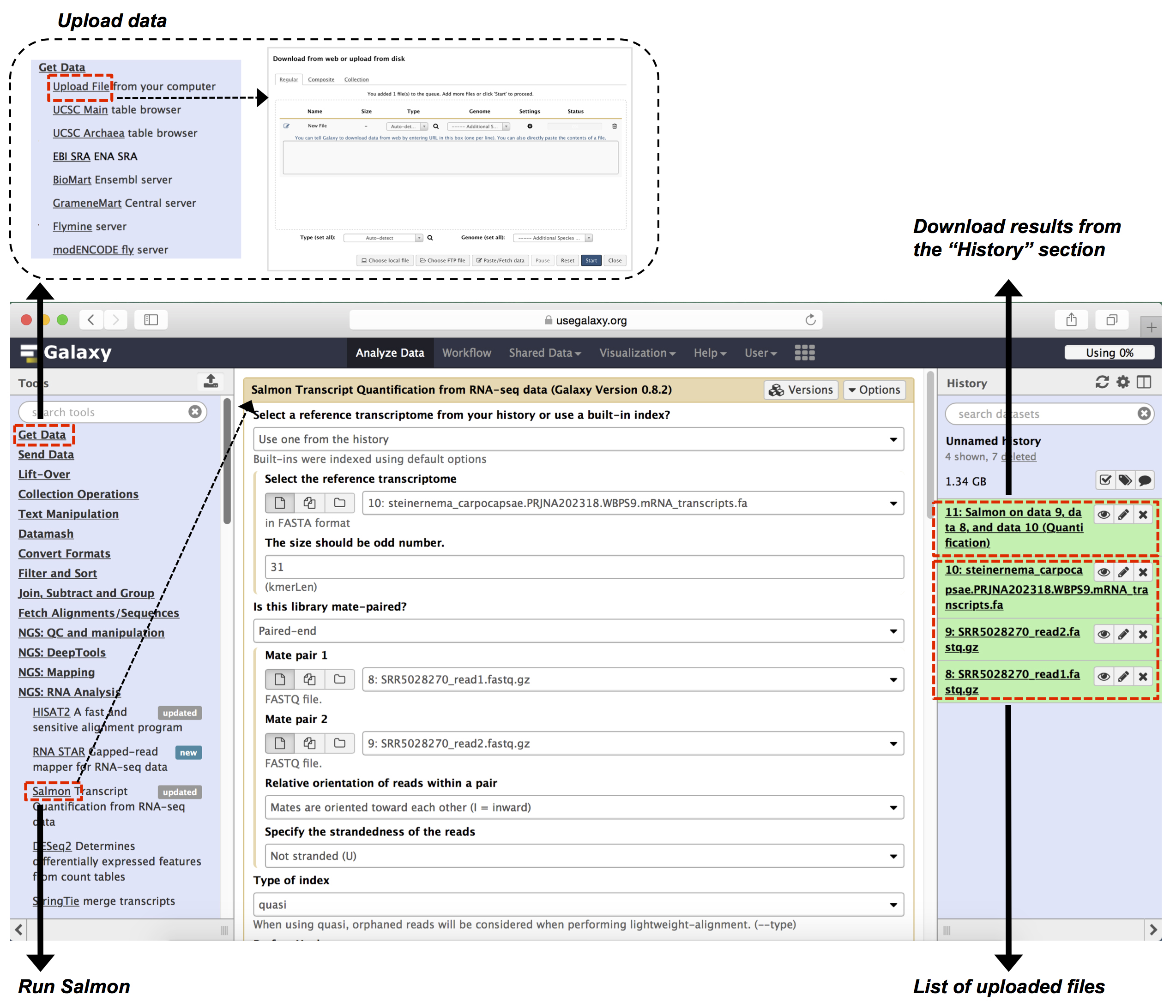
Figure 5. Galaxy interface to demonstrate sections B-C of Data analysis. User uploads data at ‘Get Data’ as indicated at top left (section B of Data analysis). Salmon is run for gene expression quantification as shown in bottom left (section C of Data analysis). Data can be downloaded from ‘History’ section on right side of page (Step C11 of Data analysis). - Data can be uploaded to Galaxy using two methods:
- Locally from your computer:
- On Galaxy webpage under ‘Tools’ menu, click on ‘Get Data’ and select ‘Upload File from your computer’.
- Click on ‘Choose local file’, and select the transcriptome fasta file. Under the ‘Type (set all):’ drop down menu, select ‘fasta’. Click ‘Start’ to upload the file.
- To upload the fastq files, click ‘Choose local file’ again and select the fastq file. Under the ‘Type (set all):’ drop down menu, select ‘fastq’. Click ‘Start’ to upload the file.
- On Galaxy webpage under ‘Tools’ menu, click on ‘Get Data’ and select ‘Upload File from your computer’.
- From a web-accessible link (preferred method)
- On the Galaxy webpage under the ‘Tools’ menu, click on ‘Get Data’ and select ‘Upload File from your computer’.
- Click ‘Paste/Fetch data’ and enter the URL of the transcriptome fasta file. Under the ‘Type’ drop down menu, select ‘fasta’. Click ‘Start’ to upload the file.
- Click ‘Paste/Fetch data’ and enter the URL of the paired-end fastq files. Be sure that each URL starts on a different line. Under the ‘Type’, drop down menu select ‘fastq’. Click ‘Start’ to upload the files.
- When the files are successfully uploaded, the background of the file name field in the history panel on the right of webpage turns green.
- On the Galaxy webpage under the ‘Tools’ menu, click on ‘Get Data’ and select ‘Upload File from your computer’.
- Locally from your computer:
- RNA-Seq analysis using Salmon requires RNA-Seq data (fastq file format) and the transcriptome (fasta file format). The transcriptome for S. carpocapsae can be downloaded from WormBase ParaSite website as follows.
- Running Salmon (Figure 5)
Salmon can be used to obtain gene expression levels from RNA-seq data by aligning reads and normalizing the aligned read counts to obtain normalized expression levels in the form of TPM (transcripts per million).- On the ‘Tools’ menu, under ‘NGS: RNA Analysis’, select ‘Salmon’.
- Under ‘Select the reference transcriptome’ in the middle panel, select the uploaded transcriptome fasta file.
- Under ‘Is this library mate-paired’, select ‘Paired-end’.
- For ‘Mate pair 1’, select the uploaded mate pair 1 fastq file.
- For ‘Mate pair 2’, select the uploaded mate pair 2 fastq file.
- The parameters for Salmon should be set according to the specific type of RNA-seq data to be analyzed which is determined by the library preparation protocol used. An important parameter that needs attention is ‘Specify the strandedness of the reads’. The Lu et al. (2017) RNA-seq data is unstranded, therefore the value for the above parameter should be set to default ‘Not stranded (U)’. Also, under both ‘Perform sequence-specific bias correction’ and ‘Perform fragment GC bias correction’, select ‘Yes’. Select default values for the remaining parameters. For a full description of the parameters please refer to Salmon manual (https://combine-lab.github.io/salmon/).
- Click ‘Execute’ to start the analysis.
- The status of the job is shown under a new field in the ‘History’ panel to the right.
- Click the refresh button to get an updated status of the job. The time for the submitted job to finish varies depending on the queue size on Galaxy and the number of reads uploaded. Usually it takes less than one hour for a Salmon job to finish.
- The job field background in the history panel on the right side of the webpage turns green when the job completes successfully.
- Once complete, a tab-delimited text file providing the gene/isoform names, a number of reads mapped to the genes/isoforms, and TPM (relative abundances) can be downloaded from the job field in the history panel.
- The gene expression results from step 11 can be further analyzed using tools provided through Galaxy such as DESeq2 (Love et al., 2014). e.g., differentially expressed genes/isoforms between experimental conditions/treatments can be identified.
- On the ‘Tools’ menu, under ‘NGS: RNA Analysis’, select ‘Salmon’.
Notes
- For embryos, we have also used 0.3% Triton-X for the lysis buffer.
- In our hands, 8 out 10 samples pass the first quality check using the BioAnalyzer.
- The lysis buffer must be made with sterile Eppendorf tubes, spatulas, and pipette tips.
- A new pipette tip should be used for each worm during collection.
- Samples that sequence poorly, with less than 1 million reads, or that correlate poorly with other biological replicates are excluded from analysis.
Recipes
- Lysis buffer stock (Shaham et al., 2006)
Notes:- Lysis buffer stock can be stored indefinitely wrapped in foil at room temperature.
- Aliquot Triton-X (100%) to a 1.5 ml Eppendorf tube and warm to 35 °C and use as described in Recipe.
20 µl Triton-X (100%)
200 µl Tween-20 (10%)
2 µl 0.5 M EDTA
1.628 ml nuclease free water
Total 1.871 ml - Lysis buffer stock can be stored indefinitely wrapped in foil at room temperature.
Acknowledgments
We would like to thank all members of the Mortazavi and Dillman labs for helpful discussions. This work was supported by an NIH New Innovator Award to A.M. (NIGMS DP2 GM111100). The authors declare that they have no conflicts of interest or competing interests.
References
- Afgan, E., Baker, D., van den Beek, M., Blankenberg, D., Bouvier, D., Cech, M., Chilton, J., Clements, D., Coraor, N., Eberhard, C., Gruning, B., Guerler, A., Hillman-Jackson, J., Von Kuster, G., Rasche, E., Soranzo, N., Turaga, N., Taylor, J., Nekrutenko, A. and Goecks, J. (2016). The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res 44(W1): W3-W10.
- Bray, N. L., Pimentel, H., Melsted, P. and Pachter, L. (2016). Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34(5): 525-527.
- Buenrostro, J. D., Wu, B., Chang, H. Y. and Greenleaf, W. J. (2015). ATAC-seq: A method for assaying chromatin accessibility genome-wide. Curr Protoc Mol Biol 109: 21 29 21-29.
- Dillman, A. R., Macchietto, M., Porter, C. F., Rogers, A., Williams, B., Antoshechkin, I., Lee, M. M., Goodwin, Z., Lu, X., Lewis, E. E., Goodrich-Blair, H., Stock, S. P., Adams, B. J., Sternberg, P. W. and Mortazavi, A. (2015). Comparative genomics of Steinernema reveals deeply conserved gene regulatory networks. Genome Biol 16: 200.
- Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., Batut, P., Chaisson, M. and Gingeras, T. R. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29(1): 15-21.
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2 (2014). Genome Biol 15(12): 550.
- Lu, D., Macchietto, M., Chang, D., Barros, M. M., Baldwin, J., Mortazavi, A. and Dillman, A. R. (2017). Activated entomopathogenic nematode infective juveniles release lethal venom proteins. PLoS Pathog 13(4): e1006302.
- Macchietto, M., Angdembey, D., Heidarpour, N., Serra, L., Rodriguez, B., El-Ali, N. and Mortazavi, A. (2017). Comparative transcriptomics of Steinernema and Caenorhabditis single embryos reveals orthologous gene expression convergence during late embryogenesis. Genome Biol Evolution 9(10): 2681-2696.
- Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. and Kingsford, C. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14(4): 417-419.
- Picelli, S., Bjorklund, A. K., Faridani, O. R., Sagasser, S., Winberg, G. and Sandberg, R. (2013). Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nature Methods 10(11): 1096-1098.
- Picelli, S., Faridani, O. R., Bjorklund, A. K., Winberg, G., Sagasser, S. and Sandberg, R. (2014). Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc 9(1): 171-181.
- Shaham, S. (2006). WormBook: Methods in cell biology. In: WormBook (Ed.). The C. elegans Research Community. WormBook.
- Trombetta, J. J., Gennert, D., Lu, D., Satija, R., Shalek, A. K. and Regev, A. (2014). Preparation of single-cell RNA-Seq libraries for next generation sequencing. Curr Protoc Mol Biol 107: 4.22.21-17.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Serra, L., Chang, D., Macchietto, M., Williams, K., Murad, R., Lu, D., Dillman, A. R. and Mortazavi, A. (2018). Adapting the Smart-seq2 Protocol for Robust Single Worm RNA-seq. Bio-protocol 8(4): e2729. DOI: 10.21769/BioProtoc.2729.
Category
Systems Biology > Transcriptomics > RNA-seq
Molecular Biology > RNA > Transcription
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.